
Simultaneous profiling of host expression and microbial abundance by spatial metatranscriptome sequencing
- Lin Lyu1,8,
- Xue Li1,8,
- Ru Feng1,8,
- Xin Zhou2,8,
- Tuhin K. Guha2,
- Xiaofei Yu3,
- Guo Qiang Chen4,
- Yufeng Yao5,
- Bing Su1,
- Duowu Zou6,
- Michael P. Snyder2 and
- Lei Chen1,7
- 1Shanghai Institute of Immunology, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China;
- 2Department of Genetics, Stanford University School of Medicine, Stanford, California 94305, USA;
- 3State Key Laboratory of Genetic Engineering, School of Life Sciences, Shanghai Engineering Research Center of Industrial Microorganisms, Fudan University, Shanghai 20082, China;
- 4Department of Pathophysiology, Key Laboratory of Cell Differentiation and Apoptosis of Chinese Ministry of Education, State Key Laboratory of Oncogenes and Related Genes and Chinese Academy of Medical Sciences, Shanghai Cancer Institute, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China;
- 5Laboratory of Bacterial Pathogenesis, Department of Microbiology and Immunology, Institutes of Medical Sciences, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China;
- 6Department of Gastroenterology, Ruijin Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China;
- 7Renji Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai 200127, China
-
↵8 These authors contributed equally to this work.
Abstract
We developed an analysis pipeline that can extract microbial sequences from spatial transcriptomic (ST) data and assign taxonomic labels, generating a spatial microbial abundance matrix in addition to the default host expression matrix, enabling simultaneous analysis of host expression and microbial distribution. We called the pipeline spatial metatranscriptome (SMT) and applied it on both human and murine intestinal sections and validated the spatial microbial abundance information with alternative assays. Biological insights were gained from these novel data that showed host–microbe interaction at various spatial scales. Finally, we tested experimental modification that can increase microbial capture while preserving host spatial expression quality and, by use of positive controls, quantitatively showed the capture efficiency and recall of our methods. This proof-of-concept work shows the feasibility of SMT analysis and paves the way for further experimental optimization and application.
Spatial transcriptomic (ST) sequencing has revolutionized the biological research field. This class of technologies combine the strength of two pillars of modern biological research: sequencing and imaging. One form of ST mapping works by capturing the messenger RNA from a permeabilized tissue slice and labeling these RNA molecules with 2D spatial barcodes (Ståhl et al. 2016; Salmén et al. 2018). Generally, this has been applied to complex human tissues such as the brain and tumors (Berglund et al. 2018; Thrane et al. 2018; Asp et al. 2019; Maniatis et al. 2019; Moncada et al. 2020; Fawkner-Corbett et al. 2021; Maynard et al. 2021). In the intestines and other organs, microbes live alongside or within close proximity to host cells, and metagenomic sequencing has long been used to study the complex microbial composition on various host body sites. These microbiome studies have greatly improved our understanding of the human biology, and microbes have been found in novel places (Nejman et al. 2020; Poore et al. 2020) and showed intriguing dynamics across solid tissue (Ha et al. 2020). However, they generally lacked spatial resolution, and attempts at addressing this limitation is just emerging (Shi et al. 2020; Duncan et al. 2021). Inspired by a recent study about COVID-19 in which virus sequences were recovered from host single-cell transcriptomic data and analyzed alongside host data (Bost et al. 2020), we tested if it is possible to capture microbial sequences in ST sequencing and showed that validated microbial information can be obtained that can shed new light on host–microbe interactions.
Results
SMT extracts microbial abundances alongside host expression
To evaluate the microbial sequence content from spatial transcriptome data, we performed ST sequencing, using the Visium kit from 10x Genomics, on both human and murine intestinal samples, in which the presence of microbes is well-known. The human samples included dissected colon samples from two colorectal cancer patients. For each patient, samples from the tumor site and histologically normal site distant from the tumor were embedded together and put in the same capture window, and two replicate windows were processed. The murine samples came from both the small intestine and large intestine, and cross-sectional slices from three mice were included in a single window (Fig. 1A; Supplemental Fig. S1C). The murine samples were rinsed so as to remove fecal material while preserving the mucus layer. The human samples were rinsed at the operation room before being transported to downstream processing. Each slice was sequenced to at least 85,000 reads per covered spots, and the captured RNAs were on average 11,000 per spot, similar to that of published results (Ståhl et al. 2016).
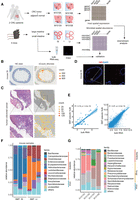
Spatial metatranscriptomics (SMT) of human and murine intestines. (A) A schema of the SMT framework and design of the first part of study. Note that for CRC, tumor and adjacent normal tissue were embedded in the same window and designated T and N, respectively. (B,C) H&E staining of the tissue sections (left) and corresponding spatial feature plot showing microbial sequence per-spot captured (right) for murine small intestine (B) and human colorectal cancer patient tissue, from patient M1016, with zoomed view (C). Representatives are shown, and an additional example can be found in Supplemental Figure S1. (D) FISH result for mouse small intestine section stained for bacteria 16S. Scale bars: 300 μm (left), 20 μm (right). (E) Correlation between SMT-generated pseudobulk measurements (x-axis) and bulk RNA-seq measurements (y-axis) for microbial abundance at genus level (left) and host gene expression (right). Spearman's correlation coefficients and P-values are shown. (F,G) Stacked bar plot showing microbial abundances for mouse (F) and human (G) samples at family level; only the top ones were shown.
