
Efficient and accurate KIR and HLA genotyping with massively parallel sequencing data
- 1Department of Data Science, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, USA;
- 2Department of Biomedical Informatics, Harvard Medical School, Boston, Massachusetts 02115, USA;
- 3Lyda Hill Department of Bioinformatics, University of Texas Southwestern Medical Center, Dallas, Texas 75390, USA
Abstract
Killer cell immunoglobulin like receptor (KIR) genes and human leukocyte antigen (HLA) genes play important roles in innate and adaptive immunity. They are highly polymorphic and cannot be genotyped with standard variant calling pipelines. Compared with HLA genes, many KIR genes are similar to each other in sequences and may be absent in the chromosomes. Therefore, although many tools have been developed to genotype HLA genes using common sequencing data, none of them work for KIR genes. Even specialized KIR genotypers could not resolve all the KIR genes. Here we describe T1K, a novel computational method for the efficient and accurate inference of KIR or HLA alleles from RNA-seq, whole-genome sequencing, or whole-exome sequencing data. T1K jointly considers alleles across all genotyped genes, so it can reliably identify present genes and distinguish homologous genes, including the challenging KIR2DL5A/KIR2DL5B genes. This model also benefits HLA genotyping, where T1K achieves high accuracy in benchmarks. Moreover, T1K can call novel single-nucleotide variants and process single-cell data. Applying T1K to tumor single-cell RNA-seq data, we found that KIR2DL4 expression was enriched in tumor-specific CD8+ T cells. T1K may open the opportunity for HLA and KIR genotyping across various sequencing applications.
Polymorphisms in immune receptor genes diversify the immune response, which strengthens the resilience of a population to diseases. In particular, the major histocompatibility complex (MHC) encoded by the highly polymorphic human leukocyte antigen (HLA) genes can present various peptides depending on the personal HLA sequences. The peptides on MHC could trigger different immune responses, thus affecting the severity of a disease like SARS-CoV-2 infections (Migliorini et al. 2021) in a person. Besides HLA genes, the killer cell immunoglobulin like receptor (KIR) gene family residing on 19q13.4 is also highly polymorphic and can modulate the activity of natural killer (NK) cells and T cells (Vilches and Parham 2002). There are 17 KIRs in humans, including eight inhibitory KIRs (KIR2DL1, KIR2DL2, KIR2DL3, KIR2DL5A, KIR2DL5B, KIR3DL1, KIR3DL2, and KIR3DL3), seven activating KIRs (KIR2DS1, KIR2DS2, KIR2DS3, KIR2DS4, KIR2DS5, KIR3DS1, and KIR2DL4), and two pseudogenes (KIR2DP1 and KIR3DP1). Regarding the regulation function, KIR2DL4 is special in that it exerts the activating signal and also has the potential for inhibition (Faure and Long 2002). Although most of the KIRs interact with MHC class I molecules, the KIR ligand space could be broad, and this involves KIRs in various immune-regulation mechanisms. For example, one immune-evasion mechanism in several tumor types is to up-regulate HHLA2, which binds with KIR3DL3 to inhibit T cell and NK cell activity (Bhatt et al. 2021). In summary, the identification of the alleles, or genotyping, of these highly polymorphic genes in a person can lead to a better understanding of infectious diseases (Walter and Ansari 2015), vaccination (Bolze et al. 2022), organ transplantation (Ruggeri et al. 2002; Zamir et al. 2022), autoimmune diseases (Li et al. 2022), and cancer (Purdy and Campbell 2009; Naranbhai et al. 2022).
Because of the importance of these polymorphic genes, researchers created the Immuno Polymorphism Database (IPD) to curate the HLA and KIR allele sequences (IPD-IMGT/HLA and IPD-KIR, respectively) (Robinson et al. 2020). IPD-IMGT/HLA is the foundation for numerous computational methods, such as seq2HLA (Boegel et al. 2012), OptiType (Szolek et al. 2014), PolySolver (Shukla et al. 2015), HLA-HD (Kawaguchi et al. 2017), Kourami (Lee and Kingsford 2018), HISAT-genotype (Kim et al. 2019), HLA*LA (Dilthey et al. 2019), and arcasHLA (Orenbuch et al. 2020), that can infer HLA alleles from RNA-seq, whole-genome sequencing (WGS), or whole-exome sequencing (WES) data. However, many of the HLA genotypers have hardwired the HLA information in the program or require specialized reference sequences, so they could not be directly applied to KIR genotyping. Besides, KIR genes have unique biological features that make the sequence analysis challenging. First, KIR genes can be lost on a chromosome except for the four framework KIR genes (KIR2DL4, KIR3DL2, KIR3DL3, and KIR3DP1) (Pende et al. 2019), whereas the HLA class I (HLA-A, HLA-B, HLA-C) and HLA class II (including HLA-DPB1, HLA-DQB1, HLA-DRB1) genes are expected to be present. Second, some KIR genes are highly similar to each other, such as KIR3DL1 and KIR3DS1, and HLA genes are more distinct (Supplemental Fig. S1). As a result, the KIR genotyper PING (Norman et al. 2016; Marin et al. 2021) could not disentangle all the KIR genes during allele predictions, and it is designed for the KIR-targeted amplified data. Other methods, such as KIR*IMP (Vukcevic et al. 2015) and KPI (Roe and Kuang 2020), focus on gene-level analysis rather than delving into allelic variations. Motivated by the biological relationship between KIRs and HLAs and the limitations in existing genotyping methods, we developed a flexible and user-friendly computational method, The ONE genotyper for Kir and hla (T1K), to accurately genotype KIR and HLA genes of a sample from the genomic or RNA sequencing data.
