
Minimal positional substring cover is a haplotype threading alternative to Li and Stephens model
Abstract
The Li and Stephens (LS) hidden Markov model (HMM) models the process of reconstructing a haplotype as a mosaic copy of haplotypes in a reference panel. For small panels, the probabilistic parameterization of LS enables modeling the uncertainties of such mosaics. However, LS becomes inefficient when sample size is large, because of its linear time complexity. Recently the PBWT, an efficient data structure capturing the local haplotype matching among haplotypes, was proposed to offer a fast method for giving some optimal solution (Viterbi) to the LS HMM. Previously, we introduced the minimal positional substring cover (MPSC) problem as an alternative formulation of LS whose objective is to cover a query haplotype by a minimum number of segments from haplotypes in a reference panel. The MPSC formulation allows the generation of a haplotype threading in time constant to sample size (O(N)). This allows haplotype threading on very large biobank-scale panels on which the LS model is infeasible. Here, we present new results on the solution space of the MPSC. In addition, we derived a number of optimal algorithms for MPSC, including solution enumerations, the length maximal MPSC, and h-MPSC solutions. In doing so, our algorithms reveal the solution space of LS for large panels. We show that our method is informative in terms of revealing the characteristics of biobank-scale data sets and can improve genotype imputation.
Human chromosomes are a mosaic of ancestral chromosomal segments, resulting from accumulated recombination events. By way of transitivity, when querying a chromosomal haplotype against a panel of haplotypes, one can view the query haplotype as a mosaic of identical-by-descent (IBD) segments with haplotypes in the panel. Such mosaics, also called haplotype threadings, are the foundational model for haplotype phasing, genotype imputation, and relatedness inference.
For many years, the state-of-the-art haplotype threading method was the Li and Stephens (LS) model (Li and Stephens 2003). The LS model produces a haplotype threading by using a hidden Markov model (HMM) in which penalties are incurred for mismatches and switching between different haplotypes in adjacent sites in the panel in the threading. When sample size is small, the matching segments, that is, the continuous streak of match with the same haplotype template in the panel, are typically short, and the LS model can be parameterized to achieve a balance between mismatch tolerance and minimizing switching. Indeed, most current phasing and imputation methods are based on the LS model (Das et al. 2016; Loh et al. 2016; Browning et al. 2018, 2021; Delaneau et al. 2019; Rubinacci et al. 2020).
However, when sample size of the panel is large, LS model becomes inefficient, as its time complexity is linear to the size of the panel, O(MN), where M is the number of haplotypes in the panel and N is the number of sites per haplotype. The latest imputation methods based on LS have used PBWT to quickly identify a subset of templates as “surrogate parents” of the query, but they still use LS HMM to sample the threading over the subset (Browning et al. 2018; Rubinacci et al. 2020).
Recent work has attempted to obtain the optimal haplotype threading in the LS model in time sublinear to the size of the panel. Lunter (2019) described “fastLS,” an algorithm that implements the LS model and obtains the optimal haplotype threading through the use of the Burrows–Wheeler transform. His algorithm obtains the optimal haplotype threading orders of magnitude faster than the Viterbi algorithm. Rosen and Paten (2019) also described an algorithm that achieved runtime orders of magnitude faster than the Viterbi algorithm. Their method achieves this using the efficient sparse representation of haplotypes and the lazy evaluation of dynamic programming. These algorithms have also been claimed to have runtime sublinear to the size of the reference panel. However, this claim has only been shown empirically.
In this work, we discuss an alternative formulation of the haplotype threading problem. This is based on the observation that when the size of the panel is large enough, approaching the size of the population, the IBD segments shared between the query and the template haplotypes can be much longer. Also, the error rates on modern large panels are very low (∼0.1%}). Therefore, the problem of haplotype threading for large panels is the LS model at the regime where the mismatch rates and switch rate are very low. In other words, it is of interest to make combinatorial formulations of the problem.
In a previous work, Sanaullah et al. (2022) introduced a new formulation of haplotype threading, the minimal positional substring cover (MPSC). The MPSC problem is, given a query haplotype z and a set of haplotypes X, to find a smallest set of segments of haplotypes in X that covers z. This formulation corresponds to the LS model with zero mismatch rate, and the only objective is to minimize the switch rate. Obviously, the MPSC formulation is too limited as it does not tolerate mismatches. In a sense, MPSC is a more general formulation of haplotype threading. The traditional haplotype threading is a special case of MPSC with nonoverlapping segments. MPSC formulation captures the fact that although the switching of templates indicates some recombination events, the exact breakpoint of the recombination events may be anywhere within the overlapping region between the templates. Furthermore, this combinatorial formulation is theoretically attractive because it can be solved in worst case O(N) time (Sanaullah et al. 2022). That is, given a PBWT of the reference panel, an MPSC haplotype threading of a query haplotype can be performed in time independent to the number of haplotypes in the reference panel. Further, a number of variations of the MPSC formulation can be solved efficiently, including leftmost and rightmost MPSCs, MPSC composed of only set maximal matches, and h-MPSC.