We developed an analysis pipeline to extract microbial information from the ST data. In short, reads unmapped to the host genome were first screened to remove low-complexity reads, then were aligned against the NCBI Nucletide database (https://www.ncbi.nlm.nih.gov/nucleotide/) using BLAST, and were subsequently assigned taxonomic IDs. When there are conflicting assignments for reads belonging to the same unique molecular identifier (UMI) group, a strategy combining majority voting and most recent common ancestor was used to resolve it. The end result is a spatially resolved microbial abundance matrix alongside the spatial host gene expression matrix generated via the standard ST analysis. We compared our procedure with Kraken 2 (Wood et al. 2019), a tool commonly used in taxonomy assignment for metagenomic reads, and found our tailored procedure produced less dubious calls (Methods) (Supplemental Table S1).
The percentage of microbial sequences in a data set varies across different samples, ranging from 10−6 in a human sample to 10−3 in murine ones. The Visium kit uses poly(T) to capture messenger RNA, and in theory, microbial sequences without a poly(A) tail would be depleted. Although this was indeed the case, some random priming did occur, and sequences without long stretches of poly(A) were still captured. This is in line with reports from the RNA-velocity work (La Manno et al. 2018), in which immature messenger RNAs, which lack poly(A) tails, account for ∼25% of the typical data from the 10x Genomics Chromium single-cell platform, which also uses poly(A) capture. Furthermore, ribosomal RNA is the most abundant RNA species in a cell, accounting for >80% of total RNA. Ribosomal RNA is also the molecular marker of choice for nucleated microbial taxonomy. Accordingly, the majority of the microbial sequences recovered in SMT were from ribosomal RNAs (Supplemental Fig. S1F). In addition, most microbial species, except airborne fungi ones, showed clear spatial patterns (agglomerated microbial signal shown in Fig. 1B,C). For these reasons, the microbial signals obtained from our SMT analysis are unlikely to be merely artifacts. For comparison, we applied our microbial sequence extraction method on single-cell sequencing data derived from unsorted intestinal samples, and the microbial signal thus produced was spread relatively evenly across different cell types and likely represents noise or contaminants (Supplemental Fig. S1A,B). This may be because only intracellular bacteria can be captured in cleaned single-cell suspension and these bacteria are rare and because the gentle treatment used inside a droplet is ineffective for lysing them.
Evaluation of contamination level and biases in SMT
To further evaluate the extent of contaminating microbial sequences during experiments versus true organ resident microbial signals, we plotted the microbial abundance across all spatial spots, regardless of whether it is covered by tissue or not (Fig. 1B,C). For both human and mouse, the luminal side of the intestines harbors more microbial signal, as is expected. The human tumor tissue showed a higher abundance of microbes and also more microbes penetrated deeper into the tissue, as would be expected from the compromised barrier function in tumors and their folded overgrowth. In addition, the pattern of microbial distribution is similar between replicates, both spatially and in terms of taxa composition (Fig. 1F; Supplemental Fig. S1D). These clear and expected patterns validated our SMT methods as noise or contaminants would show up relatively randomly across the capture window.
To compare results with alternative methods, for the mice small intestine, we visualized a neighboring section of the embedded tissue by fluorescence in situ hybridization (FISH) with the bacterial probe EUB338, targeting 16S gene (Fig. 1D). The FISH result was in agreement with SMT, showing bacteria presence concentrated in the intestinal lumen side. Furthermore, each window contained samples from three mice, and the bacteria loads of these samples differ. This difference could be a bona fide biological difference, combined with variance in sample processing, or a slight difference in sample origin, that is, more distal or proximal. Indeed, the order of the bacteria loads were observed to be consistent in proximate sections used for FISH and SMT, as were expected. All these observations lend credibility to SMT.
Next, we evaluated the biases in the microbial signals from SMT. During the standard Visium ST process, the host tissue sections are permeabilized to release its RNA, and this process is far from ideal for microbial sequence capture. This brought up the question if certain microbial species would be preferentially analyzed and represent a biased picture of the true microbiome (some biases are always expected). We were also concerned that if the capture efficiency of microbial sequences was too low, there would be a great fluctuation in the results, and they would be unreliable. To address these issues, for the embedded murine samples, we extracted RNAs from neighboring sections and performed total RNA sequencing, in which poly(A)-based enrichment was not used and only host ribosomal RNA was depleted. Similar to our SMT pipeline, we processed the bulk total RNA-seq data to generate microbial abundances, one abundance profile for one window (containing samples from three mice). We then, to compare with the bulk sequencing data, combined the SMT's per-spot abundance profiles for one window into a single pseudobulk profile and evaluated both the correlation of microbial abundances at the genus level as well as that for host genes (Fig. 1E; Supplemental Fig. S1H). The Spearman's correlation of the microbial abundances is positive and significant between the bulk and pseudobulk for the same tissue and is insignificant otherwise, again validating the SMT approach. The pattern is different for host genes, as the correlations are all significant and positive owing to similarity in overall expression. Lastly, the microbial compositions based on SMT were shown to be mostly of intestinal origin based on the literature. Known common contaminants and artifacts were present but at low levels, for example, Ralstonia and Actinomyces; the former is a well-known clinical contaminant that often causes contamination of distilled water and solutions, whereas the latter is a widespread bacteria found in soil and waterbodies (Fig. 1F,G; Ha et al. 2020; Dohlman et al. 2021).
