
Evaluation of N6-methyldeoxyadenosine antibody-based genomic profiling in eukaryotes
- 1Department of Genetics, Blavatnik Institute, Harvard Medical School, Boston, Massachusetts 02115, USA;
- 2Department of Genome Sciences, University of Washington, Seattle, Washington 98195, USA;
- 3Division of Medical Genetics, Department of Medicine, University of Washington, Seattle, Washington 98195-7720, USA;
- 4Brotman Baty Institute for Precision Medicine, Seattle, Washington 98195, USA
Abstract
Low-level DNA N6-methyldeoxyadenosine (DNA-m6A) has recently been reported across various eukaryotes. Although anti-m6A antibody–based approaches are commonly used to measure DNA-m6A levels, this approach is known to be confounded by DNA secondary structures, RNA contamination, and bacterial contamination. To evaluate for these confounding features, we introduce an approach for systematically validating the selectivity of antibody-based DNA-m6A methods and use a highly selective anti-DNA-m6A antibody to reexamine patterns of DNA-m6A in C. reinhardtii, A. thaliana, and D. melanogaster. Our findings raise caution about the use of antibody-based methods for endogenous m6A quantification and mapping in eukaryotes.
Endogenous DNA-m6A is a widespread DNA modification in bacterial genomes (Ratel et al. 2006a) and some unicellular eukaryotes (Hattman et al. 1978; Fu et al. 2015) and, more recently, has been described in multicellular eukaryotes such as Arabidopsis thaliana (Liang et al. 2018), Caenorhabditis elegans (Greer et al. 2015), Drosophila melanogaster (Zhang et al. 2015), Mus musculus (Koziol et al. 2016; Wu et al. 2016; Li et al. 2020), Homo sapiens (Wu et al. 2016; Xie et al. 2018), and others (Luo and He 2017). The level of DNA-m6A in eukaryotes has been reported to vary from as low as 6 ppm (i.e., six m6A-modified bases per million adenines) in humans and mice (Wu et al. 2016) to as high as 8000 ppm in Tetrahymena pyriformis (Hattman et al. 1978). However, the biological role of DNA-m6A in multicellular eukaryotes remains largely undefined and has been recently shown to frequently arise from contamination as opposed to endogenous DNA-m6A (Kong et al. 2022). m6A antibody–based DNA immunoprecipitation sequencing (m6A-DIP-seq) is often used for mapping m6A in these organisms, but this technology has been proven to be frequently confounded by artifactual signal when measuring low-abundance DNA-m6A (Ratel et al. 2006b; Schiffers et al. 2017; Lentini et al. 2018; O'Brown et al. 2019; Douvlataniotis et al. 2020; Kong et al. 2022). Specifically, results from anti-DNA-m6A antibodies have thus far been proven to be unreliable (O'Brown et al. 2019), with microbial and RNA contaminants thought to be a major contributor to these false measurements (Douvlataniotis et al. 2020; Kong et al. 2022). Furthermore, lot-to-lot variability in antibody quality likely detracts from the reliability of antibody-based methods for quantifying m6A. To address this, we developed a straightforward approach to systematically validate the selectivity of antibody-based DNA-m6A methods and applied this to 12 commercially available anti-m6A antibodies.
Results
To directly evaluate N6-adenine DNA immunoprecipitation–based detection methods, we sought to develop a straightforward approach for systematically testing the selectivity and sensitivity of anti-DNA-m6A antibodies for the global and locus-specific detection of DNA-m6A. To accomplish this, we leveraged a recently described approach for generating complex genomic DNA (gDNA) libraries with titratable amounts of locus-specific DNA-m6A (Stergachis et al. 2020). Specifically, we used gDNA from eukaryotic cells lacking significant endogenous DNA-m6A (negative control), as well as gDNA isolated after treating nuclei from these cells with a nonspecific m6A-methyltransferase (m6A-MTase; positive control). Importantly, this process of treating nuclei with nonspecific m6A-MTase results in the clustering of m6A-modified bases within regulatory elements and internucleosomal linker regions (Stergachis et al. 2020), a pattern that mirrors the distribution of validated endogenous m6A in eukaryotes (Kong et al. 2022). We then tested 12 commercial anti-DNA-m6A antibodies against these samples to systematically quantify the sensitivity and selectivity of these anti-m6A antibodies (Fig. 1A). We showed that commonly used cell culture lines, such as Drosophila S2-DRSC, and human K562 cells can be used for generating the positive and negative control samples, as these cells lack appreciable endogenous DNA-m6A (Fig. 1B; Supplemental Fig. S2), and treating nuclei from these cells with an m6A-MTase results in the methylation of ∼14% of all adenines across various sequence contexts (i.e., DNA-m6A amount of 140,000 ppm) (Supplemental Fig. S1; Stergachis et al. 2020).