KIR genes are selectively expressed in T cells, including CD8+ T cells (Björkström et al. 2012). The T cell is the central component in adaptive immunity, and CD8+ T cells can recognize antigens presented on MHC class I and eliminate corresponding cells such as cancer cells. Because KIR can regulate these important CD8+ T cells, it is fundamental to understand the KIR expression patterns. Therefore, we used public single-cell RNA-seq (scRNA-seq) data (Simoni et al. 2018) of the tumor-infiltrating CD8+ T cells to check whether certain KIR alleles could be related to tumor immunity.
Results
Overview of the method
The principle of T1K is to find abundant alleles and genes based on the read alignments (Fig. 1A). A similar strategy is adopted in methods like HISAT-genotype (Kim et al. 2019) and arcasHLA (Orenbuch et al. 2020). The allele sequences can be obtained from the IPD or a custom database. T1K first extracts candidate reads from the raw data FASTQ files or an alignment BAM file. T1K computes the abundance of all the input alleles simultaneously using the weighted expectation-maximization (EM) algorithm (Dempster et al. 1977) to maximize the likelihood of read alignments to the reference alleles. By modeling all the alleles together, T1K can handle the reads that are mapped to multiple highly similar KIR genes. T1K reports the allele at the allele series level (three digits for KIR and six digits for HLA by default), so it will sum the abundances of each allele within the same allele series. For simplicity, we will continue using the term allele for the genotyping results. If there are more than two valid alleles for a gene, T1K picks the pair of alleles that maximizes the total number of reads that can be aligned to all the selected alleles. Therefore, T1K reports, at most, two alleles per gene, that is, no more than 34 alleles when genotyping KIRs. T1K then applies a Poisson model to calculate the quality score for each called allele to further filter the false alleles. In addition to genotyping each sample, T1K provides postprocessing methods to extend the genotyping results. These include novel single-nucleotide polymorphism (SNP) detection on representative alleles and the report of single-cell-level allele abundances.
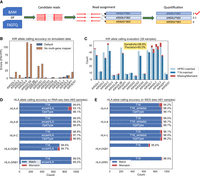
Evaluation of T1K. (A) Overview of the T1K workflow. (B) The KIR allele prediction accuracy of T1K and T1K with no multiple-gene mapped reads (mimicking arcasHLA) on simulated reads from KIR mRNAs. (FN) False negative; (FP) false positive. (C) The KIR allele prediction accuracy of T1K on WGS. (HPRC matched) The alleles inferred from the HPRC phased genome that can be found in T1K predictions; (T1K matched) the alleles predicted by T1K that can be found on HPRC genomes. (D) The HLA allele prediction accuracy of T1K, arcasHLA, and OptiType on RNA-seq data validated with 1kPG annotation. (E) The HLA allele prediction accuracy of T1K and OptiType on WES data validated with 1kPG annotation. With T1K_whitelist, T1K only reports alleles present in an allele whitelist coded in OptiType's Python script.
Performance of KIR genotyping on simulated data
We examined the KIR genotyping accuracy of T1K with 1000 simulated KIR-specific RNA-seq data. There are two types of errors, where false positive (FP) means T1K falsely reports an allele not in the ground truth, and false negative (FN) means the method misses a true allele. T1K made 33 errors (FN + FP) for the 16,038 alleles across all the simulated samples. One of the key ideas in T1K is to estimate the abundances of all the alleles together to include multiple-gene mapped reads. To test the benefit of this strategy, we removed the reads assigned to multiple genes, which is a filter adopted in arcasHLA. After the filtering, this arcasHLA-like strategy made 383 errors, supporting the importance of considering all the alleles simultaneously (Fig. 1B). In this and following evaluations, we ignored alleles with a quality score of zero in T1K.
To handle absent KIR genes or homozygous genes, T1K filters an allele if its abundance is lower than a user-specified fraction of the abundance of another allele for this gene. If the threshold is set too low, then T1K will have too many candidate alleles to have robust genotyping results. If the filter threshold is too high, it will lose true alleles. We evaluated the impact of different thresholds on allele prediction accuracy. T1K produced highly accurate predictions in a wide range of threshold settings between 0.1 and 0.25, suggesting that T1K is robust to the selection of this parameter (Supplemental Fig. S2). The default threshold is set at 0.15 as this setting resulted in the best accuracy.