The localized host cell response to cytomegalovirus infection as captured by SMT
An advantage of SMT over 16S sequencing is that by using RNA-seq, SMT captures fungi and viral sequences as well as bacteria ones. Zoonotic viruses generally have mRNA that resemble their hosts, and we can identify one such virus infection case in great spatial detail in one of the CRC patients. In both replicate windows of patient M1016, viral sequences reached as high as 3.4%. These viral sequences were detected across the tissue, possibly owing to spread of the viral particles. They were concentrated in a few spots, reaching as high as several hundred UMIs per spot. On both replicates, the locations of high viral load were the same and likely represented a single focal area of active infection. This virus, identified as Cytomegalovirus, is known to infect fibroblasts, and a deconvolution of the infected spots, which were 55 μm in diameter and usually contained many cells, showed them to be mostly composed of fibroblast (Fig. 2A). We next investigated the immediate consequence of viral infection on these fibroblasts by looking at their expression. To this end, infected spots were compared with uninfected fibroblast-rich spots. Among the differentially expressed genes, interferon-related genes are not found, potentially owing to the immune-compromised nature of the tumor microenvironment. Still, some known participators are found, including HLA-E, which is known to be up-regulated by cytomegalovirus infection, and IL32 and TNIP1, both up-regulated and involved in host defense (Fig. 2B). These genes were discovered before using cellular infection models (Fukushi et al. 1999; Ulbrecht et al. 2000; Kim et al. 2018). SMT is able to recapitulate results from these studies and provide more insights including more genes, under a more natural setting in human and in a spatially resolved way.
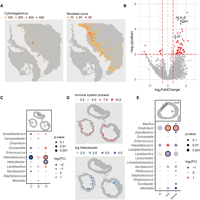
Host–microbe interactions revealed by SMT. (A) Spatial feature plot showing distribution of Cytomegalovirus in patient M1016's section as detected by SMT (left) and distribution of fibroblasts as inferred by its marker gene score (right). (B) Volcano plot showing differentially expressed genes between infected and uninfected fibroblast spots. Highly significant genes (P < 0.01 and |log2FC| > 0.5) were colored red, with several known ones labeled and highlighted in blue. (C) Dot plot showing the fold change and P-value for microbial genera that were differentially distributed in the three sample murine samples as detected by chi-square test, for the mouse large intestine; those with P < 10−8 and |log2FC| > 0.5 were circled. (D) Spatial feature plot showing module score of immune system process (top) and distribution of genus Helicobacter (left). (E) Dot plot showing fold change and P-value for microbial genera that were differentially distributed in the three regions designated “in,” “tissue,” and “out” by chi-square test, for mouse small intestine; those with P < 10−8 and |log2FC| > 0.5 were circled.
SMT identifies highly immunogenic bacteria in murine intestine
The local cellular response to infection shown above is a good testament to SMT's capability in investigating microbe–host interaction. Compared with other spatial methods, FISH for example, SMT's strength comes in its taxonomical resolution and systemic nature. For a proof of concept, we investigated the host–microbe interactions using their spatial correlations under homeostasis, using the murine data set as it included multiple animals and covered a complete cross-section of the intestines. A major challenge in this regard is the uncertainty or variation in the scale of interaction. Another technical issue is the sparse nature of the microbial signals and, to a lesser degree, that of the host genes. To address both issues, a kernel smoothing function is applied to both host expression and microbial signals first (Methods) (Supplemental Fig. S2A). The kernel width is adjustable, and first-degree neighbor was used by default. This way, small-scale spatial interaction in the 100-µm range can be investigated using spatial correlation between the two smoothened signals. However, after multitest correction, no interaction reached statistical significance. If we loosen the criteria, some suggestive interactions can be seen for the more abundant microbes like Helicobacter (Supplemental Fig. S2B). Future development with improved capture efficiency and spatial resolution is required.
Zooming out to larger spatial-scale interactions, we took a different approach wherein microbes and host pathways were evaluated on a per sample basis. There were three samples per window, and sample-level microbe abundances were compared. Specifically, microbial genera were tested for enrichment or depletion in particular samples, against background fluctuations, using the chi-square test, and the microbes with a per-window UMI count larger than five were shown (Fig. 2C). Host pathways were checked for differential enrichment among the different samples. Pathways were used instead of genes to reduce false positives. In the large intestine, Helicobacter is depleted in sample 3. On the other hand, out of the 31 top-level GO pathways under biological processes, the immune system process is also down-regulated in sample 3 (Fig. 2D; Supplemental Fig. S2C). The correlation of Helicobacter with immune response was documented before, and species in this genus were shown to be synergistic in tumor immunotherapy in a mouse model (Overacre-Delgoffe et al. 2021). Our analysis suggests its high immunogenicity stands out from other bacteria even in seemingly homeostatic states. The difference between these homogenetic animals could be owing to bona fide biological variation and/or has to do with spatial variation in sample origin—whether they were more proximal or distal in the GI track and their proximity to lymphoid structures. A few other bacteria also showed inter-sample differences, for example, Faecalibaculum in the large intestine (Fig. 2C). However, their levels did now seem to closely track any top-level pathways.