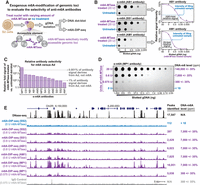
Sensitivity and selectivity of anti-DNA-m6A antibodies. (A) Schematic for assaying anti-DNA-m6A antibody selectivity and sensitivity using gDNA isolated from cell nuclei exogenously methylated with a nonspecific DNA m6A-MTase. (B) DNA dot-blots using three separate anti-DNA-m6A antibodies against gDNA from untreated S2 cells versus gDNA from S2 cells treated with m6A-MTase. To the right are quantifications of DNA dot-blot signal intensity for the 20-unit treated sample, a four-parameter log-logistic fit to these signals (purple line), and the relative signal intensity of the untreated 50-ng sample as opposed to the treated sample. (C) Bar chart quantifying the relative selectivity of each anti-DNA-m6A antibody toward DNA-m6A, as measured using DNA dot-blots. (D) DNA dot-blot and table showing the relative amount of DNA-m6A after treatment of S2 cell nuclei with increasing amounts of a nonspecific DNA-m6A-MTase. (E) Genomic locus comparing the relationship between DNase-seq and m6A-DIP-seq signal. m6A-DIP-seq performed using five separate antibodies on untreated S2 cell gDNA, or gDNA from S2 cell nuclei treated with 0.6 U or 0.075 U of m6A-MTase. Signal from IgG antibody control also displayed. The y-axis is identical for all m6A-DIP-seq and DNase-seq experiments.
We found that all 12 antibodies detected DNA-m6A, albeit with sensitivities that differed by over two orders of magnitude (Fig. 1B,C; Supplemental Fig. S2). However, the selectivity of these antibodies for DNA-m6A, as opposed to unmethylated adenine, varied between the 12 antibodies. For example, whereas the MP1 antibody showed a more than 500,000-fold selectivity toward DNA-m6A, the AM1 antibody was only 25-fold selective toward DNA-m6A as opposed to unmethylated Ad (Fig. 1B,C; Supplemental Figs. S2, S3).
To determine the limit of detection for the MP1 antibody, we titrated the amount of m6A-MTase used to generate the positive control sample, showing that the MP1 antibody can detect DNA-m6A levels down to ∼10 ppm (Fig. 1D). In contrast, as most other m6A-antibodies are only about 1000-fold more selective for m6A as opposed to unmethylated adenines, these other anti-DNA-m6A antibodies could only quantify DNA-m6A levels down to ∼1000 ppm before the DNA-m6A signal became indistinguishable from off-target antibody recognition of unmethylated adenines, precluding the use of these antibodies for quantifying endogenous DNA-m6A levels in essentially all multicellular eukaryotic organisms (Luo and He 2017). However, given challenges associated with quantifying absolute m6A abundances using DNA dot-blots, as well as the potential impact m6A clustering along individual DNA molecules may have on antibody-mediated detection, it is likely that there will be some variability in measuring the exact selectivity and sensitivity of these antibodies (Supplemental Fig. S1B).
Next, we sought to evaluate the sensitivity of antibody-based DNA-m6A methods for identifying endogenous DNA-m6A-modified genomic loci using these exogenously methylated positive control samples. Specifically, we applied DNA-m6A immunoprecipitation followed by massively parallel sequencing (m6A-DIP-seq) to gDNA isolated from S2 cell nuclei treated with 0.6 units of m6A-MTase (Fig. 1A), as this treatment results in a global DNA-m6A level of ∼7000 ppm (Fig. 1D) and the selective deposition of DNA-m6A at approximately 17,000 accessible chromatin elements genome-wide in a pattern mirroring that of DNase-seq (Stergachis et al. 2020). For this approach, we only tested five of the antibodies with varying degrees of sensitivity and specificity for DNA-m6A. Notably, only the MP1 antibody accurately detected nearly all exogenously DNA-m6A-methylated sites genome-wide, with the other four antibodies detecting only between 2% and 45% of the exogenously methylated sites (Fig. 1E; Supplemental Fig. S4), suggesting that these four antibodies show both poor selectivity and sensitivity for the detection of m6A-modified bases and genomic loci.
To test the detection limit of DNA-m6A antibodies for identifying modified genomic loci, we applied m6A-DIP-seq to gDNA isolated from S2 cell nuclei treated with only 0.075 units of m6A-MTase. This treatment results in a global DNA-m6A level of ∼300 ppm (Fig. 1D), which is similar to endogenous DNA-m6A levels in some multicellular eukaryotes (Luo and He 2017), although still two orders of magnitude higher than that recently described in D. melanogaster embryos as well as A. thaliana seedlings (Kong et al. 2022). Although the m6A-DIP-seq signal using the MP1 antibody was markedly reduced in this sample, we still identified ∼29% of the genomic loci exogenously modified with DNA-m6A (Fig. 1E), showing that appropriately sensitive and selective anti-DNA-m6A antibodies can detect low-abundance DNA-m6A-modified loci.