Performance of KIR genotyping on real data
We investigated T1K's accuracy of KIR genotyping on real data. We compared T1K against the experimentally validated KIR alleles from 1000 Genomes Project samples (1KGP) (The 1000 Genomes Project Consortium 2012), which genotyped KIR3DL1/KIR3DS1 and KIR3DL2 (Norman et al. 2016). Because this annotation did not distinguish KIR3DL1 and KIR3DS1, we focused on the comparison of KIR3DL2. We initially ran T1K on both RNA-seq and WES data (Supplemental Table S1). However, KIR genes were expressed only in a subset of immune cells. Most of the RNA-seq samples (444 out of the 463) only had fewer than two KIR genes with a positive quality score allele. We thus ignored the RNA-seq data owing to lack of information. We investigated T1K's accuracy of KIR3DL2 using WES data. The KIR3DL2 annotation was at the full digit level, so we converted the annotated alleles to the first three digits. Furthermore, because KIR3DL2 is the framework gene, they should be on both chromosomes. Hence we regarded the single-allele (homozygous) prediction of T1K as one allele showing up twice. After this, both the T1K and the 1KGP KIR3DL2 annotations had a pair of alleles per sample. For the matching criteria of a sample, we considered alleles as a pair to avoid overcounting the matches of the homozygous case. For example, when T1K predicted the single-allele KIR3DL2*001 for a sample and the annotation was KIR3DL2*001/KIR3DL2*002, we counted it as one match and one mismatch. Based on these criteria, there was no need to distinguish between sensitivity and precision, which both equal the fraction of matched alleles. In the KIR3DL2 annotation, there were alleles without available genomic sequences in the IPD-KIR database, such as KIR3DL2*003, so we excluded those samples from the analysis. For the remaining 202 WES samples, we evaluated T1K's accuracy of the 404 KIR3DL2 alleles, where T1K achieved 99.0% (400/404) accuracy.
The previous analysis only interrogated a subset of KIR2DL4 alleles, so we used 26 samples from the Human Pangenome Reference Consortium (HPRC) (Supplemental Table S2) to conduct a comprehensive evaluation of all the KIR genes. These samples have both phased reference genomes and Illumina whole-genome sequencing short reads. To identify the KIR alleles on the phased genomes, we aligned the IPD-KIR allele genome sequences to each genome. The alignment could be used to identify KIR gene regions and to select the alleles with minimal differences in the exonic region as the ground truth (Methods). We then used T1K to predict alleles from the Illumina short reads, and compared the results against the ground truth. T1K achieved high sensitivity and identified 96.8% of alleles found on the phased reference genomes (Fig. 1C). While being highly sensitive, T1K also had high precision such that ∼95.0% of the called alleles can be validated in the phased genomes. The strategy of incorporating secondary read assignments with worse alignments in the intronic region improved the precision by 2.6% in this evaluation.
The phased reference genomes also provide the ground truth of novel genomics variations. We evaluated the unambiguous SNPs in the correct alleles predicted by T1K and found that 95.0% of these SNPs inferred by T1K can be validated on the phased genome (Supplemental Fig. S3). T1K could not resolve the ambiguous SNPs and did not try to compute the SNPs in the case of hybrid genes, so in such cases, the sensitivity was low. In this evaluation, the unambiguous SNPs of T1K constituted ∼41.2% of the total novel variations found on the phased genome.
PING (Norman et al. 2016; Marin et al. 2021) is another pipeline specialized in KIR genotyping. However, this pipeline was designed for targeted sequencing data and reported errors on the above WGS data. PING provided five real data sets in the package as test examples on which we examined the results from T1K and PING. On its own data, PING could not completely distinguish KIR2DL2/KIR2DL3, KIR2DS3/KIR2DS5, KIR3DL1/KIR3DS1, and KIR2DL5A/KIR2DL5B, whereas T1K provides full resolution at the gene level. When inspecting the remaining well-separated KIR genes, 95.5% of T1K's genotyping results were supported by PING (Supplemental Table S3), and PING had five unsolved cases. With four threads (default in PING), T1K took 9 min to genotype all the samples, whereas PING spent 235 min. Even though PING reported more information such as gene copy numbers, PING's longer running time could still be mainly because its allele calling method was slower than T1K's.
Performance of HLA genotyping on real data
We compared T1K against other HLA genotypers to further evaluate T1K on real RNA-seq and WES data. Because arcasHLA and OptiType were reported as the most effective methods in several HLA genotyping benchmark studies (Orenbuch et al. 2020; Thuesen et al. 2022; Claeys et al. 2023; Yu et al. 2023), we included these two methods in our evaluations. We used the HLA annotation from 1KGP as the ground truth (Abi-Rached et al. 2018) and compared the performance on samples with both RNA-seq data and WES data (Supplemental Table S1). Because the HLA annotation of 1KGP was at the four-digit level, for example, HLA-A*01:01, we converted the results from T1K and arcasHLA from six digits to four digits. It is not likely to miss the 1KGP-annotated HLA genes on a chromosome, so we expanded the homozygous prediction of T1K as one allele showed up twice. This is the paradigm of arcasHLA, OptiType, and the 1KGP annotation. The matching criterion is similar to the case of the previous KIR3DL2 evaluation by considering the match for each allele pair. For the gene for which 1KGP had more than two annotated alleles, we selected the pair that matched the best with the benchmarked method. When evaluating HLA-DQB1, we ignored the samples with no 1KGP annotation. As before, the fraction of matched alleles would equal both sensitivity and precision and was suitable to represent the accuracy. We first evaluated the results of T1K, arcasHLA, and OptiType on the RNA-seq data (Fig. 1D). T1K achieved the highest accuracy in HLA-A, HLA-B, HLA-C, and HLA-DQB1, where its accuracies were ∼99.3% and 99.0% on HLA class I alleles and class II alleles, respectively.