SMT recapitulates the migration of Clostridium across intestinal walls
Bacteria species are generally confined in the intestinal lumen. However recent reports suggested certain bacteria, the most prominent being Clostridium, can migrate across the intestinal wall in inflammatory bowel disease and even under homeostatic status (Ha et al. 2020). SMT provides a good opportunity to validate this observation and to investigate similar dynamics in a systematic way. To this end, we used the mouse intestinal data set. For these slices, the spots under tissue, contained within the lumen, or just outside the tissue were designated into regions “tissue,” “in,” or “out,” respectively. And for all bacteria with an overall frequency of more than 20, regional enrichment or depletion was tested using a chi-square test as before. In the small intestine, Clostridium stood out as having a significant enrichment on “tissue” and “out” (Fig. 2E), in agreement with a previous report (Ha et al. 2020). In the large intestine, the Clostridium presence was too low to yield any conclusion. Instead, Enterococcus and Staphylococcus were shown to have outward migratory capabilities with borderline significance (Supplemental Fig. S2E). Nevertheless, we caution that these results are still preliminary, and more SMT samples, preferably with better capture efficiency and resolution, in addition to orthogonal assays are needed to confirm and expand the discovery.
Quantitative analysis of SMT's sensitivity and recall using positive controls
To quantitatively measure SMT's sensitivity and recall, we carried out additional experiments using positive controls and, at the same time, tested a FISH-inspired optimization for SMT. The experimental design is shown in Figure 3A. We used two windows on a Visium chip. Cultured control bacteria, including Pseudomonas aeruginosa and Staphylococcus aureus, were serial diluted and pipetted on three corners of a capture window before tissue sections from the same patient were placed. The last corner was left out as a blank control. Neighboring sections from the same tissue sample were used in the two windows. Then, standard ST procedures were carried out. For one of the windows, designated M1054B (patient M1054, replicate window B), an additional 15-min lysozyme treatment was performed after the permeabilization step (Methods). This step is commonly performed in FISH experiments and was shown to increase capture of diverse species with cell walls (Chassaing et al. 2014; Zou et al. 2018). After SMT data processing, a clear circular-shaped bacterial signal was evident at the two corners with the highest concentrations, with the next corner down in concentration still showing an elevated bacteria load over the blank corner. The reason that there were two clear circular shapes instead of three is likely the steep difference in bacteria concentration. The concentration was 20-fold higher in one corner over the next, and there was a 400-fold difference between the highest corner and the third one. Still, some diffusion of the molecules from the control bacteria did occur because they were actually placed outside the tissue (Methods) and could diffuse away from the point of origin, especially following the crevices at the outer edge of the tissue section Still, based on the strong contrast between the regions with positive control versus those immediately adjacent, one can expect that for real tissue resident microbes, the diffusion effect should be less pronounced. We then calculated, for each corner with a different control bacteria colony forming unit (CFU), the ratio between UMI count and the underlying CFU value. Factoring in the diffusion effect, the signal from one quadrant of a window was pooled together to represent the control bacteria added in that corner. As shown in Figure 3B, the UMI-to-CFU ratio, at the intermediate CFU level, is 0.55 UMI/CFU for Staphylococcus and 0.27 UMI/CFU for Pseudomonas. The trend lines stay linear between the highest two CFU levels and begin to level out owing to the diffusion effect at lower bacteria concentrations. These results suggest approximately one bacteria sequence captured per two to four bacteria cells. We caution that these estimates were crude because only two control bacteria were used and one of them, Staphylococcus, is Gram-positive and has an additional lysis step applied (discussed later), which represented confounding factors. Microbial sequences other than the applied control bacteria were also quantified, and some “spillover” in taxonomy was evident as control bacteria sequences were called incorrectly, usually assigned to closely related organisms. These spillover signals can be identified as their per-spot correlations with the control bacteria are abnormally high (Supplemental Fig. S3A). These spillover signals were calculated to be 8.13% for the Staphylococcus and 1.8% for Pseudomonas, representing a positive recall of >90%.
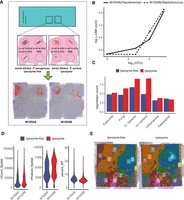
Capture efficiency and recall of SMT as measured by positive controls and evaluation of experimental optimization for SMT. (A) Study design of the positive control and optimization experiments and, shown at the bottom, spatial distribution of the microbial sequences captured by SMT superimposed on tissue histology, showing clear circular patterns. (B) Control bacteria sequences captured by SMT at each corner (measured as total microbial UMI count; y-axis) against actual count of bacteria applied at that corner (measured as CFU; x-axis). (C) Bar plot showing microbial sequence capture with or without the additional lysozyme step. (D) Violin plots of host gene UMI count, gene count, and mitochondria percentage (common measurements to QC single-cell and ST data) with or without lysozyme treatment, no significant difference could be observed. (E) Spatial dim plots showing clustering of spatial spots using host expression data with or without lysozyme treatment; the similar pattern between the two conditions indicated the effect introduced by lysozyme treatment on host gene expression was minimal.
