
Transposable element accumulation drives size differences among polymorphic Y Chromosomes in Drosophila
Abstract
Y Chromosomes of many species are gene poor and show low levels of nucleotide variation, yet they often display high amounts of structural diversity. Dobzhansky cataloged several morphologically distinct Y Chromosomes in Drosophila pseudoobscura that differ in size and shape, but the molecular causes of their large size differences are unclear. Here we use cytogenetics and long-read sequencing to study the sequence content of polymorphic Y Chromosomes in D. pseudoobscura. We show that Y Chromosomes differ almost twofold in size, ranging from 30 to 60 Mb. Most of this size difference is caused by a handful of active transposable elements (TEs) that have recently expanded on the largest Y Chromosome, with different elements being responsible for Y expansion on differently sized D. pseudoobscura Y's. We show that Y Chromosomes differ in their heterochromatin enrichment and expression of Y-enriched TEs, and also influence expression of dozens of autosomal and X-linked genes. The same helitron element that showed the most drastic amplification on the largest Y in D. pseudoobscura independently amplified on a polymorphic large Y Chromosome in Drosophila affinis, suggesting that some TEs are inherently more prone to become deregulated on Y Chromosomes.
Y Chromosomes are a fascinating part of the genome in species with heteromorphic sex chromosomes (Bachtrog 2020). Not only do Y Chromosomes determine maleness in many taxa (Bull 1983; Bachtrog et al. 2014), but their clonal and male-limited transmission is responsible for several unique evolutionary processes that shape their genomic composition (Charlesworth and Charlesworth 2000; Bachtrog 2013). Y Chromosomes are derived from ordinary autosomes, but over time, they often have diverged considerably from their former homolog, the X Chromosome (Charlesworth 1978; Bull 1983).
Y Chromosomes are transmitted through males only, which makes them an ideal location for male-beneficial genes (Rice 1984). However, Y Chromosomes also have a reduced effective population size and lack recombination; this strongly decreases the efficacy of natural selection and results in the erosion of ancestral genes on the Y (Charlesworth 1978; Bull 1983). Y Chromosomes often accumulate repetitive DNA and evolve a heterochromatic appearance (Charlesworth et al. 1994). Indeed, old Y Chromosomes of many species are typically regarded as gene-poor and repeat-rich “genetic wastelands.”
A lack of recombination and the small effective population size imply that variation should be low on Y Chromosomes, and levels of nucleotide diversity are indeed highly reduced on the Y of various species (Zurovcova and Eanes 1999; Bachtrog 2004). Yet, polymorphic Y Chromosomes in Drosophila influence a variety of traits, including male fertility (Chippindale and Rice 2001), temperature tolerance (Rohmer et al. 2004), lifespan (Griffin et al. 2015), and expression of hundreds of genes across the genome (Lemos et al. 2008; 2010). It has been speculated that structural variation involving repetitive regions influences these traits by globally modulating the genome-wide balance of heterochromatin within the genome (Francisco and Lemos 2014; Brown et al. 2020a). Yet, the molecular basis of structural Y variation has not been established.
Morphologically distinct Y Chromosomes that occur in natural Drosophila pseudoobscura males were first described almost a century ago (Lancefield 1929), and Dobzhansky categorized seven morphologically distinct Y Chromosomes that differ in size and shape (Dobzhansky 1937; 1935). The molecular basis of size variations of D. pseudoobscura Y Chromosomes and their origins is unknown, but Dobzhansky speculated that polymorphic Y Chromosomes are derived by losses of sections from the largest Y (Dobzhansky 1935, 1937). He also concluded that morphological and physiological characteristics of a male are not affected by the type of its Y Chromosome.
Here, we take advantage of recent advances in sequencing technology to revisit the large difference in Y size among D. pseudoobscura strains. We combine cytogenetic techniques with whole-genome sequencing and chromatin and transcriptome profiling to study the sequence composition of different Y Chromosomes and their phenotypic consequences.