We next compared the results of T1K and OptiType on WES data. arcasHLA was excluded from this evaluation as it was incompatible with genomic sequencing data. T1K's allele prediction accuracy was worse than that of OptiType, where T1K and OptiType had 96.1% and 98.0% average accuracy on HLA class I genes, respectively (Fig. 1E). Although OptiType did not genotype HLA class II genes, T1K reached an accuracy of 95.8% for HLA-DQB1 and 98.5% for HLA-DRB1. The T1K WES accuracy was lower than its RNA-seq accuracy for the same donors. This may be owing to the intronic sequences and the UTRs. For example, one of the errors that T1K made was the false-positive prediction of HLA-A*02:783 in two samples. There were three differences between HLA-A*02:783 and HLA-A*02:01:01:01 near the end of exon 3. Meanwhile, this part of exon 3 and the intron after exon 3 in HLA-A*02:783 were identical to the last part of HLA-U*01:03 and its UTR (Supplemental Fig. S4). As a result, many WES reads from HLA-A*02:01 and HLA-U can also be aligned to HLA-A*02:783, causing its abundance inflation. In RNA-seq data, the reference sequences directly join exons 3 and 4, which could reduce the confounding abundances from the HLA-U allele.
OptiType by default filters read alignments from rare alleles or non-HLA class I genes based on a list of frequent alleles hardcoded in its source code. As HLA-A*02:783 was not on this list, OptiType avoided the errors related to the allele. We hypothesized the lower accuracy of T1K on WES data compared with that of OptiType was owing to the complexity of the complete allele reference file. To validate the assumption, we added a T1K option to only retain read assignments from the alleles in a user-specified whitelist. Using a whitelist derived from OptiType's frequent alleles list, we improved T1K accuracy from 96.1% to 98.3%, surpassing OptiType (Fig. 1E). The observation suggests some alleles in the complete IPD-IMGT database may confuse genotyping and lead to errors. However, this whitelist strategy might cause false negatives. For example, OptiType could not predict HLA-B*35:41 as this allele was filtered in the frequent allele list, whereas T1K's default mode found it in NA12827 validated by the 1KGP annotation. A whitelist curated for T1K could probably improve the T1K accuracy further. We did not go down this path to avoid overfitting to the benchmark data set.
Speed and memory usage
As consequences of the comprehensiveness of IPD-IMGT/HLA and the high expression of HLA genes, HLA genotyping using RNA-seq data is the most time-consuming and memory-demanding task in our study. Therefore, we compared the computation efficiency of T1K, arcasHLA, and OptiType on the 10 largest RNA-seq samples that were used in the HLA genotyping evaluation (Supplemental Table S4). Both T1K and OptiType started from the raw read FASTQ file, and arcasHLA used the aligned BAM file as input. All three methods could finish each sample within 3 h given eight threads. Because arcasHLA extracted the candidate reads directly from the reads mapped to Chromosome 6 in the BAM file, its overall running time was faster than that of T1K and OptiType. When comparing the speed on the genotyping step after obtaining candidate reads, T1K and arcasHLA had similar running times. Although T1K and arcasHLA required <40 GB memory, Optitype consumed >200 GB memory on three of the benchmarked samples.
Expression of KIRs in CD8+ T cells
The advancement of scRNA-seq technology enables us to explore the KIR expression patterns in immune cells computationally. To avoid the confounding effects of dropout events from low-coverage platforms like 10x Genomics scRNA-seq data (Zheng et al. 2017), we chose Smart-seq (Hagemann-Jensen et al. 2020) data for robust genotyping and abundance estimation. We first examined the KIR allele usage across 29 immune cell types by running T1K on 118 cells with Smart-seq data from four healthy donors’ blood (Monaco et al. 2019). In each donor, the NK cell consistently expressed the highest number of KIR alleles, followed by γδT cells and effector CD8+ T cells (Supplemental Fig. S5). This observation supported that KIRs were expressed in a subset of T cells (Halary et al. 1997; Björkström et al. 2012; de Vries et al. 2023).
Because of the CD8+ T cell's important role in adaptive immunity, we next explored the KIR allele usage pattern in this cell type further. We analyzed 32 CD8+ T cells with Smart-seq scRNA-seq data (Simoni et al. 2018). These cells were from multiple donors, including cancer patients, so we first grouped the cells belonging to the same person by matching the HLA class I genes based on T1K's results. After that, we ran T1K again on the cells from the same donor for KIR genotyping and obtained the allele abundance. To compare the KIR allele expression pattern, we normalized the abundances to expression fractions within each cell (for an example, see Fig. 2A; for complete results, see Supplemental Fig. S6). We observed that KIR alleles or KIR genes were expressed selectively in each cell again (Björkström et al. 2012), and most cells expressed fewer than seven KIR alleles (Fig. 2B). KIR2DL4 and KIR3DL2 genes were detected in the most number of cells (27 and 21, respectively), and they were the only KIR genes that had both alleles expressed in a cell (Fig. 2C). These two KIR genes were two of the four framework KIR genes, whereas another framework KIR gene, KIR3DL3, was much less expressed. Furthermore, the remaining framework KIR gene KIR3DP1 was not expressed by any cells, implying that the pseudogene might not be functional in CD8+ T cells.