Experimental optimization improves capture of microbes while maintaining host expression quality
With the control signals and their spillover filtered, the remaining microbial signals were treated as residential and used to evaluate the effect of the additional lysis step in the experimental procedure. Between these two conditions, microbial signals from different kingdoms, with Gram-positive and Gram-negative bacteria separated, were compared. As expected, captured transcripts from Gram-positive and Gram-negative bacteria, prokaryotes, and fungi are all significantly increased, with bacteria transcripts increased fourfold on average (Fig. 3C). At the same time, the per-spot UMI count, gene count, and mitochondria gene percentage, which are commonly used in quality checks for single-cell or ST data, remained the same (Fig. 3D). Furthermore, the spots from the two windows were clustered together, and the resulting spatial patterns were nearly identical, some shift between the two sections notwithstanding (Fig. 3E). In summary, the added lysozyme treatment greatly increased the microbial detection with no noticeable negative effect on the host gene expression.
SMT analysis on less-colonized samples
Intestinal samples are known to harbor considerable microbes. Finally, to gain a better understanding of SMT pipeline performance, it was applied on other less-colonized tissue types including the brain and lung. For the brain, raw reads from four mouse brain samples, including two from sagittal anterior and two from sagittal posterior, were downloaded from the 10x Genomics official website (https://www.10xgenomics.com). For the lung, six samples from three lung cancer patents were downloaded from the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) accession number PRJNA827183. After the standard SMT process, the normalized bacteria loads of the samples, calculated as ratio of microbial reads among the total, were compared with that of the previous human and mouse intestinal samples. The samples from the less-colonized tissue indeed have a lower microbial load, with brain ones having the lowest and the ones for lung at slightly higher levels (Supplemental Fig. S3C). Healthy lung has long been considered sterile before the rise of the microbiome sequencing studies (Whiteside et al. 2021). Our result suggests that even in a lung cancer patient, the microbial load is low. Moreover, the microbes identified in the lung included many fungi, some of which are related to respiratory infections, including Aspergillus and Pneumocystis, and some having skin origins such as Malassezia. The top-ranking bacteria also included ones related to lung disorders, like Moraxella and Lactobacillus (Whiteside et al. 2021). We caution that this particular data set is without publication or detailed protocol as of the time of writing, and better controls would be needed to distinguish true resident microbes from air or skin contaminations introduced during sample handling. For the mouse brain sample, a spatial feature plot showing the distribution of the microbes on the capture window was also shown (Supplemental Fig. S3D). After the removal of likely endogenous virus sequences, Staphylococcus became the top microbe detected in all four brain samples and accounts for >50% of the microbial sequences in each case. Staphylococcus is a well-known tissue contaminant (Foster 1960).
Discussion
Our work showed that SMT sequencing can simultaneously profile host tissue gene expression and microbial abundance in a spatially resolved manner. Our analysis framework can extract spatial microbial abundance information from data generated by currently available commercial kits, effectively lifting suitable ST data to SMT status. We showed that, with careful experimental design and vigorous contaminant control and filtering, true signals can be discerned from background noises and that the biases were low. Moreover, comparisons with the FISH result suggested the current version of SMT method to be at a similar level of sensitivity. Nevertheless, we expect that in its current form, for significant findings in humans, SMT likely will require a large number of samples, which can be an economical burden. Part of the reason has to do with technical issues like noise, limited resolutions, etc., that will be ameliorated with future development. Another reason is shared among microbiome studies in that the microbiome is highly diverse and dynamic, especially in humans. As an example, the two tumors used in these studies showed a consistent difference in microbial load on their tumor tissue. This difference could be related to the fact that one of the tumors is tertiary lymphoid structure positive and the other negative. Tertiary lymphoid structures are lymphocyte aggregates occurring outside of the lymph node or other secondary lymphoid organs. They are places where lymphocytes are educated to mount better defenses against invading pathogens or indigenous threats. The sample from human CRC patient M1015, being TLS positive, has a microbial load of 4.2 × 10−7 (calculated as microbial read ratio), whereas for M1016, TLS negative, it is 4.4 × 10−5. Further SMT experiments or alternative assays aimed specifically at that hypothesis will be needed.
In its current form, SMT technology is not without limitations. Some of them were inherited from the ST platform it is based on, like the spreading of RNA on the capture surface and relatively poor RNA recovery compared with scRNA-seq for example, whereas some limitations were unique to SMT, like sensitivity to contamination and a general low signal-to-noise ratio. To overcome these, especially in dealing with a less-colonized tissue type, careful control of experiment condition, use of large sample number, and replication may be required. To help potential users in better evaluating possible contamination, we would like to offer the following experience. First, the sectioning action did not make a considerable contribution to the spread of the microbes as we recorded the sectioning blade direction and did not observe any noticeable pattern if considerable microbes were spread by the blade. Second, the spread of RNA on the capture surface is expected to have a minor contribution, mostly at short range, as shown in the original ST paper and from the observation of the host expression and microbial distribution, especially with replication. Third, whenever the tissues are exposed, during operation or transportation, they will collect contaminating microbes. Care should be taken to minimize this, and it can be further reduced by sectioning if care is taken at that step. The remaining contaminants were generally easy to identify as a large portion of them will likely not be associated with the studied tissue or will already be documented as common contaminants. They also show pronounced batch effects and do not restrict to tissue type or origin.