Results
Cytogenetic approaches to identify and characterize different Y Chromosomes
We generated Y-replacement lines in order to avoid confounding genomic background effects when contrasting different Y Chromosomes (Clark 1990). We crossed males from different localities with virgins from the sequenced reference strain of D. pseudoobscura (MV25), and we repeatedly backcrossed males resulting from this cross with virgin females of the reference strain for nine generations (Supplemental Figs. S1, S2). This crossing scheme should ensure that resulting fly lines carry different Y Chromosomes in an otherwise isogenic background; that is, all differences in sequence composition among lines should be attributed to the Y Chromosome. A total of 28 Y-replacement lines were generated, using Y Chromosomes from diverse geographic populations (Supplemental Table S1; Supplemental Figs. S1–S3). To identify Y Chromosomes that differ in their morphology and their overall sequence content, we resorted to classical cytogenetic approaches, taking advantage of our Y-replacement lines.
We first used microscopy to broadly classify Y's that differ in shape and size across our D. pseudoobscura Y-replacement lines (Fig. 1A; Supplemental Fig. S3; Supplemental Table S2). In particular, we measured total Y size (relative to the X Chromosome, which is derived from the sequenced reference strain in each Y-replacement line), and Y shape (ratio of long vs. short arm length) to identify different Y types (Fig. 1B; Supplemental Table S2). Consistent with Dobzhansky's studies (Dobzhansky 1935, 1937), we find large variation in Y size and shape: The Y/X ratio ranges from 0.42–0.7 (Supplemental Table S2), and we found 24 acrocentric, three submetacentric, and one metacentric Y Chromosomes (Fig. 1B; Supplemental Table S3).
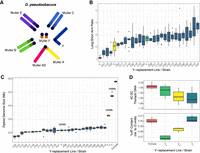
D. pseudoobscura male karyotype and Y Chromosome size variation. (A) D. pseudoobscura male karyotype. Shown are Muller elements: Muller elements A and D form the X Chromosome in D. pseudoobscura. (B) Measurements of Y Chromosome arms (ratio of long arm compared with short; y-axis) from chromosome spreads for each Y-replacement line (x-axis). Colors indicate the Y Chromosomes chosen for further investigation: blue, YL; yellow, YM; green, YS; dark blue, lines not further characterized. Dots correspond to outlier data that fall outside of Q1–1.5 × IQR or Q3 + 1.5 × IQR. (C) Diploid genome size estimations (y-axis) of Y-replacement line males (x-axis) from flow cytometry. The rightmost sample in red is the diploid genome size of females from the reference genome strain (MV25), the strain that was used to generate Y-replacement lines though backcrossing. The numbers show the inferred genome size of three males that were used for more detailed downstream analysis (and referred to as YS, YM, and YL). (D) Heterochromatin estimates in three selected Y-replacement line males. Top shows staining thoracic cells with propidium iodide, and bottom shows staining whole-brain nuclei with DAPI. Leftmost red boxplot is the sequenced/backcross female for comparison.
To further characterize the size of these Y's, we used flow cytometry to estimate the relative size of the Y against a Drosophila virilis standard (Fig. 1C; Supplemental Table S4). We found that inferred diploid genome sizes vary drastically among different Y-replacement lines, ranging from 318 to 348 Mb. Assuming that the rest of the genome is ∼291 Mb (Bracewell et al. 2019), this implies that the absolute sizes of the Y Chromosomes vary from 27 to 57 Mb.
Most of this size difference is presumably owing to differences in repetitive heterochromatin among Y Chromosomes. We chose three Y-replacement lines carrying Y's that spanned most of the spectrum of sizes for further molecular characterization. In particular, we selected a small, medium, and large Y, and we refer to them as YS, YM, and YL. Note that YS is not the smallest Y Chromosome found among our Y-replacement lines (Y-replacement line Y22 carries a Y Chromosome that is estimated to be 2.4 Mb smaller than YS) (Fig. 1C), but we chose the small Y Chromosome of line MV25 instead, because it is the Y Chromosome found in the reference genome strain of D. pseudoobscura. We used two cytometric approaches to test if males with larger Y Chromosomes harbor more heterochromatin. First, we estimated nuclear DNA of cells known to underreplicate heterochromatin. Drosophila thoracic cells show endoreplication, the process in which DNA is synthesized in S phase, but mitosis does not immediately follow, and the DNA in these cells underreplicates heterochromatin (Johnston et al. 2013). We therefore compared the amount of DNA in G1-phase cells to endoreplicated cells with propidium iodide to obtain proxy estimates for heterochromatin content across Y-replacement males (see Methods) (see Johnston et al. 2013). As a control, we compared the amount of DNA in G1-phase and G2-phase cells from brain nuclei, which replicate all the DNA, including heterochromatin. We found that YL males had a higher proportion of DNA underreplicated in thoracic cells compared with the YS males (i.e., the sequenced strain male) and females from the sequenced strain; YM had intermediate levels of underreplicated DNA (Fig. 1D; Supplemental Table S5).
