
Recent, full-length gene retrocopies are common in canids
- Kevin Batcher1,
- Scarlett Varney1,
- Daniel York2,
- Matthew Blacksmith3,
- Jeffrey M. Kidd3,4,
- Robert Rebhun2,
- Peter Dickinson2 and
- Danika Bannasch1
- 1Department of Population Health and Reproduction, University of California, Davis, Davis, California 95616, USA;
- 2Department of Surgical and Radiological Sciences, University of California, Davis, Davis, California 95616, USA;
- 3Department of Human Genetics, University of Michigan Medical School, Ann Arbor, Michigan 48109, USA;
- 4Department of Computational Medicine and Bioinformatics, University of Michigan Medical School, Ann Arbor, Michigan 48109, USA
Abstract
Gene retrocopies arise from the reverse transcription and insertion into the genome of processed mRNA transcripts. Although many retrocopies have acquired mutations that render them functionally inactive, most mammals retain active LINE-1 sequences capable of producing new retrocopies. New retrocopies, referred to as retro copy number variants (retroCNVs), may not be identified by standard variant calling techniques in high-throughput sequencing data. Although multiple functional FGF4 retroCNVs have been associated with skeletal dysplasias in dogs, the full landscape of canid retroCNVs has not been characterized. Here, retroCNV discovery was performed on a whole-genome sequencing data set of 293 canids from 76 breeds. We identified retroCNV parent genes via the presence of mRNA-specific 30-mers, and then identified retroCNV insertion sites through discordant read analysis. In total, we resolved insertion sites for 1911 retroCNVs from 1179 parent genes, 1236 of which appeared identical to their parent genes. Dogs had on average 54.1 total retroCNVs and 1.4 private retroCNVs. We found evidence of expression in testes for 12% (14/113) of the retroCNVs identified in six Golden Retrievers, including four chimeric transcripts, and 97 retroCNVs also had significantly elevated FST across dog breeds, possibly indicating selection. We applied our approach to a subset of human genomes and detected an average of 4.2 retroCNVs per sample, highlighting a 13-fold relative increase of retroCNV frequency in dogs. Particularly in canids, retroCNVs are a largely unexplored source of genetic variation that can contribute to genome plasticity and that should be considered when investigating traits and diseases.
Gene retrotransposition occurs when mRNA is reverse transcribed into DNA and inserted back into the genome, resulting in an intron-less copy of a gene referred to as a retrocopy or a processed pseudogene. This process is performed in mammals by long interspersed nuclear element 1 (LINE-1 or L1) proteins acting in trans on cellular mRNA (Jurka 1997; Esnault et al. 2000; Richardson et al. 2014). LINE-1-mediated retrotransposition results in the duplication of short (10- to 20-bp) segments of genomic DNA flanking the insertion, referred to as target site duplications (TSDs). Thousands of retrocopies have been identified in mammalian reference assemblies, although the exact number varies by species and by the annotation method applied (Rosikiewicz et al. 2017; Frankish et al. 2019). Most of these reference retrocopies are the consequence of ancestral retrotransposition events, evidenced by the accumulation of mutations that differentiate the retrocopy sequence from that of the parent gene (Rosikiewicz et al. 2017). Although these ancestral retrocopies tend to be fixed in a species (Ewing et al. 2013; Schrider et al. 2013; Kabza et al. 2015), most mammalian genomes contain active LINE-1s capable of producing novel retrocopy insertions (Penzkofer et al. 2016). These more recent retrocopy insertions may not be fixed in a species, resulting in gene copy number variation between individuals, referred to as retro copy number variants (retroCNVs) (Abyzov et al. 2013; Ewing et al. 2013; Schrider et al. 2013; Richardson et al. 2014; Casola and Betrán 2017). Although some of the retrocopies present in a reference genome assembly may be polymorphic in a species and thus may be reference retroCNVs, there are also nonreference retroCNVs that are not found in the assembly itself (Schrider et al. 2013).
RetroCNVs are a type of complex structural variant that requires specialized techniques for identification within whole-genome sequencing (WGS) data (Casola and Betrán 2017). When Illumina paired-end reads are aligned to a reference assembly, any reference retroCNV that is absent in an individual will appear as a deletion relative to the assembly. However, when an individual has a nonreference retroCNV, the reads coming from that retroCNV will align to the parent gene. Because the retrocopy lacks introns, discordant reads are observed aligning only to the exons of the parent gene. Discordant reads are also found at the 3′ and 5′ end of the gene mapping to the insertion site of the retroCNV. These two features can be used to identify nonreference retroCNVs from WGS data, with the gold standard in retroCNV discovery requiring identification of the parent gene and characterization of the insertion site (Richardson et al. 2014; Zhang et al. 2021). Estimates of gene retrotransposition rates have varied, although recent analyses using high-coverage WGS data have identified 1663 retroCNV parent genes in populations of mice (Zhang et al. 2021) and 503 in human populations (Zhang et al. 2017), indicating that gene retrotransposition is a common occurrence. There is limited information about retroCNVs in dogs (Gao et al. 2019; Kim et al. 2019; Batcher et al. 2020); however, short interspersed nuclear element (SINE) insertions, which are also mobilized in transby LINE-1 encoded proteins in a manner similar to gene retrocopies, have been shown to be highly dimorphic in dogs (Wang and Kirkness 2005; Halo et al. 2021).