In this work, we aim to expand upon the abilities of the MPSC formulation of haplotype threading greatly. We hope to explore the solution space of MPSCs, solve the length maximal MPSC variation of the MPSC problem, improve upon the previously obtained time complexity for the solution to the h-MPSC problem, and show the usefulness of the MPSC formulation haplotype threading through an imputation benchmark.
Methods
Background
By following the definitions of Sanaullah et al. (2022), a string z with N characters is indexed from zero to N − 1. The first character of z is z[0], and the last is z[N − 1]. The string z[i, j] is the substring of z that starts at character i and ends at character j.
A positional substring of a string z is a three-tuple, (i, j, z), where i and j are nonegative integers, i ≤ j + 1 ≤ |z|, and z is the “source” of the substring. The substring corresponding to the positional substring (i, j, z) is z[i, j]. If i = j + 1, then (i, j, z) corresponds to the empty string, ϵ. A nonempty positional substring (i, j, z) is contained in a string s if 0 ≤ i ≤ j < |s| and s[i, j] = z[i, j]. An empty positional substring (i, i − 1, z) is contained in a string s iff 0 ≤ i ≤ |s|. Two positional substrings, (i, j, s) and (k, l, t), are equal iff i = k, j = l, and s[i, j] = t[k, l]. The length of a positional substring (i, j, s) is j − i + 1.
A positional substring cover, C, of a string z by a set of strings X is a set of positional substrings such that every character of z is contained in a positional substring and every positional substring in the set is present in z and a string in X. The “source,” s, of a positional substring (i, j, s) in a positional substring cover can be any string s s.t. s[i, j] = x[i, j] = z[i, j] for some x ∈ X. The size of a positional substring cover is the number of positional substrings it contains. The length of a positional substring cover is the sum of the lengths of its positional substrings.
π is a projection on tuples. π1(i, j, z) = i, π2(i, j, z) = j, and π3(i, j, z) = z.
PBWT
The algorithms described in this paper require a prebuilt positional Burrows–Wheeler transform (PBWT). The PBWT is a data structure that allows memory efficient representation of a set of binary haplotypes of the same length (Durbin 2014). The PBWT also allows time-efficient outputting of matches between haplotypes in the panel. Here we provide a brief description of the PBWT and its algorithms.
The PBWT stores X, a set of M binary strings of length N. It also stores four arrays of size roughly M by N. The first array is the positional prefix array, a. The positional prefix array has size M × (N + 1); it has N + 1 columns of height M. Column i (a[i]) contains the strings in X sorted by their reversed prefixes of length i. If two strings have the same reversed prefixes of length i, their relative order is the same in a[i] and a[0]. a[i][0] contains the string with the lexicographically smallest reversed prefix of length i, and a[i][N − 1] contains the string with the lexicographically largest reversed prefix of length i. The divergence array, d, is size M × (N + 1). d[i][j] contains the starting position of the longest match between a[i][j] and a[i][j − 1] that ends at position i − 1 (inclusive). If no such match exists, d[i][j] = i. The last two arrays are the u and v arrays, each of size M × N. These arrays keep track of the position a[i][j] would have in a[i + 1] if it had zero and one at index i, respectively. That is, u[i][j] = the position in a[i + 1] that a[i][j] would have if it had a zero at index i. v[i][j] = the position in a[i + 1] that a[i][j] would have if it had a one at index i.
In this paper, we will use a variation of the PBWT that allows an alphabet of arbitrary size. The key difference is that instead of using the u and v arrays to keep track of next position, we use a three-dimensional array, w, of size M × N × |Σ|, where Σ is the alphabet. w[i][j][c] holds the position a[i][j] would have in a[i + 1] if it had c at index i. This variation of the PBWT uses O(MN|Σ|) space instead of the previous O(MN). However, the set maximal match query time complexity remains the same as before (O(N + c), where c is the number of matches found). This variation of the PBWT has been explored by Naseri et al. (2019).
We use Durbin's definitions of local and set maximal matches (Durbin 2014). This description is from Sanaullah et al. (2022). A match between two strings s and t, (i, j, s) = (i, j, t), is locally maximal if it cannot be extended in any direction and still match. (s[i − 1] ≠ t[i − 1] or i = 0) and (s[j + 1] ≠ t[j + 1] or j = N − 1). For a string 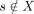 , a match between s and x ∈ X ((i, j, s) = (i, j, x)) is set maximal from s to X if there does not exist a match from s to a string in X that is larger and contains this match.
, a match between s and x ∈ X ((i, j, s) = (i, j, x)) is set maximal from s to X if there does not exist a match from s to a string in X that is larger and contains this match. 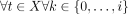
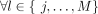 , (k = i and l = j) or t[k, l] ≠ s[k, l].
, (k = i and l = j) or t[k, l] ≠ s[k, l].
Minimal positional substring cover
Sanaullah et al. (2022) formulated the haplotype threading as the MPSC problem. The MPSC problem is, given a query string z and set of strings X, find a positional substring cover of z by X with the smallest size of all positional substring covers of z by X.