Heterochromatin is generally AT-rich in contrast to GC-rich euchromatin (Saksouk et al. 2015). Therefore, we also used DAPI, a stain that preferentially binds to AT sequences, to obtain independent estimates on heterochromatin content for YS, YM, and YL males and females from the reference strain (see Methods) (see Bosco et al. 2007). We found that both proxy measurements for heterochromatin agreed well with each other; that is, more underreplicated DNA in thoracic cells corresponds to higher %AT content (Fig. 1D; Supplemental Table S5). Thus, our cytogenetic work suggests that naturally isolated Y Chromosomes in D. pseudoobscura differ up to almost twofold in size (i.e., from 27 to 57 Mb), presumably driven mostly by different repeat composition (i.e., heterochromatin).
Assembly of a small, medium, and large Y Chromosome
We chose the same three Y Chromosomes as above (YS, YM, and YL) for further molecular characterization. We obtained high-coverage Illumina sequencing reads (15–19× coverage, obtained by standard PCR + libraries performed on gDNA from head tissue) for each strain, as well as long-read Oxford Nanopore Technologies (ONT) sequencing (Supplemental Table S6). Nanopore reads were used for de novo assembly of the different Y Chromosomes. Assemblies of repetitive regions, including the Y, are challenging, and these regions are often collapsed, highly fragmented, or missing in whole-genome assemblies (Hoskins et al. 2007; Chang and Larracuente 2019). We thus followed a previously described procedure to obtain heterochromatin-enriched assemblies, which was shown to perform superiorly to recover Y-linked fragments in Drosophila melanogaster (Chang and Larracuente 2019). We used two different assembly algorithms (Chin et al. 2016; Koren et al. 2017) and merged the resulting assemblies (Chakraborty et al. 2016) in order to produce a more contiguous and complete assembly (Supplemental Table S7). Y-Linkage of contigs was inferred using male versus female genomic coverage data (for details, see Methods and Supplemental Material 1).
Overall, we identified between 139–246 candidate Y-specific contigs across the three different Y Chromosomes (using a cutoff of Log2(male/female) >1× and >5× depth in males) (Fig. 2A; Supplemental Tables S8–S10). In the YM assembly, several Y contigs have increased male coverage, indicating that they contain collapsed sequences (i.e., the assembler presumably could not resolve two or more highly similar contigs). When we account for the inferred copy number of Y-specific contigs based on male coverage (Supplemental Tables S8–S10), the assembled sizes are YS = 38.6 Mb, YM = 43.9 Mb, and YL = 59.5 Mb. Importantly, the assembled sizes of the three Y Chromosomes are in good agreement with our estimated sizes based on flow cytometry (Table 1; Fig. 2B), suggesting that we were able to recover a large fraction of the sequence of each Y Chromosome in our assembly.
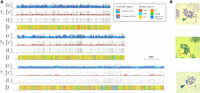
De novo assembly of three differently sized Y Chromosomes (YS, YM, YL). (A) Male and female coverage tracks of Y Chromosome assemblies with Log2(female/male) coverage and repeat landscape shown beneath. One tick mark corresponds to 5 Mb, and collapsed contigs were adjusted in the plots. Landscapes of the most abundant repeats are shown separately in Supplemental Figure S8, A through C. (B, top to bottom) Karyotypes of YS, YM, and YL males with the arrowhead denoting the Y and the arrow denoting the X.
Summary of Y Chromosome size estimations (in Mb) in D. pseudoobscura using different methods
We used optical mapping using BioNano to validate and further scaffold our assembly for YL (Supplemental Table S11; Supplemental Material 2). Optical mapping produces restriction maps for long strands of DNA, which can then be used to validate and place small contigs onto larger scaffolds. The BioNano hybrid assembly pipeline yielded 32 hybrid scaffolds, six of which contained the autosomes and the X Chromosome. The remaining 26 contained sequences from our YL Nanopore assembly. In total, 56 Nanopore contigs were fused into 17 hybrid scaffolds, and the remaining nine scaffolds contained sequences of single contigs. Overall, 63 of the 139 YL contigs are supported by optical reads, accounting for 41.7 Mb of the 55-Mb YL assembly, and only 15 contigs were cut. As expected, Nanopore contigs that were validated by optical mapping are significantly longer (mean length, 662.5 kb) versus those not covered by optical reads (mean length, 175.0 kb). Thus, optical mappings validated 75.8% of our YL assembly (Supplemental Material 2).