Although retrocopies have historically been referred to as processed pseudogenes and presumed to be nonfunctional, evidence has accumulated for retrocopy expression and functionality (Cheetham et al. 2020; Troskie et al. 2021; Zhang et al. 2021). It has been argued that retroCNVs are likely to be deleterious based on negative selection in natural populations of mice (Zhang et al. 2021), and consistent with this hypothesis, retroCNVs in humans have been shown to be involved in cancer as well as neurodegenerative, mental, or cardiovascular disorders (Ciomborowska-Basheer et al. 2021). In dogs, two recently inserted and expressed FGF4 retrocopies are associated with dominant skeletal dysplasias (Parker et al. 2009; Brown et al. 2017). Several additional FGF4 retrocopies with no known phenotypic associations were also discovered, indicating that retroCNV formation may also be a common occurrence in dogs (Batcher et al. 2020). Artificial breed selection by humans could also increase the allele frequency of functional retroCNVs, as appears to have been the case with the FGF4 retroCNVs, which are common in many breeds (Batcher et al. 2019). As such, analysis of the full landscape of retroCNVs in dogs could lead to interesting insights into retrocopy biology and may additionally help in identifying causative variants for phenotypic associations in dogs. The goal of this current study was to characterize the landscape of retroCNVs in dogs by performing retroCNV discovery on a diverse data set of canids and by further analyzing the retroCNV for evidence of function.
Results
retroCNV discovery
We used high-coverage WGS data from 293 canids (median coverage of 25.6×) aligned to the CanFam3.1 reference genome for our retroCNV discovery data set (Supplemental Table S1). Our approach to retroCNV discovery was to first identify mRNA-specific 30-mers, which are present in spliced gene sequences but absent from the CanFam3.1 reference assembly (Fig. 1A). These sequences are only found in genomic DNA when a nonreference retroCNV is present (Fig. 1B). No mRNA-specific 30-mers were identified from single-exon genes (N = 1574) or genes with recent retrocopies already present in the reference assembly (N = 75). We identified mRNA-specific 30-mers for 18,192 protein-coding genes and 10,807 long noncoding RNAs, which were then used for retroCNV parent gene discovery. In total, 1870 putative retroCNV parent genes were identified in the 293 canid data set based on the presence of these mRNA-specific 30-mers.
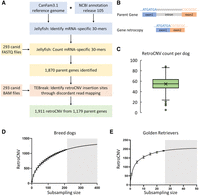
retroCNV discovery. (A) retroCNV discovery pipeline. (B) mRNA-specific sequences are formed owing to intron removal; these sequences are used to identify nonreference retroCNV parent genes in FASTQ reads from genomic DNA sequence. (C) Total nonreference retroCNV count seen in individual domestic dogs (average, 54.1). (D) Number of detected retroCNV insertions with increasing resampling sample sizes in breed dogs. Subsample sizes were selected from one to 210, with a step size of 10, and 100 replicates within each subsample. Bars represent standard deviation of the replicates at each subsample size, and the gray area shows the prediction of increasing the number of dogs beyond the number used in this study. (E) Number of detected retroCNV insertions with increasing resampling sample sizes in Golden Retrievers. Subsample sizes were selected from one to 25, with a step size of five, and 100 replicates within each subsample. Bars represent standard deviation of the replicates at each subsample size.
To resolve the insertion site of nonreference retroCNVs (Supplemental Fig. S1), we analyzed discordant paired-end reads. From the 1870 parent genes identified as having putative retroCNVs, insertion sites were resolved for 1911 total nonreference retroCNVs coming from 1179 parent genes (Fig. 2A). Many (808/1911, 42.3%) insertion sites were located within the introns of other protein-coding genes (Supplemental Table S2; Supplemental Fig. S2). Of the 1179 retroCNV parent genes, 1150 were protein coding, whereas 29 were lncRNAs. Four of the previously identified FGF4 retroCNVs were successfully identified through our approach (Supplemental Table S2). The TSD was resolved for 1676 (87.7%) of the retroCNVs, and the median TSD length was 16 bp. No insertion sites could be identified for retroCNVs derived from 691 of the putative retroCNV parent genes; however, 125 of these parent genes had discordant reads mapping between exons, which may indicate the retroCNV inserted into repetitive or unresolved regions of the CanFam3.1 reference genome (Supplemental Table S3). Further inspection revealed that 21 of the putative retroCNV parent genes had discordant reads mapping to satellite DNA, suggesting insertion of the retroCNV into a telomeric or centromeric region. Additionally, four parent genes (MITF, NME7, TXNDC12, and PPP2CB) that had discordant reads in all of the males and none of the females were identified, indicating that the retroCNVs were likely on the Y Chromosome, which is not represented in the CanFam3.1 assembly.