The ST technologies that enabled SMT are rapidly evolving (Chen et al. 2022), reaching higher resolution and obtaining more sequences per area, translating to higher sensitivity. SMT, too, will benefit from these improvements. The current SMT framework can only generate a microbial abundance table, but in the foreseeable future, by simply optimizing microbial RNA extraction or by using specifically designed capture oligos, it may become possible to actually profile the microbial transcriptomes. It seems single-cell spatial transcriptomic methods that replace poly(T) with random hexamer primers, which will benefit their application to microbial sequences, are already in development (https://www.youtube.com/watch?v=3YjGgGk8jn0). Similarly, research is ongoing in our laboratory to generate higher-resolution and more-sensitive bacteria taxonomic information by adding an adapter oligo for bacteria ribosomal RNA to the vanilla ST process. With an improvement in coverage, it will become possible to start examining microbial gene expression other than ribosomal RNA and further infer their functions. Using SMT in its current form, only the most abundant bacteria can have sufficient sequence coverage to enable functional investigation. For a proof of concept, the genic annotation for Lactobacillus, the most abundant genus from the mouse small intestine, is shown in Supplemental Figure S4.
Improvement may also come in the form of better sample preparation. For intestinal samples, mucus-preserving fixative can be used to avoid detaching of the microbes and skewing of the spatial distribution and ideally to obtain a more comprehensive overview of the microbes (Hasegawa et al. 2017). However, the 10x ST platform used in this study requires freshly frozen samples for RNA yield and is thus not readily compatible with this protocol. Further experimental optimization or alternative ST platforms are required. With these improvements and a further reduction in cost, SMT or similar technologies can become better alternatives to some of the common microbiome sequencing methods used today. For tissues in which microbes are abundant, such as the intestines, SMT enables the systemic study of host and microbe interaction. In other more sterile tissues, SMT can reveal the consequence of microbial residence or intrusion, for example, helping resolve how microbes travel to remote tumor site (as in nonintestinal solid cancers) and helping settle the debate on whether a certain body site is sterile or not, for example, fetal organs (Kennedy et al. 2021; Mishra et al. 2021).
Through the past decade, microbiome sequencing has revolutionized the way microbe and host interactions are studied, and most biological samples imaginable have had their microbiome sequenced. However, traditional microbiome sequencing is bulk in nature and suffers from the same limitations as bulk RNA-seq. SMT now enables microbiome study with fine-scale spatial resolution and the ability to study host and microbes together from the same experiment. Another interesting historical perspective was provided about a decade ago when RNA-seq was just gaining popularity: Researchers interested in infection dynamics coined the term “dual RNA-seq” to describe what was essentially meta-RNA-seq used to investigate host and microbial expression simultaneously (Westermann et al. 2012). The work presented here paves the way for future development of SMT technology, which will see further increased sensitivity and taxonomical and spatial resolution. With their high-throughput and systematic nature, SMT and technologies in the same vein will help address unmet challenges in the field of microbiomes, immunology, and beyond.
Methods
Human and murine sample processing
All human colon resection samples were obtained from colon cancer patients with written informed consent approved by the medical ethics committee of Ruijin Hospital, Shanghai Jiao Tong University. Before surgery, the patients took laxatives to clear the intestinal contents. After surgery, the tumor tissue and adjacent normal tissue were placed into conical tubes containing Roswell Park Memorial Institute (RPMI) media supplemented with 2% fetal bovine serum (FBS) and placed on ice for immediate transportation to the laboratory across the street, where they were gently brushed in cold PBS to clean the residual feces, then cut into 1- to 2-cm3 pieces, and embedded in optimal cutting temperature (OCT) sectioning media (Thermo Fisher Scientific) by submersion in isopentane (2-methylbutane, Sigma-Aldrich) precooled to −80°C in dry ice.
For murine samples, 6-wk-old male C57BL/6J mice were ordered from Shanghai SLAC Laboratory Animal. All mice were maintained in the specific pathogen-free (SPF) mouse facilities at Fudan University. All experimental procedures described in the study were approved by the institutional animal care and use committees (IACUC) of Fudan University. Feces and serum were collected and stored at −80°C. Small intestine and colon tissue were first cleaned of adipose tissue. Next, their luminal content was removed by washing it with cold PBS using a syringe. They were subsequently washed in cold PBS and then embedded in OCT sectioning media (Thermo Fisher Scientific) by submersion in isopentane (2-methylbutane, Sigma-Aldrich) precooled to −80°C in dry ice and stored at −80°C before the ST experiments.
ST experiments and sequencing
Before ST experiment, cryosections were evaluated of their RNA integrity by taking 10 slides of 10-μM thickness, went through RNA extraction by TRIzol (Invitrogen 15596026), and were analyzed by Agilent 2100. An RNA integrity number (RIN) > 7 is considered qualified. Before using a new tissue for Visium spatial gene expression libraries, the permeabilization time was optimized. The sections were fixed, stained, and then underwent permeabilization of different durations: 25 min, 18 min, or 12 min. The permeabilization times that result in the maximum fluorescence signal were selected. Once optimal conditions had been established, each section was cut at 10-μM thickness and placed onto the Visium spatial gene expression slide (10x Genomics, Visium spatial gene expression slide & reagent kit, PN-1000187). Tissue sections were fixed, stained, and then scanned on a Leica DMI8 whole scanner at 10× magnification. Then, for tissue permeabilization, the slides were placed in the slide cassette, and permeabilization enzyme was added on the slide and incubated for the time selected in optimization step. After that, reverse transcription and second-strand synthesis were on the slide, followed with cDNA quantification. The sequencing library construction was performed with a library construction kit (10x Genomics PN-1000190). The cDNA was broken into fragments of ∼200–300 bp, and then the P7 and P5 adapters were added. Sequencing was performed using Illumina NovaSeq 6000 sequencer with a sequencing depth targeting at least 50,000 reads per spot with a paired-end 150-bp (PE150) reading strategy.