Molecular characterization of repeat content of Y Chromosomes
The ancestral Y Chromosome in Drosophila consists mostly of satellite and TE-derived DNA (Pimpinelli et al. 1995; Chang and Larracuente 2019). The Y Chromosome of D. pseudoobscura is not homologous to the ancestral Y of D. melanogaster (Carvalho and Clark 2005) but instead originated from an ordinary autosome about 15 million years ago (Bracewell and Bachtrog 2020). Below, we use both assembly-free and assembly-based methods to characterize the sequence content of the three different Y Chromosomes at the molecular level.
As mentioned, cytogenetic methods suggest that YM and YL are 11 Mb and 26 Mb larger than YS, with an absolute size of YS = 33 Mb, YM = 44 Mb, and YL = 59 Mb (Table 1). Most of this difference is likely driven by repeat accumulation (Fig. 1). We used different metrics to quantify the repeat content of the three different Y Chromosomes and estimate if differences in repeat content can account for the inferred size difference among Y Chromosomes. In particular, we estimate the proportion of short reads assembling to transposable elements (TEs) using dnaPipeTE (Goubert et al. 2015), as well as the proportion of short reads mapping to a curated TE library of D. pseudoobscura (Hill and Betancourt 2018). Because the three different Y Chromosomes are isogenic at the rest of the genome, these metrics can thus be used to estimate differences in the repeat content of the different Y's.
As expected, we find that the Y-replacement line carrying YS has a larger fraction of single-copy reads (71.5%) followed by YM (69.9%) and YL (66.7%) (Supplemental Fig. S4). Using the difference in single-copy reads to estimate size differences between the Y's suggests that YM and YL have an additional 5.6 Mb and 16.8 Mb of repeats in their genomes, respectively, in comparison to YS (Table 1). We also mapped raw Illumina reads to a TE reference library from D. pseudoobscura (Hill and Betancourt 2018) to infer size differences in repeat content among Y Chromosomes by normalizing coverage to the repeat libraries by autosomal coverage. Overall, we found that YS males contained ∼85.3 Mb of repetitive DNA, YM males contained 94.0 Mb, and YL males contained 109.3 Mb (Supplemental Table S12; Supplemental Fig. S5). This suggests that YM and YL have an additional 8.6 Mb and 24.0 Mb of repeats in their genomes, respectively, in comparison to YS (Table 1).
Lastly, heterochromatin-enriched genome assemblies recover overall similar differences in repeat content. We repeat-masked our three different Y Chromosome assemblies using RepeatMasker version 4.1.0 with the D. pseudoobscura family repeat library (Hill and Betancourt 2018). Indeed, the vast majority of Y-linked sequence is repetitive in all three assemblies, with total repeat-masked bases on YS = 34.4 Mb (89%), YM = 39.1 Mb (89%), and YL = 52.4 Mb (88%). Retroelements are by far the most abundant sequence class on each of the three different Y Chromosomes (Table 2). We found that the TE families repeat-masked in our de novo assemblies corroborated the families we found in our Illumina mapping (Tables 1, 2; Supplemental Fig. S6; Supplemental Table S13). TE abundance estimated from both Illumina mappings and de novo assemblies had a significant linear correlation for all Y Chromosome variants (P-value < 2 × 10–16) (Supplemental Fig. S6).
Summary of sequences repeat-masked in D. pseudoobscura Y Chromosome assemblies
Thus, these orthogonal approaches, using both cytogenetics and high-throughput sequencing as well as assembly-based and assembly-free methods, are largely in agreement about the different Y Chromosome sizes. These results illustrate that our de novo Y assemblies are of high quality and that the differences in Y size are largely driven by differences in repeat accumulation.