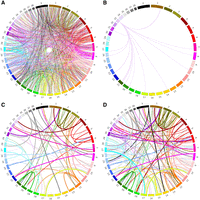
Circos plots highlighting the location of retroCNV parent genes and their insertion sites, with links colored based on the chromosome of the parent gene. (A) All recent retroCNVs identified in canids with no retroCNV-specific variants. (B) All retroCNVs of the GAP43 parent gene. (C) All retroCNVs present in a single Golden Retriever (SRR7107792). (D) All retroCNVs present in all (N = 26) Golden Retrievers. (C,D) The thickness of the link represents how common the retroCNV is within the Golden Retriever data set.
We also identified retrocopies that are present in the CanFam3.1 reference genome but missing in at least one of the 293 canids, which we refer to as reference retroCNVs. We identified 58 reference retroCNVs, which were confirmed through visual analysis of aligned WGS data (Supplemental Table S4). Several reference retroCNVs were highly prevalent in dogs (≥70%) and rare in wolves (≤10%), including the reference retroCNV AKR1B1 (retro_cfam_63), which has previously been associated with domestication (Wang et al. 2019), as well as retroCNVs of MGST3 (retro_cfam_35) and RPL27A (retro_cfam_145).
We focused all subsequent analysis on the 1911 nonreference retroCNVs with resolved insertion sites. A full matrix matching individual canids with retroCNVs is available in Supplemental Table S5. Although 880 of the retroCNV parent genes only had one retroCNV insertion site identified, 338 retroCNV parent genes had multiple insertion sites identified, including genes such as GAPDH, which had 21 retroCNV insertions (Supplemental Fig. S3; Supplemental Table S2). Most of the retroCNV parent genes (900/1179, 76.3%) had no retrocopies present in the CanFam3.1 reference assembly. Additionally, 231 retroCNV parent genes had no known retrocopies in any of the mammalian reference genome assemblies, including GAP43, which had 18 retroCNV insertion sites identified in canids (Fig. 2B; Supplemental Table S2).
Table 1 highlights some aspects of the nonreference retroCNVs by population. Dogs with an assigned breed, which we refer to as breed dogs, had 54.1 nonreference retroCNVs on average (95% CI 52.5–55.7) (Fig. 1C). Within the 227 breed dogs, there were 325 private (unique to an individual dog) retroCNVs, or 1.4 per breed dog on average (95% CI 1.2–1.7), whereas the 43 free-ranging dogs had 214 private retroCNVs and 5.0 each on average (95% CI 4.0–6.0). Most nonreference retroCNVs were identified in a small number of dogs and at a low allele frequency in the entire population (Supplemental Fig. S4). There were also 689 retroCNVs exclusive to breed dogs, 247 retroCNVs exclusive to the 18 wild canids, and 197 retroCNVs exclusive to the three African wild dogs (Table 1). We also observed that individuals that were sequenced at a higher depth tended to have more retroCNV insertion sites resolved (Supplemental Fig. S5). Although samples at greater than 30× coverage had an average of 57.9 retroCNVs (95% CI 56.2–59.6), samples between 10× and 30× coverage had 54.8 (95% CI 52.3–57.3) and samples less than 10× coverage had 40 (95% CI 35.2–44.8) on average.
Population summary of nonreference retroCNVs
retroCNV-specific gene variants
To estimate how recently the retroCNVs inserted, we identified variants that occurred in the retroCNV after insertion through the analysis of variants at the parent gene locus. Most retroCNVs had not acquired any new variants after insertion, as retroCNV-specific variants were only identified in 153/1390 (11.0%) of the retroCNVs analyzed, and only eight of 1390 (0.6%) retroCNVs had high impact mutations (Supplemental Table S6). Highlighting their more recent origin, only 92/1212 (7.6%) of the dog exclusive retroCNV had any retroCNV-specific variants, whereas 61/178 (34.3%) of the retroCNVs that were shared across canids had retroCNV-specific variants.
Resampling
We expected that more retroCNVs may be detected in even larger data sets. To test this, we performed random resampling of individual breed dogs. Although we identified 1140 retroCNVs in the 227 breed dogs, extrapolation from random resampling indicated that we would expect to identify over 1300 retroCNVs in a data set of 400 breed dogs (Fig. 1D). We similarly performed resampling within Golden Retrievers and observed that smaller sample sizes (N = 26) can sufficiently capture the majority of retroCNVs within a single breed (Figs. 1E, 2C,D). Resampling within the free-ranging dogs indicated that a large number of retroCNVs remains to be discovered, highlighting the heterogeneous nature of the free-ranging dogs (Supplemental Fig. S6).
retroCNV validation
We expected that some of the nonreference retroCNVs, although absent from the CanFam3.1 reference genome assembly, would be present in the alternative canid genome assemblies. This would confirm them as true retroCNVs and validate our discovery method. To test this, we first applied our retroCNV discovery pipeline to Illumina WGS data generated from four canids that have been used to create alternate canid reference assemblies, and identified 248 nonreference retroCNVs (Table 2). We then directly confirmed the presence of 173 of the 248 (69.8%) retroCNVs within their respective assemblies (Supplemental Table S7). We also identified the insertion site for six retroCNVs that had not been resolved through discordant read mapping (Supplemental Table S7). Most of the retroCNVs were full length with respect to the parent genes, with 153/179 (85.5%) containing the entire parental gene coding sequence.