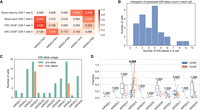
KIR alleles in CD8+ T cells. (A) KIR allele expression fractions in four cells from a colorectal cancer (CRC) patient. (B) The number of expressed KIR alleles in a cell. For example, seven cells expressed two KIR alleles. (C) The number of cells that express the KIR gene, splitting by the cases of single-allele expression or both-allele expression. Only detected KIR genes are displayed. (D) Comparison of the KIR allele fractions between CD39− CD8+ T cells and CD39+ CD8+ T cells. Each line connects a KIR allele in the CD39− cell and the CD39+ T cell from the same patient. The P-values are computed with a Wilcoxon signed-rank test and have been adjusted by the Benjamini–Hochberg procedure.
We next investigated the KIR allele expression pattern in different phenotypes of the tumor-infiltrating CD8+ T cells, where CD39+ T cells are tumor-specific T cells and CD39− T cells are bystanders (Simoni et al. 2018). Because a donor contributed, at most, one CD39− cell and one CD39+ cell in these data, we considered each KIR allele expression fraction of a patient as a pair (CD39− vs. CD39+) and ignored the patients without paired cells. When comparing the allele expression fractions, KIR2DL4 alleles were significantly enriched in the tumor-infiltrating CD39+ T cells (two-sided Wilcoxon signed-rank test, raw P-value = 5.5×10−4, adjusted P-value = 0.006) (Fig. 2D). The enrichment of KIR2DL4 remained significant when applying the unpaired two-sided Mann–Whitney U test (P-value = 0.013). Previous work has reported activating function of KIR2DL4 in NK cells (Faure and Long 2002), yet its role in the functionally similar cytotoxic CD8+ T cells remains elusive. Our observations necessitate future exploration of KIR2DL4 functionality in a subset of CD8+ T cells and tumor immunity.
Discussion
We have conducted comprehensive evaluations to show that T1K is a highly accurate genotyping method. For example, we used phased genomes from HPRC to validate the allele predictions. These approaches are computation-based, and they might be less reliable than experimental techniques, such as using PCR with allele-specific primers to examine the alleles. To increase our confidence in T1K, we investigated the HLA genotyping by evaluating with 1KGP annotations, which were experimentally validated extensively in the original study. As KIR genes have their own unique features, experimental validation might still be needed for KIR genotyping, but this is beyond the scope of the current study.
Although HLA and KIR allele sequences are well curated by IPD, there are other polymorphic genes. Some polymorphic genes, like CYP2D6 relating to drug metabolism (Wang et al. 2009), are cataloged by the PharmVar database (Gaedigk et al. 2018). T1K provides flexible and user-friendly modules to create custom references to genotype these non-IPD-curated genes. In addition, KIR genes are in the leukocyte receptor complex (LRC), and LRC contains other immune receptor gene families such as LILR and LAIR. It has been shown that a subset of the regulatory receptor genes LILR on neutrophils is genetically diverse in the population (Lewis Marffy and McCarthy 2020). To analyze the polymorphisms in the gene without a curated database, one could retrieve the allele sequences from resources like the HPRC genomes and create a custom database for T1K to genotype new samples. Similarly, we could find novel HLA and KIR alleles from these genome studies, such as KIR3DL2*003's genome sequences.
There could be structural polymorphisms in KIR loci that are too complex to process with the current implementation, such as hybrid or duplicated KIR genes (Traherne et al. 2010). T1K could not align the reads to hybrid KIR genes owing to the high divergence to the reference sequences. This might decrease the allele prediction accuracy and the power to detect variations. In addition, the quality score filter and the abundance fraction filter might suppress a true allele if the other haplotype has copy number gains. Future works like graph representations of the KIR gene sequences at the exon level and specialized statistical models for copy number inference could resolve these complex variations.
In addition to showing T1K's functionality, we explored the KIR expression patterns in CD8+ T cells. We observed that KIR2DL4 was enriched in tumor-specific CD39+ CD8+ T cells, suggesting that KIR2DL4 might be related to tumor immunity. This analysis was based on a Smart-seq data set with 32 cells, so more data need to be collected to validate and study the role of KIR2DL4 in CD8+ T cells. We did not explore KIR genes on 10x Genomics scRNA-seq data owing to the concern over dropouts, but the analysis might be feasible and reliable at the cell cluster level. With the expanding knowledge of KIRs’ function, we expect the genotypes inferred from T1K could be a valuable resource for biologists to study and find appropriate cancer treatment strategies in the future.