Sanaullah et al. (2022) provided an algorithm that outputted an MPSC of z by X in O(N) time given a PBWT of X, where N is the length of z. They also provided algorithms for outputting the leftmost, rightmost, and set maximal match–only MPSCs in O(N) time given a PBWT of X. Last, they provided an algorithm that outputted an h-MPSC of z by X in 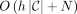 time, where C is the outputted h-MPSC.
time, where C is the outputted h-MPSC.
A leftmost MPSC of z by X is an MPSC in which each ith positional substring starts as early as the ith positional substring of every MPSC of z by X. Similarly, a rightmost MPSC of z by X is an MPSC in which every ith positional substring ends as late as every ith positional substring in an MPSC of z by X. A set maximal match–only MPSC of z by X is an MPSC of z by X such that every positional substring it contains corresponds to a set maximal match from z to X. Last, an h-MPSC of z by X is the smallest positional substring cover of z by X such that all of the positional substrings it contains are contained in h strings in X. By the definitions on positional substring covers, the size of an MPSC is the number of positional substrings it contains, and the length of an MPSC is the sum of the lengths of its positional substrings.
MPSC solution space
Although a MPSC of a query haplotype z by a panel X is an inference for haplotype threading, there may be many possible MPSCs of z by X. The MPSC outputted may not be the most accurate haplotype threading. Therefore, we consider the set of all possible MPSCs of z by X.
We begin the exploration of the solution space of MPSCs of z by X by attempting to bound it. We already have two useful bounds that can be efficiently obtained, namely, the leftmost and rightmost
MPSCs. The starting point of every ith positional substring in an MPSC of z by X must be at least 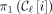 , the starting point of the ith positional substring of
, the starting point of the ith positional substring of 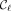 , the leftmost MPSC. Likewise, the ending point of every ith positional substring in an MPSC of z by X is at most
, the leftmost MPSC. Likewise, the ending point of every ith positional substring in an MPSC of z by X is at most 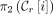 , the ending point of the ith positional substring of
, the ending point of the ith positional substring of 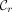 , the rightmost MPSC. Sanaullah et al. (2022) proved the existence of leftmost and rightmost MPSCs for any z and X for which there exists an MPSC of z by X. Formally, an MPSC
, the rightmost MPSC. Sanaullah et al. (2022) proved the existence of leftmost and rightmost MPSCs for any z and X for which there exists an MPSC of z by X. Formally, an MPSC 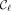 is leftmost if
is leftmost if 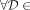 {MPSCs of z by X},
{MPSCs of z by X}, 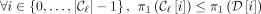 , although an MPSC
, although an MPSC 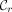 is rightmost if
is rightmost if 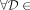 {MPSCs of z by X},
{MPSCs of z by X}, 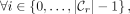
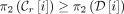 .
.
We have even tighter bounds of the solution space of MPSCs. Sanaullah et al. (2022) showed that if the ith positional substring in a leftmost MPSC of z by X begins at index j, every i − 1th positional substring in an MPSC of z by X contains the index j − 1. Here, we show a similar property for rightmost MPSCs. For the proof, see the Supplemental Material.
Every i + 1th positional substring in any MPSC of z by X contains index 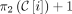 , where C is a rightmost MPSC of z by X.
, where C is a rightmost MPSC of z by X.
For every ith positional substring in a MPSC of z by X, we now have two positions that it is guaranteed to contain: 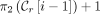 and
and 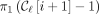 , where
, where 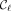 is a leftmost MPSC of z by X, and
is a leftmost MPSC of z by X, and 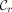 is a rightmost MPSC of z by X. Furthermore, there are MPSCs of z by X where the positions just after and before these positions are not contained in the ith positional substrings (rightmost and leftmost MPSCs of z by X with no overlap, respectively). Therefore, we have the exact range of sites common to all ith positional substrings in MPSCs of z by X. Call it the ith required region. The ith required region is contained in every ith positional substring in an MPSC of z by X. The ith required region is exactly the first site after the i − 1th positional substring in a rightmost MPSC to the last site before the i + 1th positional substring in a leftmost MPSC. For a depiction of required regions and how they are obtained, see Figure 1. The ith required region is defined in Lemma 1. For the proof, see the Supplemental Material.
is a rightmost MPSC of z by X. Furthermore, there are MPSCs of z by X where the positions just after and before these positions are not contained in the ith positional substrings (rightmost and leftmost MPSCs of z by X with no overlap, respectively). Therefore, we have the exact range of sites common to all ith positional substrings in MPSCs of z by X. Call it the ith required region. The ith required region is contained in every ith positional substring in an MPSC of z by X. The ith required region is exactly the first site after the i − 1th positional substring in a rightmost MPSC to the last site before the i + 1th positional substring in a leftmost MPSC. For a depiction of required regions and how they are obtained, see Figure 1. The ith required region is defined in Lemma 1. For the proof, see the Supplemental Material.