Next, using a highly selective and sensitive anti-DNA-m6A antibody (e.g., MP1), we reexamined endogenous DNA-m6A levels across three diverse eukaryotic organisms reported to contain endogenous DNA-m6A at similar levels: Chlamydomonas reinhardtii (Hattman et al. 1978; Fu et al. 2015), A. thaliana cauline leaves (Liang et al. 2018), and D. melanogaster 45-min embryos (Fig. 2A; Zhang et al. 2015). Overall, we found that whereas C. reinhardtii showed endogenous DNA-m6A levels consistent with prior reports, both the A. thaliana and D. melanogaster samples showed barely detectable endogenous DNA-m6A levels (Fig. 2A,B), consistent with a recent reevaluation of m6A levels in these organisms using quantitative deconvolution of single-molecule sequencing data (Kong et al. 2022), further establishing a more limited presence of endogenous DNA-m6A in these organisms.

Detection of endogenous DNA-m6A across three nonmammalian eukaryotes. (A) DNA dot-blot quantifying the relative amount of DNA-m6A in samples from three eukaryotes, as well as from S2 cell nuclei treated with increasing amounts of m6A-MTase. To the right are the observed amount of DNA-m6A in samples relative to Figure 1D. (B) Genomic locus from C. reinhardtii comparing the relationship between m6A-DIP-seq signal and input control signal. The y-axis is identical for both experiments. (C) Histogram (left) and pie chart (right) showing the distribution of identified C. reinhardtii DNA-m6A peaks relative to TSSs and repetitive elements. (D) Genomic locus from A. thaliana comparing the relationship between m6A-DIP-seq signal and input and IgG control signal. The y-axis is identical for all experiments. (E) Histogram (left) and pie chart (right) showing the distribution of identified A. thaliana DNA-m6A peaks relative to TSSs and repetitive elements.
Performing m6A-DIP-seq on C. reinhardtii cells with the MP1 antibody revealed 17,451 DNA-m6A sites genome-wide, mirroring previously published data (Fig. 2C,D; Supplemental Fig. S5; Fu et al. 2015). Notably, m6A-DIP-seq performed using the SS1 antibody, which is the antibody used in the prior C. reinhardtii study (Fu et al. 2015), resulted in a high background signal with no discernable m6A-modified peaks (Supplemental Fig. S5), highlighting that some anti-DNA-m6A antibodies may show large lot-to-lot variability, thus explaining some of the poor reproducibility associated with anti-DNA-m6A antibodies (Wu et al. 2016; Xie et al. 2018).
Performing m6A-DIP-seq on A. thaliana cauline leaves with the MP1 antibody identified 2063 endogenous DNA-m6A sites genome-wide. These peaks were enriched at gene and transposable element transcriptional start sites (TSSs) (Fig. 2E,F), consistent with prior reports (Liang et al. 2018). In contrast, performing m6A-DIP-seq on D. melanogaster 45-min embryos identified only 161 DNA-m6A peaks genome-wide, many of which overlapped likely false-positive m6A-DIP-seq peaks in S2 cells (Fig. 2G; Supplemental Fig. S6). These findings indicate that the minimal amount of DNA-m6A we detected in the D. melanogaster embryos either is not site specific within the mappable portion of the genome or results from residual bacterial contamination of the sample (Fig. 2A,B).
Finally, using our high-quality maps of endogenous DNA-m6A loci in A. thaliana and C. reinhardtii, we sought to determine whether similar sequence motifs are enriched within the DNA-m6A-modified loci in these organisms. Three distinct classes of DNA-m6A MTase motifs have been described to date: the ApT motif, which is methylated by the MTA1c complex in ciliates (Beh et al. 2019); the GAGG motif, which is methylated by the DAMT-1 enzyme in C. elegans (Greer et al. 2015); and the RRACH motif, which is methylated in ssDNA by the METTL3-METTL14 complex in H. sapiens (Woodcock et al. 2019). Although, DNA-m6A appears to be only mediated by MTA1c at VATB motifs in C. reinhardtii (Kong et al. 2022), all three of these motifs appeared to be enriched in the C. reinhardtii DNA-m6A-modified loci (Supplemental Fig. S7), likely reflecting the co-occurrence of the GAGG and RRACH motifs within the same genomic loci as the VATB motifs in C. reinhardtii. In contrast, none of these motifs were significantly enriched in A. thaliana DNA-m6A-modified loci. Rather, A. thaliana DNA-m6A-modified loci showed a separate highly enriched motif, indicating that distinct pathways are likely generating DNA-m6A in A. thaliana and C. reinhardtii.
Discussion
In conclusion, we show a robust approach for systematically evaluating the selectivity of anti-DNA-m6A antibodies using a controlled system that enables the titratable DNA-m6A methylation of thousands of loci genome-wide across various sequence contexts. Importantly, this approach mirrors the clustered DNA-m6A patterns that are observed endogenously in eukaryotes. Using this approach, we show that most commercially available anti-DNA-m6A antibodies show poor selectivity toward DNA-m6A, with selectivity that would limit their utility for all endogenous m6A quantification applications in eukaryotic species. Notably, this includes one of the most commonly used anti-DNA-m6A antibodies (Fu et al. 2015; Greer et al. 2015; Koziol et al. 2016; Wu et al. 2016; Liang et al. 2018; Xie et al. 2018; Hao et al. 2020). These findings highlight the role of poor antibody selectivity, in addition to bacterial and RNA contamination (Kong et al. 2022), to prior reports of abundant DNA-m6A across diverse eukaryotic organisms, and potentially hold broader implications beyond DNA-m6A genomic profiling as many commercial anti-m6A antibodies are used interchangeably with DNA- and RNA-m6A methodologies.