In sum, we have implemented the novel computational method T1K that can genotype KIR genes or HLA genes from various sequencing platforms, including RNA-seq, WES, and WGS data. T1K showed that the EM algorithm–based approach was highly accurate and efficient even when inferring all of the thousands of alleles in the database simultaneously. We showed that KIR2DL4 alleles were more expressed in tumor-specific CD8+ T cells than the bystander tumor-infiltrating CD8+ T cells. We expect T1K's versatile framework will contribute to KIR, HLA, and other polymorphic gene studies in the future.
Methods
Sequencing data
We generated 100 simulated samples using Mason (Holtgrewe 2010). Each sample was generated with the options “illumina -i -s 17 -sq -mp -n 100 -ll 500 -hs 0 -hi 0 -pi 0 -pd 0 -pmmb 0.0005 -pmm 0.001 -pmme 0.003 -nN -N read_count.” Each sample selected six to nine random KIR genes and two random alleles (allowing the same) for each KIR gene. The parameter “read_count” in the mason command was set to have 50 read pairs for each allele on average. The read length is 100 bp.
For the real data set in the KIR genotyping evaluation, we used 26 samples (Supplemental Table S1) from the Human Pangenome Reference Consortium (HPRC) (Wang et al. 2022; Liao et al. 2023) with both haplotype-resolved reference genomes and an Illumina WGS paired-end short read of 150-bp read length. For the HLA genotyping comparison, we downloaded 463 samples from 1KGP that have annotated HLA genotype, RNA-seq, and WES-seq data (Supplemental Table S3). We used STAR (Dobin et al. 2013) with default parameters to align the RNA-seq data to the human reference genome hg38. Two of the WES samples (HG00104 and NA18487) had damaged FASTQ files, so we excluded them from the WES genotyping analysis. For the Smart-seq analysis, we downloaded the scRNA-seq data from the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession numbers PRJNA418779, PRJNA453180, and PRJNA453183.
Reference sequences
IPD curates a comprehensive set of allele sequences for both HLA and KIR genes, so T1K builds the reference sequences based upon it as many other methods. In this work, T1K used IPD-KIR v2.10.0 for KIR genotyping and IPD-IMGT/HLA v3.44.0 for HLA genotyping. OptiType has its own processed HLA reference sequence in the package, and we ran arcasHLA with IPD-IMGT/HLA v3.44.0. In this study, we used T1K v1.0.2, PING (GitHub commit 1d1b5d1), arcasHLA v0.2.5, and OptiType v1.3.5.
The T1K reference database is prepared differently depending on whether the input sequences contain introns. For RNA-seq data, T1K concatenates the exons parsed from the EMBL-ENA formatted DAT file for each allele. For WGS or WES, we added the 200 bp from each flanking intron on two sides of every exon. If two exons’ flanking introns overlap, we will merge the intervals. For the long introns, we add a one-character “N” at the boundary to indicate the gap in the intronic region. For all data types, we padded the sequences with 50-bp sequences from 3′ and 5′ UTRs to give more anchors for the read alignment. The DAT file also annotates whether a sequence is partial, and T1K will ignore those partial sequences.
When the UTR is exonized, such as HLA-C*04:09N and KIR2DL3*010, the allele contains sequences that are missing in other alleles. As a result, reads from the UTR region are more likely to be mapped to the UTR-exonized alleles and cause inflation of the abundance estimation. To alleviate the mapping bias, T1K trims the last exon of an allele if the allele is longer than the common allele length of the gene. The trimming is only applied to the last exon, and the trimmed last exon length equals this gene's most frequent last exon length. In this way, T1K will keep the sequence if there are insertions in the middle of the allele. Although this strategy will remove allele-specific sequences, the remaining variations, including the stop codon, might be sufficient for genotyping. For example, T1K still correctly identified HLC-C*04:09N in NA12718 and NA12777 in the 1KGP evaluation.
Candidate reads
The first step of T1K is to extract the reads from the interested genes provided in the reference sequences, such as KIR and HLA genes. For user convenience, T1K is compatible with both aligned BAM input and FASTQ input, and the choice of input format does not affect the genotyping outcome in our tests. For reads that are aligned to alternate contigs, that are unmapped, or that are in the FASTQ file, the extraction algorithm is similar to the overlap detection algorithm in TRUST4 (Song et al. 2021) by checking the colinear seeds hit. Suppose the total length of the reference sequences is L, and the seed length used in this step is log4L + 1. For example, in the RNA reference sequence of HLA genotyping, L is about 16 million, so seed length equals 13. As a result, a k-mer in a read hits about once to the reference sequence in expectation. Because reference sequences are highly redundant, the seed length overestimates the hit probability and will not incur much computation overhead. A read will be a candidate if the chain covers 20% of the read length. When given the BAM input, most of the reads mapped outside the genotyped genes can be directly ignored, so T1K was ∼30% faster than the FASTQ input.
Read assignment
To conduct abundance estimation, T1K assigns each read, or a read fragment, to its best-aligned alleles. The best alignments are the ones with the most matched nucleotides between the reference sequence and a read. The reference sequences are highly redundant, and a read can be aligned to thousands of alleles. This becomes the computation bottleneck in RNA-seq analysis, where HLA genes can express millions of reads. We notice that the ultrahigh coverage will create a large number of identical reads. Therefore, T1K will first sort the reads by the nucleotide sequence and then only conduct the alignment for the duplicates once. For paired-end data, T1K conducts the same procedure by regarding each read fragment as two independent read ends and then selects the two compatible read-end alignments, resulting in the optimal read pair alignment score.