Required regions for MPSC of z by X. The ith required region is bounded by the i + 1th positional substring in the leftmost MPSC and the i − 1th positional substring in the rightmost MPSC.
There exists a contiguous nonempty range of sites for every 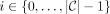 , such that
, such that 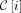 contains this range of sites for all MPSCs C of z by X. Call the largest such range the ith required region. For
contains this range of sites for all MPSCs C of z by X. Call the largest such range the ith required region. For 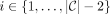 , this range is
, this range is 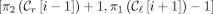 , where
, where 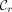 and
and 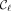 are rightmost and leftmost MPSCs of z by X, respectively. The required region for
are rightmost and leftmost MPSCs of z by X, respectively. The required region for 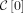 is
is 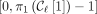 ; for
; for 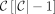 ,
, 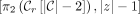 .
.
MPSC graph
We attempt to represent the solution space of MPSCs as a graph. Positional substrings are vertices in the graph, and edges occur between positional substrings that are adjacent or overlapping. Consider the naive graph G = (V, E), where the set of nodes V is the set of positional substrings that are contained in both z and X with z as the source string. That is, v = (i, j, z) ∈ V ⇔ (i, j, z) is present in z and x ∈ X. There is an edge between two positional substrings u and v if u starts before v and u and v are adjacent or overlapping (π1(u) ≤ π1(v) and π2(u) ≥ π1(v) − 1). In this graph, all shortest paths from ( − 1, 0, z) to (N − 1, N, z) correspond to MPSCs of z by X. Furthermore, all MPSCs are represented by a shortest path from ( − 1, 0, z) to (N − 1, N, z). However, there are possibly O(N2) nodes and O(N4) edges in this graph, so a shortest path–finding algorithm would run in O(N4) time. Therefore, we attempt to simplify the graph. We begin with the following observations. Their proofs can be found in the Supplemental Material.
Every nonempty positional substring m = (i, j, z) contained in z and a string in X is contained in a set maximal match from z to X.
For any two set maximal matches from z to X, m = (i, j, s) and n = (k, l, t), i = k ⇔ j = l.
Therefore, we consider a graph in which the set of nodes is the set of set maximal match positions from z to X. Every possible nonempty match is contained in at least one of these nodes. There is an edge between nodes u and v with the same conditions as before: π1(u) ≤ π2(v) and π2(u) ≥ π1(v) − 1. Call the node with index 0 the source node, s. Call the node with index N − 1 the sink node, t. s and t are unique by Claim 3. There is a one-to-one correspondence between the shortest paths from s to t in this graph and MPSCs of z by X composed of set maximal matches. Furthermore, there is a one-to-one correspondence between the shortest paths in the graph from s to t and MPSCs of z by X. The ith positional substring in the MPSC is contained in the ith node in the path to which it maps. By Claim 3, the number of nodes in this graph is O(S)⊆ O(N). However, the number of edges in this graph may be O(S2); therefore, a shortest path–finding algorithm on this graph would still take O(S2)⊆ O(N2) time. We simplify the graph of the solution space again, this time considering the ith required regions.
We will construct a graph in which the set of nodes is the set of set maximal match positions from z to X. However, we will also consider the information gained from the ith required regions. For two nodes u, v, there is an edge from u to v in the graph if the previous requirements are fulfilled, u contains the complete ith required region, and v contains the complete i + 1th required region. The following property is useful in this construction. A proof can be found in the Supplemental Material.
For every positional substring that is contained in z and a string in X, if it contains a full required region, it does not contain sites from any other required region.
If u contains the ith required region and v contains the i + 1th required region, then u starts before v. Therefore, there is an edge from u to v in this graph iff u contains the ith required region, v contains the i + 1th required region, and u and v are adjacent or overlapping. For a depiction of this graph for z and X, see Figure 2. All paths from s to t in this graph correspond to a MPSC of z by X composed of only set maximal matches. Furthermore, all MPSCs of z by X correspond to a path from s to t in the graph, where the ith positional substring is contained in the ith set maximal match position in the path.
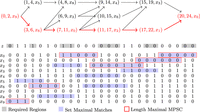
Constructed MPSC graph of z by X. In gray are the required regions for z and X. Highlighted in blue are the set maximal matches from z to X. Highlighted in red is the length maximal MPSC of z by X.
This graph can be constructed very efficiently. As before, there are O(S) ⊆ O(N) nodes in the graph. These nodes can be found in O(N) time. All ith required regions can be obtained in O(N) time. This is performed by first calculating a leftmost MPSC and a rightmost MPSC of z by X in O(N) time (Sanaullah et al. 2022). Then, each ith required region can be obtained in constant time by Lemma 1. The set maximal match positions can be obtained in O(N) time (Sanaullah et al. 2021). The last step is the creation of the edges between nodes. Although there may be O(S2) ⊆ O(N2) edges in this graph, we avoid the explicit construction of all of them by exploiting the following property. Call Ri the set of nodes that contained the ith required region. Call Ri[j] the node in Ri with the jth smallest starting position, j ∈ {0, …, |Ri| − 1}. Then,
The set of nodes Ri[j] has an edge to is a subset of the set of nodes Ri[j + 1] has an edge to.