Overall, our results raise caution with using antibody-based approaches for evaluating species with very low endogenous DNA-m6A levels, even when using well-validated antibodies. Specifically, we find that lot-to-lot variability can drastically impact the selectivity of anti-DNA-m6A antibodies and that m6A-DIP-seq can often result in unreliable peak calls from simple repeats or RNA contamination. For example, even when using a well-validated anti-DNA-m6A antibody, the peaks identified using m6A-DIP-seq in D. melanogaster 45-min embryos were largely nonspecific, indicating that antibody-based approaches are likely not sensitive and selective enough for species with similar levels of endogenous DNA-m6A.
In addition, our findings in C. reinhardtii highlight the challenges with calling m6A motifs when using m6A-DIP-seq, as enriched motifs may be bystanders as opposed to the direct targets of m6A-MTases. Of note, although this approach provides a powerful tool for evaluating antibody selectivity and relative m6A abundance, the absolute quantification of DNA-m6A using dot-blots is subject to confounding, and absolute m6A abundance is likely best performed using either mass spectrometry–based approaches or more recently described SMRT-sequencing quantitative deconvolutional methods (Kong et al. 2022). Overall, our approach provides a tractable system for continually reassessing the selectivity of anti-DNA-m6A antibodies as new lots and antibodies become commercially available to ensure the accuracy of antibody-based approaches for detecting DNA-m6A-modified loci in eukaryotes. If antibodies are going to be regularly released for endogenous DNA-m6A mapping in eukaryotes, it is paramount that vendors and researchers consistently and reproducibly reevaluate these antibodies for their selectivity and sensitivity.
Methods
Production of nonspecific DNA-m6A-MTase
The nonspecific DNA-m6A-MTase Hia5 was produced as previously described (Stergachis et al. 2020). Of note, the method presented in this paper can also be performed using other nonspecific DNA-m6A-MTases, including the commercially available EcoGII (New England Biolabs M0603S). We have previously shown that at least five nonspecific DNA-m6A-MTases can be used interchangeably (Stergachis et al. 2020).
Treatment of S2 cell nuclei with m6A-MTase and gDNA isolation
Drosophila S2-DRSC cells were grown and isolated as previously described (Stergachis et al. 2020). Specifically, 3 million S2 cells were pelleted at 250g for 5 min and then resuspended in 60 μL of buffer A (15 mM Tris at pH 8.0, 15 mM NaCl, 60 mM KCl, 1 mM EDTA at pH 8.0, 0.5 mM EGTA at pH 8.0, 0.5 mM spermidine). Sixty microliters of cold 2× lysis buffer (0.1% IGEPAL CA-630 in buffer A) was added to each sample and mixed by gentle flicking and then kept on ice for 10 min. Samples were pelleted for 5 min at 350g and 4°C, and the supernatant was removed. Nuclei pellets were gently resuspended with wide-bore pipette tips in 57.5 μL buffer A and moved to a 37°C thermocycler; 1.5 μL SAM (0.8 mM final) and Hia5 m6A-MTase diluted in buffer A were added and then carefully mixed by pipetting with wide-bore tips. The specific reaction conditions are as follows: 20 U Hia5 for 10 min, 0.6 U Hia5 for 10 min, 0.4 U Hia5 for 10 min, 0.2 U Hia5 for 10 min, 0.1 U Hia5 for 10 min, 0.075 U Hia5 for 10 min, 0.05 U Hia5 for 10 min, and 0.025 U Hia5 for 10 min. The reactions were incubated at 37°C and then stopped with 3 μL of 20% SDS (1% final) and transferred to new 1.5-mL microfuge tubes. The sample volumes were increased by adding 130 μL buffer A and an additional 7 μL of 20% SDS. All samples were mixed with 3 μL Proteinase K (New England Biolabs P8107S) and incubated for 1 h at 50°C. The DNA was purified by adding 200 μL (1:1) of phenol:chloroform:isoamyl alcohol (25:24:1; saturated with 10 mM Tris at pH 8.0, 1 mM EDTA), then mixed by vigorous tube inversions, and incubated for 10 min at room temperature (RT). Extractions were centrifuged for 10 min at 17,900g, and the upper aqueous phase was transferred to new microfuge tubes and the DNA precipitated by adding 0.5 μL GlycoBlue coprecipitant (Invitrogen AM9515), 0.1 volume of 3 M sodium acetate (pH 5.2), and 2.5 volumes ice-cold 100% ethanol. The DNA was pelleted by centrifuging at 20,000g for 10 min at 4°C and washed by repeating the centrifugation with 1 mL of ice-cold 70% ethanol. The tubes were inverted over a tube rack and air-dried for 15 min before resuspension in 200 μL of buffer A. All samples were mixed with 3 μL of RNase A (Invitrogen AM2271) and incubated for 1 h at 37°C. The samples subsequently underwent an additional phenol:chloroform:isoamyl alcohol purification, and residual phenol was removed with a second extraction by adding 200 μL chloroform:isoamyl alcohol (24:1) to all samples and repeating the extraction procedure. The DNA ethanol precipitation, washing, pelleting, and drying were repeated before resuspension in 54 μL of 10 mM Tris (pH 7.5).