Analysis of retroCNVs in alternative canid genome assemblies
Genome assembly involves the collapsing of heterozygous haplotypes, a process that could have resulted in heterozygous retroCNVs being excluded from the assembly sequence (Feng and Li 2021). Therefore, we also analyzed Pacific Biosciences (PacBio) long-read data from each sample aligned to the CanFam3.1 assembly for evidence of retrocopy insertions (Supplemental Data S1). We found evidence for 236/248 (95.1%) of the retroCNVs within the long-read data (Supplemental Table S8). A poly(A) tail ≥10 bp was observed in 211/236 (89.4%) of the retroCNVs, with median length of 29 bp. A TSD was identified for 218/236 (92.4%) of the retroCNVs, with median length of 14 bp. Overall, 244/248 (98.4%) of the retroCNVs were validated either within their respective genome assembly or in the long-read data (Table 2).
We also developed PCR assays for nine of the predicted retroCNVs and validated them through Sanger sequencing (Fig. 3A). In individuals positive for the retroCNV, we observed a poly(A) tail at one end of the insertion site and the 5′ gene sequence of the retroCNV parent gene on the other end of the insertion site (Fig. 3C), which matched the expectation from the discordant reads and confirmed the presence of a retrocopy. For each retroCNV sequenced, the TSD was identified as the duplicated genomic sequence present at both ends of the insertion (Fig. 3C; Supplemental Table S9). The TSD identified through Sanger sequencing for these nine retroCNVs matched the predicted TSD from the discordant reads. We additionally identified dogs lacking the retroCNVs (Fig. 3B), confirming the retrocopies as polymorphic insertions. The list of retroCNVs validated through PCR and Sanger sequencing and the associated breeds is available in Supplemental Table S9.
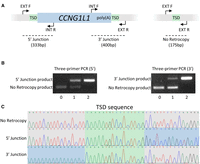
retroCNV validation. (A) Primer design for retroCNV genotyping, with CCNG1L1 as an example. When CCNG1L1 is present, the EXT_F and INT_R primers produce a 333-bp product at the 5′ junction, and the INT_F and EXT_R primers produce a 400-bp product at the 3′ junction. When the retrocopy is absent, the EXT_F and EXT_R primers produce a 175-bp product. (B) Three-primer PCR results for CCNG1L1 at the 5′ and 3′ junctions for individuals with zero, one, and two copies of CCNG1L1. The two external primers EXT_F and EXT_R are included in both reactions, as well as one of the internal primers, INT_F (5′ Junction) or INT_R (3′ Junction). (C) Sanger sequencing results for the CCNG1L1 retroCNV. The TSD is identified as the genomic sequence from the insertion site, which is present at both the 5′ and 3′ ends of the retroCNV.
retroCNV selection and expression
We calculated a fixation index (FST) for the retroCNVs across breed clades (Parker et al. 2017) and identified 97 retroCNVs that had significantly elevated FST (see Methods). This included the two previously identified FGF4 retroCNVs (Table 3; for full list and clade distribution, see Supplemental Tables S10, S11).
RetroCNVs with the highest FST between breed clades
To determine if the retroCNVs showed evidence of expression, we first performed WGS and retroCNV discovery in six Golden Retrievers and then RNA-seq using testes from the same six Golden Retrievers. There were 113 total nonreference retroCNVs identified in the six individuals. RetroCNV-specific variants were present in 24 of the 113 retroCNVs, allowing for the distinction between parent and retroCNV-derived transcripts, and confirmed the expression for two retroCNVs. Among the retroCNVs that had inserted within the introns of another gene, chimeric reads between a retroCNV parent gene and a gene at the insertion site were observed in four of 42. This included the COILL2 retroCNV, which is inserted within the 5′ UTR of LOC100686934, producing a novel chimeric transcript in two of the six Golden Retrievers (Supplemental Fig. S7). However, five of the 42 insertion site genes were not sufficiently expressed in testes (fewer than one transcript per million in all samples) to allow for the evaluation of chimera formation with the retroCNVs. Additionally, discordant reads mapping to the parent gene loci were observed at 11 of the insertion sites. Overall, at least one form of evidence for expression was observed for 12.4% (14/113) of the retroCNVs in six Golden Retriever testes (Supplemental Table S12). The expressed retroCNV FARSBL1 was present in 60% of all dogs and only a single wild canid.
retroCNV discovery in humans
To determine the rate of retroCNV occurrence in another species for comparison, we performed retroCNV discovery in 78 individuals from 26 populations using The 1000 Genomes Project Consortium phase 3 high-coverage data set (The 1000 Genomes Project Consortium 2015; Byrska-Bishop et al. 2021). We resolved insertion sites for 46 nonreference retroCNVs from 44 parent genes in the 78 samples (Supplemental Table S13). Of the 44 retroCNV parent genes, 40 have been previously identified in human data sets, whereas 34 of the 46 retroCNV insertion sites had been previously identified. A full analysis of which human retroCNVs have been identified in previous studies is available in Supplemental Table S14. Individuals in this data set had 4.2 retroCNVs on average (95% CI 3.9–4.6).