ST data processing and analysis
Raw reads were aligned to the human transcriptome GRCh38-3.0.0 reference or mouse transcriptome mm10-3.0.0 reference using 10x Genomics SpaceRanger v.1.0.0, and exonic reads were used to produce mRNA count matrices for these samples. HE histology images were also aligned with mRNA capture spots using SpaceRanger.
The matrix file and HE image were then imported into R (R Core Team 2020) and analyzed using Seurat package (version 4.0.1) (Butler et al. 2018). Downstream analysis was carried out following Seurat's recommended procedure. Briefly, SpaceRanger output was imported into Seurat object, followed by normalization, scaling, variable feature identification, dimension reduction, and clustering to find the general variation of the spots. Distribution of the major cell types was explored using the AddModuleScore function with cell type–specific marker genes.
SMT data analysis
An in-house pipeline was developed to take the BAM file generated by SpaceRanger as input and output a microbial spatial abundance matrix. This matrix, together with the host expression one, can be loaded into a Seurat object, and then spatially resolved microbe–host interaction can be explored using our custom codes. The SMT data analysis was described in the following steps.
Spatial abundance matrix generation
Unmapped reads were extracted from the SpaceRanger BAM file and filtered to remove low-complexity sequences. The filtered reads were then aligned against the nt database with BLASTN 2.10.1+. The BLAST outputs were formatted to preserve query ID, taxID, subject title, alignment length, e-value, identity, coverage, as well as UMI and spatial barcode, which were extracted from the original BAM file. Taxa levels from kingdom to species were called for an individual mapped read using its corresponding taxID with the taxa table downloaded from the NIH taxonomy portal (https://www.ncbi.nlm.nih.gov/taxonomy). One UMI usually contains several reads. To make taxonomy calls for UMIs with conflicting read calls, we use the following strategy. If there is a >80% majority, this majority call is used. Otherwise, a last common ancestor of the conflicting calls is used. This way, a taxID-spatial barcode matrix could be generated.
Removal of environmental contaminants
Before further analysis, microbial abundance matrices were denoised to remove likely contaminants that are distributed across the capture window in a random fashion. In detail, for each evaluated taxon, the total number of spots that contain this taxon and the median distance between these spots were calculated; then a number of permutations, default to 10,000, were performed in which the same number of spots was randomly chosen and the median distance between them calculated. An empirical P-value was generated by calculating the proportion of permutations that produced equal or smaller median distances than real data. A P-value of ≥0.05 indicates likely contaminants. The proportion of removed microbial data at UMI level ranged from 37% in the human samples, which were cleaned better, to 6% in the mouse intestine. This step would only remove taxa that are highly likely to be contaminants, and taxa without enough sequence would be left to be on the conservative side. Some known contaminant taxa that do not reach high abundance can still be excluded from analysis later. We optionally kept a known list of contaminants that can be used to filter the abundance matrix directly.
Importing and using the abundance matrix
The abundance matrix and the host expression matrix were loaded into the same Seurat object for analysis. Specifically, a regular ST Seurat object was generated using the host expression first. The microbial abundance matrix was then processed into a phyloseq object (phyloseq 1.32.0) (McMurdie and Holmes 2013). This phyloseq object was then incorporated into the Seurat object as a separate assay. Custom codes were written that can generate an abundance table at a specified tax level on the fly and add that as an additional assay. This way, the spatial distribution of the microbes could be investigated and visualized in the same way as host gene features.
Spatial correlation between host genes and microbes
For spatial correlation analysis, expression data and microbial counts were first smoothened using a kernel method (Supplemental Fig. S2A). With a kernel radius of one, the UMI count for the center spots was expanded to its nearest six spots with a weight of 0.25. Then correlation between microbes and genes could be carried out using Spearman's correlation or Kullback–Leibler divergence. When investigating interaction at a larger spatial scale, for example, at the level of intestine sections or animals, the spatial spots were first designated into several regions and a chi-square test was used to find microbes that were significantly enriched or depleted in certain regions. In a similar fashion, enriched host expression features could be identified. Alternatively for top-level host pathways (31 of them for GO biological process), highly variable ones can be identified by simple visualization.
Comparison with Kraken 2
To compare our SMT analysis pipeline with a commonly used tool, Kraken 2, data from M1016 were used for evaluation. The same unmapped reads were extracted from BAM file from the SpaceRanger output directory with SAMtools to generate a FASTA file. After that, SMT and Kraken 2 were used separately for metatranscriptomic profiling. The identity and coverage of SMT were set to 80% and 60%, respectively, whereas Kraken 2 was run with default parameters.
We used the read-per-UMI metric as an indicator for the reliability of taxa assignment. As reads used for this purpose (unmapped to host) were enriched with low-quality ones, and the low-quality reads would generally have a low read-per-UMI count, whereas bona fide microbial UMI would have read-per-UMI counts similar to that of host gene UMIs (Supplemental Table S1).