Treatment of K562 cell nuclei with m6A-MTase and gDNA isolation
Human K562 cells were grown and isolated as previously described (Stergachis et al. 2020). Reaction conditions were similar to those described for the S2 cells above, with the following differences: (1) 1 million K562 cells were used per reaction; (2) 2× lysis buffer consisted of 0.05% IGEPAL CA-630 in buffer A; (3) 400 U Hia5 was used; and (4) the Hia5 reaction was performed for 10 min at 25°C before being stopped with the addition of SDS.
A. thaliana cauline leaf gDNA extraction
Cauline leaves were collected from three separate flowering col-0 A. thaliana plants and combined together. Two biological replicates were obtained from plants grown 3 mo apart from each other. Approximately 1g of cauline leaves was combined with 1.2 mL of buffer A supplemented with 1% sodium dodecyl sulfate (Sigma-Aldrich L3771), 0.5% polyvinylpyrrolidone (Sigma-Aldrich PVP40), and fresh 1% 2-mercaptoethanol (Sigma-Aldrich M3148) in a Dounce homogenizer. Tissue was ground using a “B” pestle until homogenous. The lysate was transferred in 200-μL aliquots to 1.5-mL microfuge tubes, and all samples were mixed with 3 μL Proteinase K and incubated for 1 h at 50°C. The DNA was purified by adding 200 μL (1:1) of phenol:chloroform:isoamyl alcohol (25:24:1; saturated with 10 mM Tris at pH 8.0, 1 mM EDTA), then mixed by vigorous tube inversions, and incubated for 10 min at RT. Extractions were centrifuged for 10 min at 17,900g, and the upper aqueous phase was transferred to new microfuge tubes and the DNA precipitated by adding 0.5 μL GlycoBlue coprecipitant (Invitrogen AM9515), 0.1 volume of 3 M sodium acetate (pH 5.2), and 2.5 volumes ice-cold 100% ethanol. The DNA was pelleted by centrifuging at 20,000g for 10 min at 4°C and washed by repeating the centrifugation with 1 mL of ice-cold 70% ethanol. The tubes were inverted over a tube rack and air-dried for 15 min before resuspension in 200 μL of buffer A. All samples were mixed with 5 μL of RNase A (Invitrogen AM2271) and incubated for 1.5 h at 37°C. The DNA purification with phenol:chloroform:isoamyl alcohol was repeated, and residual phenol was removed with a second extraction by adding 200 μL chloroform:isoamyl alcohol 24:1 to all samples and repeating the extraction procedure. The DNA ethanol precipitation, washing, pelleting, and drying were repeated before resuspension in 25 μL of 10 mM Tris (pH 7.5) and pooling of all samples.
C. reinhardtii gDNA extraction
C. reinhardtii (cc-5155, Chlamydomonas Resource Center) was grown in TAP media until log phase (2 × 106 to 6 × 106 cells/mL) under continuous illumination at 100 μM photons/m2/sec. Cells were pelleted, and DNA was extracted from the cell pellet as described above for A. thaliana cauline leaves.
D. melanogaster 45-min embryo gDNA extraction
D. melanogaster early embryos were collected from young (<7 d) male and female wild-type flies. Two biological replicates were collected 2 mo apart. Approximately 100–200 flies were transferred to large embryo collection cages and fed baker's yeast paste for 3 d before collection. Fresh 100-mm molasses agar plates were exchanged daily. Timed egg lays were used to stage embryos by adding a fresh molasses agar plate with yeast paste to the collection cage for 30 min; this was then replaced with a fresh plate for the next collection cycle. The plate was submerged in 1× PBS, and the eggs were dislodged with a paint brush, then inverted over a funnel into an egg basket, and rinsed with additional PBS. The egg basket was then inverted over a 50-mL conical tube, and the eggs were collected with PBS and pelleted for 3 min at 300g. The PBS was removed, and the eggs were transferred with a wide-bore pipette tip in ∼1 mL of PBS to a 1.5-mL microfuge tube and pelleted for 2 min at 500g. The PBS was removed, and the egg pellet was flash-frozen in liquid nitrogen and stored at −80°C. On average, the embryo pellets were frozen 15 min after removal of the agar plate from the collection cage, yielding embryos between 15 and 45 min of development (referred hereafter as 45-min embryos). The pellets were transferred to 1.2 mL of buffer A supplemented with 1% sodium dodecyl sulfate, 0.5% polyvinylpyrrolidone, and fresh 1% 2-mercaptoethanol, and DNA was extracted as described above for A. thaliana cauline leaves.