Rate of retroCNV formation in canids
We observed a total of 214 nonreference retroCNVs in the three African wild dogs (Lycaon pictus), which had 173 retroCNVs each on average. Most of these retroCNVs were private to the African wild dogs, and only 17 retroCNVs were shared between the African wild dogs and any other canid, indicating that most of the retroCNVs identified in either species inserted after the species had diverged. Similarly, we identify 194 retroCNVs that are exclusive to gray wolves and 1010 retroCNVs exclusive to breed dogs. Genetic analyses have indicated that domestication in dogs occurred ∼25,000 yr ago, whereas breed formation largely occurred within the last 200 yr (Ostrander et al. 2019). If the 1010 breed dog–specific retroCNVs inserted after domestication, we can estimate the rate of retroCNV accumulation at approximately four per 100 yr. Alternatively, within our data set of Golden Retrievers (N = 26), we identify 10 retroCNVs that are exclusive to the breed and not found in any other breed dogs or canids. Because the Golden Retriever breed was formed ∼200 yr ago, 10 Golden Retriever–exclusive retroCNVs would indicate a similar rate of retroCNV accumulation at five per 100 yr.
Discussion
Previous analyses of canid retrocopies have focused on those retrocopies present in the CanFam3.1 reference assembly, which was produced from a single dog (Hoeppner et al. 2014; Rosikiewicz et al. 2017; Gao et al. 2019). In this study, we characterized a rich landscape of retroCNVs in canids, consistent with an active LINE-1. By applying a novel approach to retroCNV discovery on a diverse data set of 293 canids, we identified 1911 retroCNVs, most of which had inserted recently. Domestic dogs have 54.1 nonreference retroCNVs each on average, and as many of the retroCNVs are private to a single individual, we also expect to find additional retroCNVs in larger discovery data sets. We observed many retroCNVs that appear under selection and that, even within a single tissue type, 12% of the retroCNVs were expressed or forming novel chimeric transcripts with nearby genes, indicating that some of the retroCNVs may have functional and phenotypic consequences in canids.
Our approach to retroCNV discovery successfully resolved a large number of retroCNVs in canids. In humans, previous studies using low-coverage WGS data sets have underestimated the rate of retrocopy insertion (Richardson et al. 2014). When long-read assemblies were analyzed, the estimated rate of retroCNV formation in humans was increased from 39 events per 939 individuals to 40 events per 22 individuals, or 4.1 per individual on average (Feng and Li 2021). Similar to the analysis by Feng and Li, we found that humans had on average 4.2 retroCNV insertions using our discovery method, which identified 46 total retroCNV insertions in 78 human genomes. Although most of the retroCNV parent genes have been reported in previous analyses of human genomes, eight of the insertion sites have not been previously identified (Zhang et al. 2017). Previous analysis of canine SINEs, which are also mobilized via LINE-1 proteins acting in trans, highlighted a rate of SINE insertions 10- to 100-fold higher than that observed in humans (Wang and Kirkness 2005). Although phenotypic associations with retroCNVs are rare, SINE and LINE-1 insertions are a significant source of phenotypic variability in dogs, being responsible for 10% of the phenotype-associated variants identified to date (Online Mendelian Inheritance in Animals [OMIA] Sydney School of Veterinary Science, March 29, 2022; https://omia.org/), whereas in humans, transposable elements are responsible for only 0.27% of all disease mutations (Callinan and Batzer 2006). In a recent analysis of the CanFam3.1 and UMICH_Zoey_3.1 reference assemblies, 16,221 dimorphic SINEs and 1121 dimorphic LINE-1s were identified, which represented a 17-fold increase in SINE differences and an eightfold increase in LINE-1 differences compared with the number found in humans (Halo et al. 2021). Domestic dogs, at 54.1 retroCNVs on average, also have a 13-fold increase in retroCNVs relative to humans, data that are consistent with either a highly active or a highly promiscuous LINE-1 in dogs. We also identified 231 retroCNV parent genes that have no known retrocopies in any other mammalian species, which might indicate that canine LINE-1 proteins are less selective. Among these retroCNV parent genes was GAP43, which had 18 retroCNV insertions and which may have implications in cognitive function in dogs (Routtenberg et al. 2000).
In this study, we only considered retroCNVs with identifiable insertion sites as valid, although we provide evidence for the presence of 125 additional retroCNVs with unresolved insertion sites. Our method of retroCNV parent gene discovery also cannot identify single-exon genes or genes with recent retrocopies present in the CanFam3.1 reference assembly. Still, we estimated the rate of retroCNV formation in domestic dogs at around four retroCNVs per 100 yr, which is still an underestimate as our data set of 228 breed dogs does not capture every retroCNV in the entire population. A recent analysis of retroCNVs in mice estimated their rate of retroCNV formation at two per 100 yr (Zhang et al. 2021). This would indicate that domestic dogs have a rate of retroCNV formation even 2× greater than mice, which are known to have a large number of active LINE-1s (Penzkofer et al. 2016) and have been shown to have fivefold as many retroCNVs as humans (Ewing et al. 2013). In natural populations of mice, however, retroCNVs were also shown to be under negative selection owing to deleterious effects, in which retroCNVs are quickly removed from the population (Zhang et al. 2021). Also, in human populations, retroCNV insertions are not found in evolutionarily conserved regions, which indicates that highly deleterious retroCNVs are under negative selection (Zhang et al. 2017). We observed that many retroCNVs appear to be under positive selection in dogs, with significantly elevated FST values. Like the FGF4 retrogenes, other retroCNVs may be under positive selection by breeders owing to their phenotypic effects (Batcher et al. 2019), although they may also be neutral variants whose frequencies differ owing to the dynamics of breed formation and low genetic diversity within breeds (Bannasch et al. 2021).