In terms of speed, for a typical data set of 400 million reads, Kraken 2 took 25 sec to finish the alignment of unmapped reads from the FASTA file, whereas SMT took 21,000 sec. This was all done using 48 threads on a dual Epyc 7552 server with 1 TB of memory.
Fluorescence in situ hybridization
OCT embedded tissue slides were fixed with methyl alcohol at −20°C. Slides were stained using the FISH kit (Guangzhou Exon Biotechnology) following the manufacturer's protocol. Cy3-labeled probes (EUB338-GCTGCCTCCCGTAGGAGT) were hybridized overnight at 37°C. Sections were washed at room temperature and 60°C in washing buffer I for 5 min, followed by two additional rounds of washing for 5 min at 37°C in washing buffer II. Staining was visualized with the Leica TCS SP8 confocal microscope at 20×/0.7 lens magnification and 63×/1.40 oil lens magnification. The images were analyzed using Imaris 9.7 microscopy imaging software.
Bulk RNA-seq processing and analysis
OCT embedded tissue slices were used for RNA extraction with TRIzol reagent (Invitrogen 15596026) followed by library construction using a Illumina TruSeq stranded total RNA kit (Illumina 20020596) and then went through RNA sequencing using the Illumina NovaSeq 6000 system (150-bp paired-end reads). The raw reads were aligned to the mouse reference genome (version mm10) and human reference genome (version hg38) using HISAT2 RNA sequencing alignment software (Kim et al. 2019). The alignment files were processed to generate read counts for genes using SAMtools (Danecek et al. 2021) and HTSeq. Read counts were normalized and underwent differential analysis using R package DESeq2 (Love et al. 2014). P-values obtained from multiple tests were adjusted using the Benjamini–Hochberg correction.
Experimental optimization of SMT and related analysis
As ST experiment is not designed for microbial RNA capture, an experimental optimization following microbial FISH assay was tested. Our optimization followed the method used previously (Chassaing et al. 2014). Specifically, after the tissue permeabilization step, a lysozyme treatment of 15 min was added (Beyotime Biotechnology ST206) before the reverse transcription and second-strand synthesis.
To compare the effects of lysozyme treatment, counts of resident microbes (positive controls removed) were normalized with global UMI counts and scaled to counts per 107. A Wilcoxon test was performed to compare the difference of microbial sequence abundance at the kingdom level between lysozyme-free and lysozyme-treatment groups. Host expression qualities were evaluated by common metrics such as feature per spot, UMI per spot, and percentage of mitochondrial sequences, all using the Seurat R package. Finally, host expressions under the two conditions were integrated and the spots clustered by the expression profiles, and the two conditions were visualized by the Seurat SpatialDimPlot function.
Using known bacteria as positive controls and related analysis
To validate and quantify SMT results, we used two control bacteria: a Gram-positive S. aureus and a Gram-negative P. aeruginosa. The bacteria were grown overnight for 16–18 h at 37°C on a shaker, 250 rpm, in LB broth supplemented with kanamycin. Bacteria were centrifuged at 5000 rpm for 5 min, washed twice, and resuspended in sterile PBS. The OD600 was measured to estimate bacterial density, and serial plating was performed on LB agar plates to quantify the use dose by counting CFUs after an overnight incubation at 37°C. The aim was to apply three different amounts of control bacteria at the three corners of a Visium capture window, 200,000 CFU, 10,000 CFU, and 500 CFU, respectively, and the last corner was left blank. Accordingly, the bacteria suspension was serial diluted, and 0.5 µL of bacteria suspension was applied at the corners of capture area and let to dry before the ST experiments. During the ST process, the areas of application were generally covered by tissue.
For each Visium window with control bacteria added, the rectangular capture area was split evenly into four quadrants, and each part was related to a particular gradient of added bacteria. During sequence alignment, sequences from one organism could potentially map to other related organisms, which were termed “spillovers” in this paper. When known control bacteria were used, this spillover effect was relatively easy to identify. To get rid of spillovers caused by ambiguous mapping, Spearman's correlation analysis was performed, and those whose correlation coefficient with added control, for example, Pseudomonas or Staphylococcus, was larger than 0.5 were considered spillovers and were removed. The efficiency of the control microbe detection was estimated by dividing total microbial UMI count from the second quadrant by its corresponding CFU count (10,000). This intermediate CFU was used as a plateauing of capture efficiency was observed at higher CFU.
Data access
The mouse data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA789453.
The human data generated in this study have been submitted to the Genome Sequence Archive of China National Center for Bioinformation (GSA; https://ngdc.cncb.ac.cn/gsa-human) under accession number HRA002678. Raw data can be obtained by following the guidelines of Genome Sequence Archive for noncommercial use (https://ngdc.cncb.ac.cn/gsa-human/document/GSA-Human_Request_Guide_for_Users_us.pdf). The code used for the analysis in this paper is available as Supplemental Code and is uploaded to GitHub (https://github.com/affinis/SMTa).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank the Ruijin Hospital of Shanghai for their help with tumor tissue preparation and specimen acquisition. We also thank the sequencing core of Shanghai Institute of Immunology for library construction and sequencing. This study was supported by Shanghai Science and Technology Commission special grant 20JC1410100 awarded to G.Q.C. and the Innovative Research Team of High-level Local Universities in Shanghai SHSMU-ZLCX20211600 awarded to L.C.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277178.122.
- Received August 4, 2022.
- Accepted January 31, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