Antibodies
The primary antibodies used in this paper are as follows:
-
Rabbit polyclonal anti-N6-methyladenosine antibody (MilliporeSigma ABE572–RRID:AB_2892213), referred hereafter as “MP1”;
-
Rabbit polyclonal anti-N6-methyladenosine antibody (MilliporeSigma ABE572-I–RRID:AB_2892214), referred hereafter as “MP2”;
-
Mouse monoclonal anti-N6-methyladenosine antibody (Active Motif 61756–RRID:AB_2793759), referred hereafter as “AM1”;
-
AbFlex N6-methyladenosine (m6A) antibody (rAb) (Active Motif 91261–RRID:AB_2892216), referred hereafter as “AM2”;
-
N6-Methyladenosine (m6A) antibody (pAb) (Active Motif 61495–RRID:AB_2793658), referred hereafter as “AM3”;
-
Rabbit polyclonal anti-N6-methyladenosine antibody (Synaptic System Ab. 202 003–RRID:AB_2279214), referred hereafter as “SS1”;
-
Mouse monoclonal anti-N6-methyladenosine antibody (Synaptic System Ab. 202 111–RRID:AB_2619891), referred hereafter as “SS2”;
-
Rabbit polyclonal anti-N6-methyladenosine antibody (Abcam ab151230–RRID:AB_2753144), referred hereafter as “AB1”;
-
Anti-N6-methyladenosine (m6A) antibody, rabbit monoclonal (Sigma-Aldrich SAB5600251–RRID:AB_2892215), referred hereafter as “SA1”;
-
N6-Methyladenosine (m6A) antibody (Diagenode C15200082-50–RRID:AB_2892212), referred hereafter as “DG1”;
-
N6-Methyladenosine (m6A) (D9D9W) rabbit mAb (Cell Signaling Technology 56593–RRID:AB_2799515), referred hereafter as “CS1”; and
-
EpiMark N6-methyladenosine enrichment N6-methyladenosine antibody rabbit mAb (New England Biolabs E1611AVIAL–RRID:AB_2923416), referred hereafter as “NB1.”
The secondary antibodies used in this paper are as follows:
-
Anti-rabbit IgG HRP-linked (Cell Signaling Technology 7074–RRID:AB_2099233) and
-
Anti-mouse IgG HRP-linked (Cell Signaling Technology 7076–RRID:AB_330924).
DNA-m6A dot-blot
DNA sample dilutions were made in a 20× SSC buffer in a 96-well plate followed by denaturation for 10 min at 95°C. Nitrocellulose membrane was wetted in 20× SSC buffer, secured in a HYBRI-DOT manifold (Life Technologies), and then washed under vacuum with 20× SSC buffer, followed by the addition of denatured DNA samples to each well. Membrane was then placed on a dry Whatman filter paper and cross-linked with 125 mJ in a GS gene linker UV chamber (Bio-Rad) using the C-L setting. The membrane was then washed with 20 mL of 1× TBS-T (10 mM Tris at pH 7.5, 0.25 mM EDTA, 150 mM NaCl, 0.1% TWEEN-20) and blocked in 15 mL of 1× TBS-T + 5% nonfat dry milk for 1 h at RT. All primary antibodies were diluted 1:1000 in 10 mL 1× TBS-T + 5% nonfat dry milk, and primary incubation was performed overnight at 4°C on a slow shaker. The blot was washed three times in 20 mL of 1× TBS-T for 15 min total. Secondary antibodies were diluted 1:2000 in 15 mL 1× TBS-T + 5% nonfat dry milk and incubated with the blot for 1 h at RT. Three washes were performed, and the blot was developed with Pierce ECL plus western blotting substrate (Thermo Fisher Scientific 32132) and imaged using film or the Azure Biosystems imager.
DNA-m6A dot-blot image analysis
Imaged dot-blots were converted to 8-bit TIFF files, and total summed pixel intensity was recorded using ImageJ. For each blot, intensity values were normalized relative to the highest treated DNA concentration. For samples with sufficient data points, a dose response model (drm) was generated from the normalized data using the drm function found in the R package “drc” (R Core Team 2022). A four-parameter log logistic function (“LL.4”) was used, with fixed parameters set to (NA, 0, 1, NA). EC50 values were generated from the resulting drm, using the effective dose (“ED”) function. Relative antibody selectivity was calculated by backfitting untreated sample data to the drm generated by the corresponding treated sample and normalizing to the input DNA concentration (see Data access).