Recent repeat accumulation drives size differences among Y Chromosomes
The difference in sizes among Y Chromosomes could be due to some repeats being mobilized on the Y or due to structural mutations (such as duplications and deletions) resulting in size variation. Indeed, Dobzhansky hypothesized that smaller Y Chromosomes originated via multiple deletions from the largest Y (Dobzhansky 1935, 1937). To identify which repeats contribute to the observed size differences among Y Chromosomes, we used both assembly-free and assembly-based methods. We mapped short reads to a repeat library to infer different abundances of repeats on the different Y Chromosomes (Fig. 3A; Supplemental Table S12), and also identified repeats on our assembled Y Chromosomes (Fig. 3B; Supplemental Table S13). We found that only a handful of repeats were responsible for the majority of additional DNA of YL and YM (Table 3), and most of the TE families responsible for additional DNA of YL are not causing the size increase of YM.
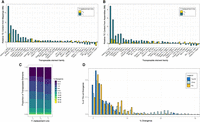
Repeat abundance and age suggest recent TE mobilization contributes to size differences among polymorphic Y's. (A) TE abundance in YM and YL relative to YS from paired-end mappings to the TE library. TEs for which the absolute difference is >200 kb are shown. For details, see Methods section “Repeat content estimation by reference library.” (B) TE abundance in YM and YL chromosome assemblies relative to the YS assembly. TEs for which the absolute difference is >200 kb are shown. For details, see Methods section “Repeat content estimation by de novo assemblies.” (C) Divergence in TE copies from whole-genome Illumina sequencing across the Y's as calculated by dnaPipeTE. (D) Divergence in TE copies from the YL assembly based on the top 50 most abundant TEs (relative to YS), other TEs, and rolling-circle transposons as calculated by RepeatMasker. Percentages of TEs sum to 100% for each category.
Size contribution (in base pairs) and inferred copy number of top repeats responsible for size differences of in D. pseudoobscura Y Chromosome
A few distinct repeats account for most of the size increase in the large Y Chromosome (Table 3). In particular, we find that the Helitron-1_DPe transposon has notably higher copy numbers on YL compared with YM and YS. Read coverage suggests that this TE (which is ∼7.2 kb in size) has roughly 70 copies in the YS and YM strains but 1090 copies in YL, resulting in an extra 7.4 Mb of sequence on YL (Fig. 3A; Table 3; Supplemental Fig. S7). A similar excess of this transposon is found in the assembled Y Chromosomes: ∼200 kb and 150 kb are repeat-masked on YS and YM for Helitron-1_DPe but 4.7 Mb on YL (Supplemental Table S13). As expected for an active TE, we find copies of Helitron-1_DPe distributed across many different Y-linked scaffolds (Supplemental Figs. S7, S8). Helitron TEs replicate through a rolling-circle mechanism, which can result in the generation of tandem copies of the element (Pritham and Feschotte 2007). Indeed, genomic distribution patterns suggest that this element can occur in clusters of tandem arrays across the large Y Chromosome (Supplemental Figs. S8, S9). BLAST searches using queries consisting of the last 50 bp of the Helitron-1_DPe element fused to the first 50 bp yielded 50 hits on our YL assembly, and mate-pair violations using paired-end sequencing data further support the presence of partial copies of tandemly repeated elements (Supplemental Fig. S9; Supplemental Table S15). Because many of the tandem copies of Helitron-1_DPe on YL are partial (Supplemental Fig. S9), our BLAST search yields a conservative estimate for the number of copies in tandem. We find eight nearly full-length copies (using an 70% alignment cutoff) of Helitron-1_DPe on the assembled autosomes and X (and three copies on unmapped scaffolds). This is in contrast to the 289 near-full length copies we find in the YL assembly and the 10 and 14 copies in the YM and YS assemblies, respectively.
The repeat that is responsible for most of the size increase of YM, and ranked second on YL, is a 267-bp-long satellite sequence that is homologous to the intergenic spacer region (IGS) of the rDNA cluster. In many Drosophila species, including D. melanogaster, the rDNA cluster is found on both the X and Y Chromosome and functions as a pairing site for the sex chromosomes during meiosis (McKee et al. 1992). A previous study has shown that the Y of D. pseudoobscura, which is not homologous to the Y of D. melanogaster, lacks functional rDNA clusters on the Y Chromosome (Larracuente et al. 2010). Instead, in situ hybridization revealed four clusters bearing only the IGS of the rDNA repeats on the Y Chromosome of the D. pseudoobscura strain investigated (Larracuente et al. 2010). We infer roughly 13,300 copies of the IGS satellite on YS, 19,800 copies on YM, and 26,100 copies on YL, which adds ∼1.7 Mb of DNA to YM, and 3.4 Mb of DNA on YL (Fig. 3A,B; Supplemental Tables S12, S13). The IGS repeats are highly clustered in our assemblies, as expected for satellite sequences, and are consistent with the in situ results (Larracuente et al. 2010).