T1K further supplements the read alignment by strategically incorporating assignments with lower scores. In the reference sequences for genomic sequencing data, we only include part of the introns flanking the exons and mark the remaining parts as gaps. This strategy could create alignment bias near the boundary. For example, if allele A has a deletion at position x before the gap, allele A's intron will contain the first nucleotide of the corresponding gap in other alleles. As a result, a read aligned after position x and partially overlapping with the gap will have one more base matched with allele A than other alleles. To reduce this bias, T1K considers the alignment part extending in the gap as all matches. Furthermore, the IPD-KIR is much less comprehensive than IPD-IMGT/HLA, and the inadequate information on intron sequences could lead to false negatives. Therefore, we add the option to incorporate the read assignments with one more mismatch in the nonexonic region than the best alignment.
To avoid low-quality reads and false-positive read assignments, T1K filters the assignments with a low alignment identity. In the case of KIR genotyping, we require at least 80% of matched bases. For HLA genotyping, we have tested several cutoffs and found 97% gave a good performance, where 97% was also the threshold proposed in OptiType. For WGS data, because reads can come from the whole genome and are more likely to introduce false positives, we need to use a more stringent alignment similarity threshold at 90%. We have made these settings in the “‐‐preset” option of T1K. Additionally, T1K can take a user-specified allele whitelist to only retain the assignments from the listed alleles. In the evaluation of HLA genoptying, the whitelist in T1K_whitelist was created by iterating through the alleles in T1K default database and only keeping the ones whose first four digits showed up in the OptiType's frequent allele list.
Abundance estimation
T1K estimates the abundances of all the alleles by maximizing the likelihood of read assignments. The log-likelihood function
is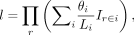 where θi is the abundance fraction or probability of allele i among all alleles, Li is the length of allele i, and Ir∈i is one if allele i is one of read r’s alignment target and zero otherwise. If Li is much less than (500 bp by default) than the most common allele length of the gene, we will force Li to be the mode of the allele length. We apply the EM algorithm to find the solution by introducing the latent variable zr,i indicating read r is from allele i. So, the conditional expectation of the log-likelihood function can be written as
where θi is the abundance fraction or probability of allele i among all alleles, Li is the length of allele i, and Ir∈i is one if allele i is one of read r’s alignment target and zero otherwise. If Li is much less than (500 bp by default) than the most common allele length of the gene, we will force Li to be the mode of the allele length. We apply the EM algorithm to find the solution by introducing the latent variable zr,i indicating read r is from allele i. So, the conditional expectation of the log-likelihood function can be written as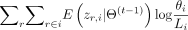 in E-step at iteration t, where
in E-step at iteration t, where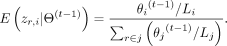 In M-step, we compute the updated abundances that maximize the conditional expectation, which is
In M-step, we compute the updated abundances that maximize the conditional expectation, which is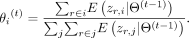 In the above equations, we use i, j to index alleles and r to index the read. The initial abundance for each allele is proportional to the allele series frequency in the database.
For example, HLA-A*01:01:01:01 is in the HLA-A*01:01:01 series and the size of this series is 91 in the reference sequences
for RNA-seq data, so its initial abundance is 91 times higher than the singleton HLA-A*01:82 allele. The intuition is that
the large allele series suggests that it is better studied and could be more common in the population. The iterations terminate
when the update changes the Θ by less than 10−5 in total or the number of iterations exceeds 1000. Finally, the abundance for allele i is
In the above equations, we use i, j to index alleles and r to index the read. The initial abundance for each allele is proportional to the allele series frequency in the database.
For example, HLA-A*01:01:01:01 is in the HLA-A*01:01:01 series and the size of this series is 91 in the reference sequences
for RNA-seq data, so its initial abundance is 91 times higher than the singleton HLA-A*01:82 allele. The intuition is that
the large allele series suggests that it is better studied and could be more common in the population. The iterations terminate
when the update changes the Θ by less than 10−5 in total or the number of iterations exceeds 1000. Finally, the abundance for allele i is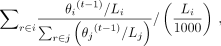 which is the definition of the fragments per thousand bases (FPK).
which is the definition of the fragments per thousand bases (FPK).
T1K implements two methods to expedite the execution of the EM algorithm. First, we group the reads assigned to the same set of alleles together. Therefore, the EM algorithm is weighted. Second, we adopt the SQUAREM algorithm (Varadhan and Roland 2008), which has a faster convergence rate than the vanilla EM algorithm. To impose sparsity, we implement the heuristics that removes the low abundant alleles every 10 iterations based on the abundances estimated in that iteration. This is similar to the sparsity method in arcasHLA.