DNA-m6A immunoprecipitation sequencing (m6A-DIP-seq)
gDNA was sheared using a Covaris M220 focused-ultrasonicator (peak power: 75.0; duty factor: 15.0%; cycles/burst: 200; duration: 720 sec; water bath temperature: 20°C) to achieve a fragment length of ∼100 bp. Libraries were constructed from these sheared DNA samples using the NEBNext Ultra II DNA library prep kit for Illumina (New England Biolabs E7645S) and NEBNext multiplex oligos for Illumina index primers sets 1 and 2 (New England Biolabs E7335S and E7500S). To increase input DNA above single library limits (1 μg) for samples with the lowest DNA-m6A abundance (D. melanogaster embryos and A. thaliana cauline leaves), duplicate end prep and adaptor ligation reactions were prepared and combined into single immunoprecipitations (including IgG controls). The samples were end-repaired by combining 1 μg of DNA in 50 μL of 10 mM Tris (pH 8.0) with 7 μL of end prep reaction buffer and 3 μL of end prep enzyme mix in PCR strip tubes. Samples were mixed and placed on a thermocycler using the end repair program (30 min at 20°C, 30 min at 65°C, and then hold at 4°C). Adaptors were ligated to samples by adding 2.5 μL of the adaptor for Illumina, followed by 30 μL of the ligation master mix, and then 1 μL of the ligation enhancer. Samples were incubated for 15 min at 20°C, and then 3 μL of USER enzyme was added and incubated for 15 min at 37°C. For cleanup, 116 μL (1.2×) of Agencourt AMPure XP beads (Beckman Coulter A63880) was added to each sample and mixed, followed by 10 min of RT incubation and 10 min of magnetic separation. The supernatant was discarded, and the beads were washed twice on the magnet with 200 μL of fresh 80% ethanol and then dried for 4 min. Samples were removed from the magnet, and 50 μL of 10 mM Tris (pH 7.5) was added followed by 10 min of RT incubation and 10 min of magnetic separation. The samples were transferred to new 1.5-mL LoBind tubes, combined with 20 μL of 100 μM blocking oligo (AGATCGGAAGAGCGTC; duplicate reactions are pooled and combined with 40 μL of blocking oligo), and incubated for 10 min at 95°C to denature the DNA. Tubes were then transferred to an ice/water slush mix for 10 min for rapid cooling. To each sample, 325 μL 0.1× TE buffer (255 μL for pooled samples) and 100 μL 5× IP buffer (50 mM Tris at pH 7.5, 750 mM NaCl, 0.5% IGEPAL CA-630) were added in addition to 5 μg of anti-N6-methyladenosine antibody (see above for catalog numbers) or normal rabbit IgG (MilliporeSigma NI01-100UG). Samples were incubated for 12 h on a rotator at 4°C. Twenty-five microliters of Protein A Dynabeads (Invitrogen 10001D) per sample was prepared by removing supernatant after magnetic separation and washing four times in 1× IP buffer (10 mM Tris at pH 7.5, 150 mM NaCl, 0.1% IGEPAL CA-630) followed by resuspension in 25 μL 1× IP buffer. The washed Protein A beads were added to each sample and rotated for 4 h at 4°C. Samples were then washed with 750 μL 1× IP buffer six times, with each wash consisting of 5 min of rotation at 4°C. DNA was then eluted by adding 48 μL of Proteinase K digestion buffer (20 mM HEPES at pH 7.5, 1 mM EDTA, 0.5% SDS) and 2 μL Proteinase K to the beads and incubating for 1 h at 50°C with 1200 rpm of shaking. Samples were separated on a magnet, and the supernatant was transferred to new strip tubes. For cleanup, 90 μL (1.8×) of AMpure XP beads was added to each sample following the same procedure above but with elution in 17 μL 10 mM Tris (pH 7.5) followed by quantification with the Qubit ssDNA assay kit (Invitrogen Q10212). For PCR enrichment, 15 μL of each sample was transferred to a new strip tube and combined with 5 μL of Universal PCR primer, 5 μL of Index primer (both provided in NEBNext Multiplex Oligos for Illumina index primers sets 1 and 2), and 25 μL NEBNext Ultra II Q5 master mix (PCR cycling conditions: 30 sec at 98°C; five to seven cycles for 10 sec at 98°C and then for 75 sec at 65°C; final incubation for 5 min at 65°C). For cleanup, 60 μL (1.2×) of AMpure XP beads was added to each sample following the same procedure above, but with elution in 33 μL of 10 mM Tris (pH 7.5). Libraries were quantified using a Qubit dsDNA HS assay kit (Q32851), and size distribution was checked on an Agilent Bioanalyzer high-sensitivity DNA chip. Libraries were sequenced using an Illumina HiSeq 4000 to a read depth of about 20 million reads using paired-end 76-bp read lengths.
Input control samples
gDNA from S2 cells, A. thaliana cauline leaves, D. melanogaster 45-min embryos, and C. reinhardtii cells were sheared as above and underwent library construction as above. These samples were then amplified by adding the NEBNext Ultra II Q5 master mix as above (Program: 30 sec at 98°C; three cycles for 10 sec at 98°C and then for 75 sec at 65°C; final incubation for 5 min at 65°C) and cleaned using 1.2× AMpure XP beads as above. Libraries were sequenced using an Illumina HiSeq 4000 to a read depth of about 20 million reads using paired-end 76-bp read lengths.