The next most common repeat on YL is the Polinton-1_DPe repeat. Polintons are large DNA transposons (Polinton-1_DPe is 15.8 kb) that encode several proteins necessary to replicate themselves (Krupovic and Koonin 2015). Read coverage analysis suggests that there are about 67 copies of this element on YS and YM but 236 copies on YL, adding an additional 2.7 Mb of sequence (Supplemental Table S12). This element is distributed widely across different Y-linked scaffolds (Supplemental Fig. S8), and even read-coverage across the element suggests that many copies are full length on the different Y Chromosomes (Supplemental Fig. S7). The next most common element is the T122 repeat, a 1.5-kb-long uncharacterized element with roughly 70 copies on YS and YM, but almost 1700 copies on YL (Supplemental Table S12), adding an additional 2.5 Mb of sequence. T122 repeats are often found in tandem clusters, mostly separated by HelitronN-1_DPe sequences, which may indicate coamplification of T122_X by Helitron-initiated gene-capture. The next most common repeat is the Copia2-I_Dpse element (4.2 kb in size) that has 270 and 330 copies on YS and YM, respectively, but 670 on YL, thereby adding 1.7 Mb, and three different gypsy elements that are two to three times more abundant on YL than YS and YM, and each adding about another 1 Mb of sequence to YL. Thus, a total of only eight repeats are responsible for >20 Mb of additional DNA on YL (i.e., >85% of the total size gain), with Helitron-1_DPe by far being the largest contributor to the size gain of YL.
On the other hand, a much larger number of TEs contribute to the more modest size gain of YM, with the top eight expanded repeats contributing to <50% of size gain on YM. As mentioned, the repeat leading to most of the size increase on YM is the IGS satellite (adding ∼1.7 Mb of DNA to YM). The next most common one is the 1.1-kb-long HelitronN-1_DPe element and the related 4.6-kb-long MINIME_DP element (Supplemental Fig. S10), which have roughly 6400 (HelitronN-1_DPe) and 900 (MINIME_DP) copies on both YS and YM, but 7100 (HelitronN-1_DPe) and 1000 (MINIME_DP) copies on YM (thereby adding 730 kb and 360 kb of sequence to YM). Gypsy17-I_Dpse adds another 320 kb to YM, and all other amplified TE families add <200 kb sequence each to YM (Supplemental Table S12).
We also used our assembled Y Chromosome contigs and used RepeatMasker to annotate repeats and infer differences in abundance (Fig. 3B; Supplemental Fig. S11; Supplemental Tables S13, S14). Because some contigs in our assemblies were collapsed, we calculated differences in repeat content by accounting for the copy number of assembled contigs. Similar to our assembly-free method, we found that YM and YL had 5.4 and 19.2 Mb extra TEs, respectively, in comparison to YS (Fig. 3B). Again, the same TE families (Helitron, 267-bp IGS, Copia, Polinton) as found in the coverage-based approach are responsible for the size differences between Y Chromosomes based on repeat-masking our assemblies. Thus, both methods suggest that only a small subset of repeats drive most of the observed size differences on YL and that there is overall little correspondence in the types of TEs that have accumulated on the differently sized Y Chromosomes.
Individual copies of recently active TEs are more similar to each other than TEs that are older, and the amount of sequence divergence of individual copies from the consensus can be used to infer the age of when a particular TE family proliferated (Makałowski et al. 2019). Consistent with the size increase on polymorphic Y's being driven by the recent expansion of a few TEs, YM and especially YL contain younger TEs. We find that YM and YL have 4.9% and 23.3% more of their genome-wide TEs within 0%–1% divergence from the consensus compared with Ys, respectively (Fig. 3C). Further, the subset of TEs that are the main contributor to the larger sizes are younger than other TEs (Fig. 3D; Supplemental Fig. S12), and amplified TEs cluster on phylogenetic trees (Supplemental Fig. S13). We further leveraged our assemblies to characterize divergence of TEs strictly located on the Y's. As expected, we found many copies of TEs from different families, especially rolling circle elements (RC; to which the Helitron element belongs), were young in the large Y Chromosome (Fig. 3D). Thus, our de novo genome assemblies provide orthogonal confirmation that TEs have recently amplified on the large Y. Together, these results show that the large Y Chromosome is the derived state in D. pseudoobscura and that DNA accumulation on these larger Y Chromosomes is due to recent TE activity.