Although gene retrocopies were historically considered nonfunctional pseudogenes, more recently, it has been recognized that retrocopies, which are a form of structural variant, are often functional through a variety of mechanisms that have been explored in recent reviews (Cheetham et al. 2020; Ciomborowska-Basheer et al. 2021). We found evidence for expression in 14 out of 113 retroCNVs in testes tissue from six Golden Retrievers, including four novel chimeric transcripts. However, this is likely an underestimate owing to our use of a single tissue type for transcriptional assessment. Additionally, many of the retroCNVs were indistinguishable from their parent genes and thus cannot be effectively queried for evidence of expression through RNA-seq analysis alone. Comparison of overall gene expression in larger RNA-seq data sets including individuals with and without specific retroCNVs may be required to determine expression of retroCNVs that are identical to their parent genes. When such analyses were performed in mice, differences in retroCNV parent gene expression were found between individuals with and without the respective retroCNVs, including many cases in which overall expression was significantly reduced in the individuals with the retroCNVs (Zhang et al. 2021). Our data confirms that the AKR1B1 reference retroCNV, which has previously been shown to be expressed and associated with dog domestication, is common in dogs and rare in wolves (Wang et al. 2019). Similarly, the MGST3 and RPL27A reference retroCNVs and the expressed FARSBL1 retroCNV are both common across dogs and rare in wild canids and may play roles in domestication. AKR1B1, MGST3, and FARSB are also involved in metabolism (Jakobsson et al. 1997; Crosas et al. 2003), whereas the RPL27A retrogene is inserted within NCOA3, a coactivator of transcription involved in thyroid function (Nolan et al. 2021).
The four Y Chromosome retroCNVs were present in all male canids and thus were not true retroCNVs, and their sequence deviation from their parent genes of origin indicates that they are likely ancestral. Two of the Y Chromosome retroCNVs had been previously identified through the identification of autosomal variants that were actually sex-linked and owing to the retroCNV insertion (Tsai et al. 2019). In this study, we also identified variants at the retroCNV parent gene loci, which are likely attributable to nonreference retroCNVs. These variants would normally be attributed to variation in the parent genes, potentially hindering any analyses looking for causative mutations; it is noteworthy that some variants identified in the coding sequences of genes may in fact be attributable to nonreference retroCNVs elsewhere in the genome.
Domesticated dog breeds have undergone artificial selection, which has led to extreme phenotypic diversity between breeds as well as breed predispositions to many heritable disorders (Wayne and Ostrander 2007; Asher et al. 2009; Summers et al. 2010; Gough et al. 2018; Bannasch et al. 2021). In particular, many dog breeds are at a substantially higher risk for specific cancers compared with human populations (Schiffman and Breen 2015). We have shown that dogs have many retroCNVs, consistent with a highly active LINE-1, and we also observed that some retroCNVs are under selection or are also capable of expression or insertional mutagenesis through the formation of novel chimeric transcripts with nearby genes. These functional retroCNVs are likely a contributing factor to the phenotypic diversity seen in canids and are also strong candidates for disease associations in dogs, including susceptibility to cancers. We hope this list of retroCNV insertion sites will be a useful resource for the canine research community and that further assessment of the retroCNVs for evidence of function will provide insight into the genetics of phenotypic traits under selection in dogs.
Methods
Data selection
Illumina sequencing data aligned to the CanFam3.1 reference were downloaded from the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) (Hoeppner et al. 2014). The data set included 227 dogs from 76 different breeds (referred to as “breed dogs”), 43 free-ranging dogs (marked as either “village” or “indigenous” by the data provider), three dingoes, 10 gray wolves, two red wolves, five coyotes, and three African wild dogs. A full list of samples used in this study and their accession numbers is available (Supplemental Table S1). Ten samples were removed from the analysis owing to a low number of retroCNV insertion sites being resolved, possibly owing to the quality of the sequencing data.