DNase-seq
Previously published DNase-seq data were used (accessed from the NCBI Gene Expression Omnibus [GEO; https://www.ncbi.nlm.nih.gov/geo/] under accession number GSE146942) (Stergachis et al. 2020).
Absolute quantification of DNA-m6A levels
The absolute quantity of DNA-m6A in S2 cell nuclei treated with 20 U of Hia5 for 10 min was determined using Pacific Biosciences (PacBio) circular consensus sequencing. These PacBio data were previously published by our group (GEO accession number GSE146942) (Stergachis et al. 2020). Specifically, we restricted our analysis to reads with 50 or more circular consensus passes, which enables the highly accurate assessment of absolute m6A levels (O'Brown et al. 2019). The methylation profile on each read was determined as previously described (Stergachis et al. 2020), and the average adenine methylation level was then calculated, which was 14%. The absolute quantity of DNA-m6A in other samples was derived by comparing the relative dot-blot signal intensity to that of the 20 U Hia5 sample.
Identification of DNA-m6A-modified genomic loci
Reads were mapped to their respective genome as previously described (Stergachis et al. 2020). Genome builds used were dm6 (D. melanogaster), TAIR9 (A. thaliana), and creinhardtii_281_v5.0 (C. reinhardtii). Signal tracks were generated using BEDOPS (Neph et al. 2012), and signal was normalized to 1 million reads. For the Drosophila S2 samples, DNA-m6A peaks were identified using the MACS2 software (Zhang et al. 2008) using the IgG control sample as the background. For the D. melanogaster 45-min embryo and A. thaliana cauline leaf samples, DNA-m6A peaks were identified using the MACS2 software (Zhang et al. 2008) using both the IgG control sample and the input control sample as the background. For the C. reinhardtii samples, DNA-m6A peaks were identified using the MACS2 software (Zhang et al. 2008) using the input control sample as the background. A Q-value cutoff of 0.01 was used for each sample. Biological replicates were performed for the D. melanogaster 45-min embryo and A. thaliana cauline leaf samples, but only a single replicate was used for analyses.
DNA-m6A loci genomic positions
A. thaliana gene and transposable element TSSs were identified using the “TAIR9_GFF3_genes_transposons.gff” annotation file downloaded from The Arabidopsis Information Resource (Huala et al. 2001). C. reinhardtii gene TSSs were identified using the “Creinhardtii_281_v5.5.gene_exons.gff3” annotation file, and C. reinhardtii repeat elements were identified using the “Creinhardtii_281_v5.5.repeatmasked_assembly_v5.0.bed” annotation file, both of which were downloaded from the Phytozome JGI (Merchant et al. 2007). The position of the center of each DNA-m6A peak to each of these element classes was determined using BEDOPS (Neph et al. 2012).
DNA-m6A motif enrichment
The ApT motif was derived from Beh et al. (2019). The GAGG motif was derived from Greer et al. (2015). The RRACH motif was derived from Woodcock et al. (2019). Motif enrichments were calculated by comparing the number of motifs identified in DNA-m6A peaks from a given species versus the number of motifs identified in shuffled sequences of DNA-m6A peaks from that same species. Motif instances were identified using FIMO (Grant et al. 2011) with a P-value cutoff of 0.001 for the GAGG and RRACH motifs and 0.01 for the ApT motif. De novo motif discovery using MEME suite (Bailey and Elkan 1994) was performed on A. thaliana cauline leaf DNA-m6A peaks. Motif instances within A. thaliana cauline leaf and C. reinhardtii DNA-m6A peaks were identified using FIMO and a P-value cutoff of 0.0001.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE197747. All code used in quantifying DNA dot-blots is available at GitHub (https://github.com/StergachisLab/m6A-Antibody-Analysis) and as Supplemental Code.
Competing interest statement
A.B.S. has filed a patent application related to the work described here. The title of the patent application is “Methods, Compositions, and Kits for Identifying Regions of Genomic DNA Bound to a Protein.” The U.S. Application was filed on April 2, 2021, PCT/US2021/025644.
Acknowledgments
We thank Aaron Aker for kindly providing C. reinhardtii cells. We thank Jennifer Bush for kindly providing A. thaliana plants. We thank Richard Binari for kindly providing D. melanogaster flies. We thank Eric Haugen and Richard Sandstrom for computational assistance. We thank L. Stirling Churchman and John A. Stamatoyannopoulos for helpful comments on the manuscript. This research was supported by National Institutes of Health grants DP5-OD029630 (Office of the Director) and GM007748 (National Institute of General Medical Sciences). A.B.S. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund and is a Pew Biomedical Scholar.
Author contributions: A.B.S. and B.M.D. designed the experiments. A.B.S., B.M.D., and B.J.M. performed the experiments. A.B.S. performed the computational analyses. A.B.S. and B.M.D. wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276696.122.
- Received March 2, 2022.
- Accepted February 2, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