See the proof of this Lemma in the Supplemental Material. Therefore, during the construction of our graph, we do not directly construct all O(N2) edges. For every node Ri[j + 1], we only construct edges to nodes that Ri[j] does not have an edge to. Although, there may be O(N2) edges in the complete graph, we fully encode them with O(S)⊆ O(N) edges. With these edges in place and the property in Lemma 2 in mind, we have constructed a graph that fully describes the solution space of MPSCs of z by X in O(N) time. The combination of this graph and the PBWT supports many efficient queries through the exploitation of Lemma 2. This includes the counting of the number of set maximal match–only MPSCs of z by X, the counting of the number of MPSCs a positional substring is an element of, and the counting of length maximal MPSCs, all in O(N) time. Furthermore, each of these sets of MPSCs can be enumerated in O(N + Sc) time, where Sc is the number of MPSCs outputted. These problems are very useful for the efficient and accurate imputation and phasing of haplotypes.
Length maximal MPSC
The length maximal MPSC problem is, given a set X of M strings of and a string z, find a MPSC of z by X that has the largest length out of all MPSCs of z by X. Note that by claim 2 of Sanaullah et al. (2022), the length of any MPSC of z by X is less than or equal to 2N. Lemma 3 is proven in the Supplemental Material.
Every positional substring in any length maximal MPSC of z by X is a set maximal match from z to X.
Considering Lemma 3, we begin with the MPSC graph in Figure 2 and modify it in the following fashion. For every edge (u, v), we assign it a weight of the length of u. In this directed acyclic graph, every directed path from s to t still corresponds to a MPSC of z by X composed of only set maximal matches. As before, every MPSC of z by X composed of only set maximal matches corresponds to a directed path in the graph from s to t; there is a one-to-one correspondence. Therefore, the length maximal MPSC of z by X corresponds to a path in the graph by Lemma 3. In fact, there is a one-to-one correspondence between longest directed paths in the graph from s to t and length maximal MPSCs of z by X (where the length of a path is the sum of the weights of its edges).
Therefore, we can obtain a length maximal MPSC by constructing this graph and finding a longest path in it from s to t. This is easy given a PBWT of X. We begin by finding leftmost and rightmost MPSCs of z by X. This can be performed in O(N) given a PBWT of X (where N is |z|). This was shown by Sanaullah et al. (2022). We can then obtain all required regions in 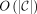 time, where
time, where 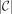 is the size of an MPSC of z by X. The calculation of each required region takes constant time. They can be calculated simply using the leftmost and rightmost
MPSCs and Lemma 1. Next, we output all set maximal match positions and one string in X that each contain each set maximal match in O(N) time. This can be performed with a simple modification of the set maximal match query algorithm by Sanaullah et al. (2021). We maintain the sorted order of the set maximal match positions provided by the query algorithm. Lastly, we keep track of
which set maximal matches contain which complete required regions. This can be performed in a simple sweeping fashion in O(S) time owing to Claim 4. So far, the algorithm has taken O(N) time.
is the size of an MPSC of z by X. The calculation of each required region takes constant time. They can be calculated simply using the leftmost and rightmost
MPSCs and Lemma 1. Next, we output all set maximal match positions and one string in X that each contain each set maximal match in O(N) time. This can be performed with a simple modification of the set maximal match query algorithm by Sanaullah et al. (2021). We maintain the sorted order of the set maximal match positions provided by the query algorithm. Lastly, we keep track of
which set maximal matches contain which complete required regions. This can be performed in a simple sweeping fashion in O(S) time owing to Claim 4. So far, the algorithm has taken O(N) time.
We now have all the required information to build the graph. We create the nodes of the graph in O(S) time, where S is the number of set maximal match positions, S ≤ N. We create the edges leaving s and entering t in O(S) time. Last, for every set maximal match position, if it fully contains a required region, we create an edge from it to nodes whose positional substrings fully contain the next required region and are adjacent or overlapping. This last step of construction of the graph takes O(S2)⊆ O(N2) time. We can find the longest path in this directed acyclic graph in O(V + E) = O(S + E)⊆ O(S2)⊆ O(N2) time. This can be performed using a shortest path–finding algorithm on the graph with the weights negated. The overall time complexity for this method of outputting a length maximal MPSC is O(N + S2). However, we have yet to exploit the property of the graph shown in Lemma 2.