Allele selection
The EM algorithm calculates the abundance for each allele with all digits. Because T1K does not focus on the variations in introns, it reports the alleles at a higher level with fewer digits. For example, T1K reports KIR and HLA alleles at three-digit and six-digit levels, respectively. The abundance for each higher-level allele series is the summation of the allele abundances in the same series. For simplicity, we still indicate the allele series as an allele. T1K selects the allele with the highest abundance as the dominant allele and filters other alleles with abundances less than a user-specified fraction (default, 15%) of the dominant allele.
Although the abundance filter can remove the noise allele in most cases, there could still be more than two alleles passing the abundance filter. T1K refines the selection by picking a pair of alleles that can improve the number of valid read assignments. Starting from the two most abundant alleles for each gene, T1K changes one of the alleles to find the allele that increases the read assignments the most. T1K repeats this procedure based on the pairs of alleles selected in the previous iteration until the number of explained read assignments could not be improved. This strategy favors the two most abundant alleles and is faster than enumerating all the allele pairs in most cases.
Genotyping quality score
T1K also scores each called allele to represent the confidence. The principle is to conduct the statistical test that compares the computed abundance with the noise abundance. The noise abundance for one allele A of gene Gi is calculated as Σj≠i αAbund(Gj)sim(Gi, Gj) + βAbund(B), where Abund(Gj) is the abundance of gene j, sim(Gi, Gj) is the sequence similarity between genes Gi and Gj, Abund(B) is abundance for the other allele of gene Gi if it is heterozygous, and α, β are user-defined constants to control the magnitude of noise. We model the abundance as a Poisson distribution and compute the P-value by testing the observed abundance under the hypothesis of noise Poisson distribution. The quality score is reported as −log10(P-value). Similar to the MAPQ in read alignment software like BWA-MEM (Li and Durbin 2009), we set 60 as the upper limit of the quality score.
SNP detection
Even though T1K reports the genotyping results at the allele-series level, it keeps track of the actual alleles in the process and reports one allele per series as the representative. This information can be used to identify SNPs that are missing from the reference database. For SNP detection, we realign the reads to the representative alleles. We process a position i of allele A with a high mismatch rate; that is, the number of alignments supporting an alternative nucleotide is at least half of the number for the reference nucleotide. Because of allele similarities, these reads could still be equally good assigned to other alleles. Therefore, T1K uses the multiple-mapped reads to incorporate other alleles and piles the allele sequences around position i. This procedure can be thought of as read alignment–guided multiple sequence alignment. The SNP could happen to any of the incorporated alleles. To select the alleles containing SNPs, T1K follows the law of parsimony by introducing the least number of SNPs to explain the most read assignments (Supplemental Fig. S7). In more detail, T1K tries all the nucleotide combinations at position i across the piled alleles to check how many reads can have matched bases at position i in any of the alleles. T1K then determines the SNP based on the nucleotide combination with the most reads having matched base at position i. In case of a tie, T1K will select the combination with the least number of SNPs. If the tie could not be broken by the SNP number rule, T1K will report all the equivalent results and mark them as ambiguous. Suppose there are N alleles found for position i, the enumeration step will consider 4N combinations. Because N is small in practice, the enumeration step is still very fast.
Single-cell processing
T1K supports scRNA-seq data such as Smart-seq data. Because cells could have cell-specific allele expression patterns, we conducted the genotyping for each cell individually and then merged the results. The merge step filters alleles with too low total quality scores and selects the two alleles with the highest quality scores for each gene. After obtaining the allele calling consensus, T1K will remove irrelevant alleles from the reference sequences and reconduct the genotyping on the reduced reference sequences.
Allele annotation on HPRC phased genomes
To identify the KIR alleles on a phased reference genome from HPRC in our benchmark study, we align the KIR genomic (exon and intron) sequences from the IPD-KIR to the genome. The first step is to conduct the alignment using minimap2 (Li 2018) for quick KIR region discovery. Each KIR region must have at least 99% of bases covered in the alignment interval. Second, we realign the sequences from IPD-KIR to each KIR region with BWA-MEM to obtain accurate base-level alignments. Finally, the alleles with the least number of differences in the exonic sequences are selected.
Software availability
The T1K source code and evaluation code for this work are available at GitHub (https://github.com/mourisl/T1K and https://github.com/mourisl/T1K_manuscript_evaluation, respectively) and as Supplemental Code.
Competing interest statement
X.S.L. conducted the work while being on the faculty at DFCI and is currently a board member and CEO of GV20 Therapeutics. H.L. is a consultant of Integrated DNA Technologies.
Acknowledgments
We thank Dr. Paul Norman and the colleagues from X.S.L. group and H.L. group for helpful discussions. This work is supported by National Cancer Institute 1R01CA245318 (B.L.), 1R01CA258524 (B.L.), National Institutes of Health R01HG010040 (H.L.), U01HG010961 (H.L.), R01HG011139 (H.L.), and U01CA226196 (H.L.) and by National Institute of General Medical Sciences P20GM130454 (Dartmouth).
Author contributions: L.S., X.S.L., B.L., and H.L. conceived the project. L.S. developed the method. L.S., G.B., B.L., and H.L. conducted the evaluations and data analysis. All the authors agree on the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277585.122.
- Received December 11, 2022.
- Accepted May 4, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