Parent gene discovery using mRNA-specific 30-mers
The nucleotide sequence of a gene retrocopy resembles the processed mRNA transcript of its parent gene, with unique nucleotide sequences formed at the exon–exon junctions. These unique nucleotide sequences are only observed in genomic DNA when a retrocopy insertion is present and thus can be used to identify the parent genes of retroCNVs from WGS data. We used Gffread (Pertea and Pertea 2020) to obtain the spliced gene sequences for each gene transcript found in the NCBI CanFam3.1 annotation release 105 (https://www.ncbi.nlm.nih.gov/genome/annotation_euk/Canis_lupus_familiaris/105/). For each spliced gene sequence, we created a set of mRNA-specific 30-mers that are absent from the CanFam3.1 reference assembly using Jellyfish count (Marçais and Kingsford 2011). This identified all unique 30-mer sequences within the transcriptome that are absent from CanFam3.1 owing to intron removal, giving a maximum of 29 mRNA-specific 30-mers per exon–exon junction. We attributed mRNA-specific 30-mers from alternatively spliced gene transcripts to their respective parent gene. To reduce false positives owing to sequencing errors, we filtered out 30-mers with an edit distance of two substitutions from the reference genome using mrsFAST (Hach et al. 2014). We removed any gene that had fewer than five mRNA-specific 30-mers from further analysis, which included 1574 single-exon genes and 75 genes with recent retrocopies in the CanFam3.1 assembly. In total, 5,884,280 30-mers from 30,792 genes (median per gene, 106) were retained for retrocopy parent gene discovery. WGS data were then queried for the presence of the mRNA-specific 30-mers using Jellyfish. Genes that had at least five mRNA-specific 30-mers and at least 10% of the total 30-mers for that gene identified were considered as putative retroCNV parent genes for further analysis.
retroCNV insertion site discovery through discordant read analysis
Gene retrocopies are derived from processed mRNA transcripts and thus lack introns. When Illumina paired-end reads containing retroCNV sequences are aligned to a reference genome, they align to the parent gene locus, resulting in discordant read pairs that align only to the exons of the parent gene. Discordant read pairs can also be observed at the 5′ and 3′ ends of the parent gene, wherein one read aligns at the parent gene loci, and the other read aligns at the insertion site elsewhere in the genome. Discordant reads can thus be used to verify the presence of a retroCNV as well as identify the insertion site. We performed discordant read analysis on aligned WGS data using TEBreak (Carreira et al. 2016). The “‐‐disc_only” option was used to obtain a list of discordant read clusters of at least four reads (“‐‐min_disc_reads 4”) mapping from a putative parent gene to elsewhere in the genome via “‐‐disco_target.” Putative insertion sites for retroCNVs were visually confirmed in the Integrative Genomics Viewer (IGV) (Supplemental Fig. S8; Thorvaldsdóttir et al. 2013). A retrocopy insertion site was considered valid if discordant reads were observed mapping to the same genomic locus from both the 3′ and 5′ end of the parent gene or if discordant reads were found mapping from either the 3′ or 5′ as well as exon–exon discordant reads at the parent gene. Any discordant reads mapping from parent genes to known CanFam3.1 reference retrocopy insertion sites were ignored, as reference retroCNVs were analyzed separately. The TSD sequence was identified as the overlap between forward and reverse discordant reads at the insertion site. The 5′ and 3′ junction sequences for the retroCNV insertions were resolved using TEBreak and are available in Supplemental Table S15. Visual representations of retroCNV insertion sites were produced using Circos (Krzywinski et al. 2009). As has been proposed by Cheetham et al. (2020), we chose, rather than “pseudogene,” a term that does not make functional inferences for the retrocopies; for example, retrocopies of the FGF4 gene were labeled FGF4L1, FGF4L2, etc.
CanFam3.1 reference assembly retroCNVs
We also examined retrocopies present in the CanFam3.1 assembly for evidence of being retroCNVs; an individual lacking a CanFam3.1 reference retrocopy would appear to have a deletion at those loci when aligned to CanFam3.1. A list of reference retrocopy locations was downloaded from RetrogeneDB (Rosikiewicz et al. 2017). The data set of 293 canids previously aligned to CanFam3.1 was analyzed using DELLY with default settings to identify structural variants within 1 kb of reference retrocopies (Rausch et al. 2012). All deletions were confirmed visually in IGV. Additionally, aligned sequence data from a gray wolf and a coyote were visually analyzed in IGV at all recent canine RetrogeneDB retrocopy loci (>95% identity with parent gene) to identify any retroCNVs that may have gone undetected by DELLY.
retroCNV-specific variant identification
Sequence variants between a retroCNV and its parent gene sequence are owing either to germline variants present within the parent gene or to new polymorphisms unique to the retroCNV that occurred after insertion, which we refer to as retroCNV-specific variants. As all retroCNV-derived reads align to the parent gene loci, we analyzed variants at the parent gene loci in order to identify retroCNV-specific variants. We first identified variants at the retroCNV parent gene loci using BCFtools mpileup (Danecek et al. 2021). We then compared variant allele frequencies between individuals positive or negative for each retroCNV, and variants that only appeared in individuals with the retroCNV were considered unique to the retroCNV. RetroCNVs that were unique to wild canids were excluded from this analysis as the wild canids contained many unique variants that could not be easily differentiated between variation within the parent genes or the retroCNV sequences. retroCNVs that had multiple insertions from the same parent gene were also excluded. Variant effect prediction was performed using the UCSC Genome Browser Variant Annotation Integrator tool (Hinrichs et al. 2016).
retroCNV validation
We performed retroCNV discovery using Illumina data aligned to CanFam3.1 on four individuals that were previously used to generate additional dog genome assemblies: UMICH_Zoey_3.1 (Halo et al. 2021), UU_Cfam_GSD_1.0 (Wang et al. 2021), Canfam_GSD (Field et al. 2020), and CanLup_DDS (Field et al. 2022). We then assessed the presence of the retroCNVs within their respective assemblies using BLAST (Madden 2013).