We will now show that the longest path in the graph can be obtained in time linear to the number of nodes in the graph, O(S), through the exploitation of Lemma 2. Therefore, a length maximal MPSC of z by X can be outputted in O(N) time. Call the set of set maximal match positions (or the corresponding nodes) containing the ith required region Ri. The key idea is the following. Given the longest paths from all nodes in Ri+1 to t, the longest paths from all nodes in Ri to t can be obtained in O(|Ri+1| + |Ri|) time. This is despite the fact that there may be |Ri+1| × |Ri| edges between these nodes. We denote the set maximal match of Ri with the jth smallest starting position as Ri[j]. That is, Ri[0] is the set maximal match in Ri with the smallest starting position, Ri[1] has the next smallest starting position, etc. The calculation of the longest paths from Ri to t in linear time depends on Lemma 2. The set of nodes Ri[j] has an edge to is a subset of the set of nodes Ri[j + 1] has an edge to. Given this property, we can calculate the longest paths in a straightforward fashion that evaluates the longest path from each node in Ri+1 to t at most once. This is performed by calculating the longest paths of nodes in order of starting position of the corresponding set maximal match, from least to greatest. For a depiction of this process, see Figure 3. The pseudocode of this process is provided in Algorithm 8 in the Supplemental Material.
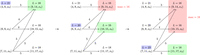
Obtaining the longest paths of the set maximal matches in Ri from the longest paths of the set maximal matches in Ri+1 in O(|Ri| + |Ri+1|) time. Subgraph of Figure 2, i = 2.
After the lengths of the longest paths from each node to t is calculated, the longest path from s to t can be calculated in a simple linear backtracking step. Start with s. For each ith required region for 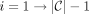 , take the first node with length of the longest path to t equal to the length of the longest path to t of the i − 1th node in the path so far minus the length of the substring of the i − 1th node in the path so far. For the pseudocode of this process, see Algorithm 6 in the Supplemental Material. With this process for outputting the longest path in the graph from s to t in O(S) time, we can find the longest path in the graph without explicitly constructing the graph. Therefore, given a PBWT of X, we output a length maximal MPSC of z by X in O(N) time. The pseudocode of this algorithm can be seen in Algorithm 1 in the Supplemental Material.
, take the first node with length of the longest path to t equal to the length of the longest path to t of the i − 1th node in the path so far minus the length of the substring of the i − 1th node in the path so far. For the pseudocode of this process, see Algorithm 6 in the Supplemental Material. With this process for outputting the longest path in the graph from s to t in O(S) time, we can find the longest path in the graph without explicitly constructing the graph. Therefore, given a PBWT of X, we output a length maximal MPSC of z by X in O(N) time. The pseudocode of this algorithm can be seen in Algorithm 1 in the Supplemental Material.
h-MPSC
An h-MPSC of a query string z by a set of strings. X is a smallest set of positional substrings that forms a positional substring cover of z by X and are each contained in at least h strings in X. An MPSC of z by X is an h-MPSC of z by X where h = 1. Sanaullah et al. (2022) provided an algorithm for outputting an h-MPSC, C, of z by X in 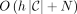 time given a PBWT of X. We suspect their algorithm outputs a “leftmost” h-MPSC. Here we describe an algorithm that improves on the previous h-MPSC algorithm running time. It outputs an h-MPSC of z by X in O(N) time. We believe the h-MPSC it outputs is “rightmost.”
time given a PBWT of X. We suspect their algorithm outputs a “leftmost” h-MPSC. Here we describe an algorithm that improves on the previous h-MPSC algorithm running time. It outputs an h-MPSC of z by X in O(N) time. We believe the h-MPSC it outputs is “rightmost.”
The original h-MPSC algorithm traversed the PBWT from index N to zero. Here, we traverse in the opposite direction, from zero to N. We begin at index zero and keep track of the block of strings that match with z on the range [i, j). The block is denoted by [f, g), where f is the first string in the block in the prefix sorting and g is the first string not in the block after f. If f = g, the block is empty. At site zero, f and g are initialized to f = 0, g = M. i and j are initialized to zero. The f and g blocks are updated in constant time per site using the w array between sites. Once the block at site j + 1 has fewer than h strings, the positional substring of the previous block (i, j − 1, z) is added to the h-MPSC. This is repeated until all sites are covered by a positional substring contained in h strings in X. This algorithm runs in O(N) time because the is traversed from index zero to N, and each site takes constant time to update the f and g block. Furthermore, the addition of a positional substring to the h-MPSC can happen at most once per site and takes constant time. A proof that the positional substring cover is an h-MPSC can be seen through a fairly simple modification of Lemma 2 in (Sanaullah et al. 2022). The pseudocode of this algorithm can be seen in Algorithm 9 in the Supplemental Material.
Results
Haplotype threading properties
We explored the properties of haplotype threadings of the MPSC formulation. The data set used was the UK Biobank (UKB) (Bycroft et al. 2018). The UKB has 974,818 haplotypes and around 700,000 sites (microarray). We used Chromosome 21, which has 9793 sites. For each haplotype in the UKB, we run our method to identify an MPSC of it using all other haplotypes in the UKB as the reference panel. We also evaluate a rudimentary method of handling mismatches in the MPSC formulation of haplotype threading by using P-smoother (Yue et al. 2022). P-smoother is a method for smoothing out sporadic mismatches in otherwise well-matched PBWT blocks to attempt to remove very rare mutations and genotyping errors from the British-only UKB panel. P-smoother was run on the default settings and flipped the alleles of ∼1.4% of the data. The smoothed panel is expected to tolerate mismatches, resulting in longer match segments and smaller MPSC sizes.
We found that the MPSC segment count of an individual has an overall distribution because not everyone's relatives are sampled evenly. However, the mode of MPSC segment count is inversely correlated with the number of templates with closely related ethnic background (Fig. 4). This behavior is even more clear when we run the experiment with varied sample sizes (Fig. 5). We plot the number of segments in each haplotype threading by self-reported ethnic background in Figure 4. The x-axis is the number of segments in the haplotype threading, and the y-axis is the frequency of that number within each self-reported ethnic background. The plotted ethnic backgrounds are the four most commonly reported ethnic backgrounds in the UKB: British, Irish, Indian, and Caribbean (860,584, 25,436, 11,320, and 8598 haplotypes, respectively). A haplotype is classified as one of these ethnic backgrounds if it was the first ethnic background the individual who owns the haplotype reported themselves as. We also plot MPSC size distributions for random subsets of the UKB of varying sizes. No threading was found for (7.3%, 0.32%, 0.003%, and 0%) of the haplotypes for M = (1000, 10,000, 100,000, and 860,022), respectively. M = 10 and 100 were also tried, but no haplotype threadings were found in either panel. These haplotypes are left out of the frequency calculation.
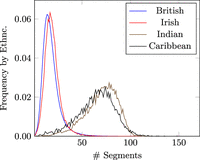
Frequency within self-reported ethnic background of the number of segments in haplotype threading of haplotypes in UK Biobank (UKB). Haplotype threading is generated with a reference panel as the rest of the UKB. Frequency is plotted by self-reported ethnic background (abbreviated “Ethnc.” on y-axis label) for the four most common in the UKB: British, Irish, Indian, and Caribbean, with 860,584, 25,436, 11,320, and 8598 haplotypes, respectively.
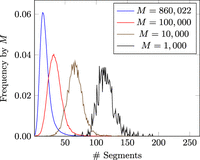
Frequency by panel size of the number of segments in British-only haplotype threading of haplotypes in the UKB . Haplotype threading is generated with a reference panel random subset of the British-only haplotypes in the UKB. Plotted by panel size for M ∈ {1000, 10,000, 100,000, 860,022}. Note that frequencies exclude haplotypes for which no threading was found.
Another property we consider is the MPSC solution space. This is the number of possible coverings of the query with equally small number of switches, which would be all considered a Viterbi solution for the LS model. We plot the count of set maximal match–only MPSCs for each haplotype in the British-only UKB data set (860,022 haplotypes) in Figure 6 in ascending order. The median solution space size was 120. The 90th and 99th percentile solution space sizes were 72,756 and 3.4 × 109, respectively. As expected, the median solution space size of the P-smoothed panel was 40. The fact that the number of solutions in MPSC solution space is small validated that UKB belongs to the regime where MPSC could be a much more efficient alternative for the fully parameterized LS model.
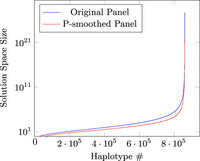
The number of set maximal match–only MPSCs per haplotype in British-only UKB panel in ascending order.
The last property of the MPSC formulation of haplotype threading we explore is the distributions of lengths of the length
maximal MPSCs. Note this length l is N ≤ l ≤ 2N, and thus, the size of the overlap region, l − N or 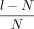 , provides one way of quantifying the potential computations (much more than a linear factor!) saved by MPSC compared with
enumerating solutions of standard LS. We plot this length distribution of length maximal MPSCs for British-only haplotypes
in Figure 7. P-smoother results in a smaller length maximal MPSC length. The length maximal MPSC provides about at average 1.21× (1.20×
after smoothing) coverage of the chromosome, indicating ∼20% of the genome belongs to the overlap regions of length maximal
MPSC, where location of recombination breakpoints cannot be unequivocally determined.
, provides one way of quantifying the potential computations (much more than a linear factor!) saved by MPSC compared with
enumerating solutions of standard LS. We plot this length distribution of length maximal MPSCs for British-only haplotypes
in Figure 7. P-smoother results in a smaller length maximal MPSC length. The length maximal MPSC provides about at average 1.21× (1.20×
after smoothing) coverage of the chromosome, indicating ∼20% of the genome belongs to the overlap regions of length maximal
MPSC, where location of recombination breakpoints cannot be unequivocally determined.
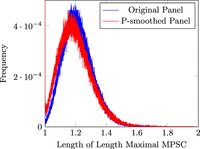
The distributions of lengths of length maximal MPSCs for all British-only haplotypes in the UKB. Length is represented as a ratio of genome length. The min ratio of an MPSC is one, and the max ratio is two. The min and max ratios observed are one and 1.974.