PacBio data were also examined for evidence of the retroCNV insertions. Individual long-read FASTQ files were aligned to the CanFam3.1 reference with minimap2 version 2.17 (Li 2018). Alignment files were sorted, merged, and indexed with SAMtools version 1.5 (Danecek et al. 2021). The predicted retroCNV insertion sites ±100 bp were analyzed using a modified version of a pipeline designed to detect LINE-1 insertions in long-read sequenced genomes. The pipeline extracts the raw long reads, which align to a locus of interest, and uses a combination of Canu and wtdbg2 to assemble the reads into contigs (Koren et al. 2017; Ruan and Li 2020). The contigs are then polished using Racon (Vaser et al. 2017), aligned to the reference using minimap2 version 2.20, and put in orientation with the reference. Precise breakpoints were identified using AGE (Abyzov and Gerstein 2011).
We developed three primer PCR assays for retroCNVs using Primer3 software (Untergasser et al. 2012), with forward and reverse primers flanking the insertion site and internal primers at the 5′ or 3′ ends of the parent gene. A panel of 10 dogs from a breed identified as carrying each retroCNV were selected at random from the Bannasch laboratory DNA repository for testing (Batcher et al. 2019). A list of the primers used in this study and their expected product sizes is available in Supplemental Table S16. Sanger sequencing was performed on an Applied Biosystems 3500 genetic analyzer using a BigDye terminator sequencing kit (Applied Biosystems).
Population analysis
For FST calculations, dog breeds were placed into the multibreed clades identified by Parker et al. (2017). Only clades containing individuals from at least three breeds were included in this analysis. Additionally, only three Golden Retrievers were selected at random to include in the retriever clade. FST between clades was calculated as described by Zhang et al. (2017), including the calculation of a null distribution from 1000 fake population sets generated through shuffling individual labels for significance estimates. retroCNVs for which 1000 fake population sets never produced an equal or higher FST than the real population were considered significant.
WGS and RNA-seq
Adult Golden Retriever testes were obtained from routine castration procedures. Tissue samples were flash-frozen in liquid nitrogen and stored at −80°C. Genomic DNA was extracted using a Gentra Puregene DNA extraction kit (Qiagen), and RNA was extracted using an RNeasy fibrous tissue mini kit (Qiagen). Library preparation and NovaSeq S4 Illumina paired-end sequencing were performed at the UC Davis Genome Center. Reads were aligned to the CanFam3.1 reference assembly using minimap2 (Li 2018). PCR duplicate reads were removed, and the aligned files were sorted and indexed using SAMtools (Danecek et al. 2021). Evidence for chimeric transcripts in the RNA-seq data set was found through visual analysis of the retroCNV insertion sites and nearby genes in IGV. Because of the small sample size and heterogeneity of retroCNVs between individuals, evidence of expression was determined visually through examination of the insert site, the 5′ UTR of the parent gene, and insertion site genes for chimeric transcripts. For retroCNVs that contained any retroCNV-specific variants, the parent gene loci were examined for evidence of the retroCNV-specific variant, which would indicate expression of the retroCNV. A minimum of two discordant reads was used to consider a retroCNV expressed.
Human comparative analysis
We performed retroCNV discovery in a subset of individuals from The 1000 Genomes Project Consortium high-coverage phase 3 data set (Sudmant et al. 2015; The 1000 Genomes Project Consortium 2015). We selected three individuals from each of 26 human populations at random for this analysis (Supplemental Table S8) and used the GRCh38 annotation release 109.20210514 for 30-mer construction. We then performed retroCNV discovery in the same manner as was performed on the canid data set, and compared the retroCNVs identified by our approach to those identified in a previous study that used the same individuals (Zhang et al. 2017). We compared the retroCNVs identified in this study to those retroCNVs identified in the same 78 individuals by Zhang et al. as well as retroCNVs identified in four other studies that used different data sets (Abyzov et al. 2013; Ewing et al. 2013; Schrider et al. 2013; Feng and Li 2021).
Data access
The WGS and RNA-seq data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA776905. The source code is available as Supplemental Code. The retroCNV insertion sites in bigBed format are available in the Supplemental Material, and at a track hub for the UCSC Genome Browser available at GitHub (https://github.com/klbatcher/retroCNV_insertions).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Laura Kirby for helpful discussions related to the use of long-read data to detect retrogene insertions. We thank Guide Dogs for the Blind, Inc., San Rafael, California, for the samples. This work was supported in part by the Lodric Maddox Graduate Fellowship, Maxine Adler Endowed Chair funds, and the Center for Companion Animal Health. Additional financial support was provided by the Students Training in Advanced Research (STAR) program through a UC Davis School of Veterinary Medicine Endowment Fund. M.B. and J.M.K. were supported by the National Institute of General Medical Sciences of the National Institutes of Health under award numbers R01GM140135 and T32GM007544.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276828.122.
- Received April 8, 2022.
- Accepted July 19, 2022.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