Run time
We measure the run time of obtaining the length maximal MPSC for various M and N. Note that the run time of obtaining a length maximal MPSC is strictly larger than the run time of obtaining an MPSC, a leftmost MPSC, a rightmost MPSC, and a set maximal match–only MPSC. This is because the first three algorithms are subroutines of the length maximal MPSC algorithm. The run time of obtaining a length maximal MPSC is strictly larger than the run time of obtaining a set maximal match–only MPSC because a length maximal MPSC is a set maximal match–only MPSC. The results of these tests can be seen in Supplemental Figure S1. We provide a brief theoretical comparison of the runtime of sublinear-LS (Rosen and Paten 2019) in the Supplemental Material.
Imputation benchmark
We implemented an imputation benchmark to show the usefulness of the MPSC formulation of haplotype threading. The imputation algorithm is naive outside of the haplotype threading: Given a haplotype threading of the haplotype to be imputed at a site, obtain the positional substrings adjacent to it in the haplotype threading. For every haplotype that contains one of these positional substrings, vote once per positional substring it contains toward the allele this haplotype has at the site to be imputed. Impute an allele if the votes are unanimous. We also evaluate the use of a P-smoothed panel. In this method, the haplotype threading is generated using the P-smoothed panel and query haplotype. Votes are counted for haplotypes who have the positional substring in the P-smoothed panel. However, allele votes are counted using the original panel.
This imputation method generalizes to all of the variations of MPSC haplotype threading; therefore, we test it on many of the MPSC variations, including length maximal MPSC, voting by set maximal MPSC solution space (SM MPSC SS), and the h-MPSC with various choices of h. The imputation benchmark was performed in the following fashion: Select 1000 random haplotypes from the UKB British-only data set (Chromosome 21). Remove a random 8814 sites (90%) from these haplotypes. Impute these haplotypes using the rest of the UKB British-only haplotypes (M = 859,022) as the reference panel using the above-mentioned MPSC-based imputation methods and Beagle (version 5.4), a state-of-the-art imputation method (Browning et al. 2018).
As shown in Table 1, all tested MPSC-based imputation methods have an imputation accuracy comparable to Beagle. Although MPSC-based methods do not cover all sites, the sites they covered are mostly “easier” sites as Beagle also has higher accuracies on those sites than the average of all sites. Over the covered sites, MSPC-based methods have roughly the same accuracies compared to Beagle. Importantly, in a few cases, MPSC-based methods are more accurate than Beagle on the sites imputed by the MPSC imputation method. This is especially the case for the SM MPSC SS–based imputation method, which is more accurate than Beagle on the imputed sites when either the original panel or the P-smoothed panel is used for imputation. This is useful because one can immediately improve the current Beagle results by repainting the Beagle imputation results with MPSC results where MPSC methods cover. Our results also offer a way of studying behaviors of LS-based methods. It seems when we increase the h in h-MPSC, the coverage of MPSC is gravitated toward “easier” regions where both Beagle and MPSC methods have high accuracy. Thus MPSC's h offers a measure of imputation confidence. Finally, the P-smoothed panel increases the power and accuracy of every MPSC threading imputation method.
Imputation performance in percentages on a random 1000 British-only UKB haplotypes
Discussion
In this paper, we have advanced the MPSC formulation of haplotype threading. We introduced the MPSC graph, a method of representing the solution space of MPSC haplotype threadings of a haplotype that allows many efficient algorithms. These include the enumeration of all MPSCs and of set maximal match–only MPSCs in optimal time. It also allows the counting of the number of MPSCs a particular positional substring is part of in O(N) time. Furthermore, we introduced a new MPSC formulation, the length maximal MPSC, that maximizes the sum of the lengths of the segments of the haplotype threading over all MPSC haplotype threadings. We provided an algorithm that outputs a length maximal MPSC of z by a reference panel X in O(N) time given a PBWT of X, linear to the number of sites and independent to the number of haplotypes in the panel. We also provided an improved algorithm for the h-MPSC problem, where each segment in the threading must be contained in h haplotypes in the reference panel. Table 2 summarizes the major algorithmic contributions of this paper.
Summary of algorithms on haplotype threading
Beyond algorithmic contributions, our algorithmic developments established the theoretical basis for linking PBWT and LS-style haplotype threading. When using these algorithms for analyzing properties of MPSC haplotype threadings for the UKB data set, we show the usefulness of the MPSC haplotype threadings through an imputation benchmark. We showed that, although the simple MPSC-based imputation does not impute all sites, when it does, the accuracy may be higher than the state-of-the-art imputation method Beagle. Especially, the variations of MPSC algorithms that offer solution space, length maximal, and high h (≥16) h-MPSC can outperform Beagle. In addition, our imputation results verified that P-smoother can be leveraged to soften the mismatch-intolerant MPSC solutions and make MPSC more robust to real data. We believe further developments based on our sets of algorithms will empower practical applications based on LS such as phasing and imputation.
Software availability
The source code is available at genome.ucf.edu/MPSC and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Institutes of Health grants R01 HG010086 and R56 HG011509. This research has been conducted using the UK Biobank Resource under application number 24247.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277673.123.
- Received January 6, 2023.
- Accepted June 6, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