Phenotypic consequences of repeat accumulation
Although repetitive DNA is often viewed as a “genetic wasteland,” recent studies have found that differences in repeat content can have profound phenotypic consequences. In particular, expression of hundreds of genes was shown to differ in D. melanogaster males with polymorphic Y Chromosomes (Lemos et al. 2008; 2010). The D. melanogaster Y Chromosome was shown to be involved in diverse phenomena related to transcriptional regulation including X-linked rDNA silencing and suppression of PEV phenotype (Zhou et al. 2012). Additionally, the presence and the number of Y Chromosomes strongly influences genome-wide enrichment patterns of repressive chromatin modifications (Brown and Bachtrog 2014; Brown et al. 2020b), with additional Y Chromosomes diminishing the heterochromatin enrichment at highly repetitive regions such as pericentromeres. Larger, more repeat- and gene-rich Y Chromosomes were also linked to an up-regulation of transposable elements in males (Wei et al. 2020; Nguyen and Bachtrog 2021). These results are generally interpreted as the Y Chromosome acting as a heterochromatin sink that redistributes repressive chromatin marks genome-wide (Francisco and Lemos 2014). The integrity of heterochromatin deteriorates as individuals age (Sun et al. 2018), and the Y Chromosome in D. melanogaster has been shown to contribute to heterochromatin loss and shorter longevity in males (Brown et al. 2020b).
To test for phenotypic consequences of Y Chromosome repeat content, we measured longevity of males that carried a small or large Y Chromosome (YS and YL) versus females, and we assayed global transcriptome and chromatin profiles from YS and YL males. We collected H3K9me3 profiles from brains of young YS and YL males in order to compare the global heterochromatin landscape in males with different Y Chromosomes. The heterochromatin sink effect of the Y would predict that global H3K9me3 levels may be lower for repeats in males with a larger Y Chromosome. Figure 4A shows that H3K9me3 enrichment is similar for TEs located on the X and autosomes in males carrying differently sized Y Chromosomes; however, TEs located on the Y Chromosome show significantly lower H3K9me3 levels in YL males than YS. Thus, the higher repeat content of YL may indeed dilute heterochromatin components in males with a large Y. We find that H3K9me3 levels are generally much lower for TE copies located on the Y Chromosome compared with autosomes and the X, irrespective of the Y variant (Fig. 4A,B; Supplemental Figs. S14, S15). This is consistent with previous findings showing lower levels of heterochromatin enrichment for Y-linked TEs (Wei et al. 2020; Nguyen and Bachtrog 2021), and repeat-rich Y Chromosomes were found to be associated with the up-regulation of transposable elements in males (Wei et al. 2020; Brown et al. 2020b; Nguyen and Bachtrog 2021).
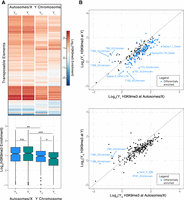
Decreased H3K9me3 enrichment on YL. (A) Heatmap with corresponding boxplot showing H3K9me3 enrichment at TEs by chromosome (autosomes/X vs. Y Chromosomes). Significance values calculated: (*) <0.05, (**) <0.01, (***) <1 × 10−5, Wilcoxon test. (B) Scatterplot of H3K9me3 enrichment at TEs by chromosome group for YS and YL (P < 0.05, two-sample t-test). The gray line indicates similar enrichment levels for both chromosome groups.
We obtained replicate expression data from heads of young and old YS and YL males to quantify TE expression between differently sized Y Chromosomes and during aging. Although global expression across all TE families is not significantly different between Y-replacement males, irrespective of age (Supplemental Fig. S16), we find that a larger number of TE families are significantly up-regulated in YL males compared with YS (12 vs. zero, at least 50% higher expression, Wald test, P-value < 0.05) (Fig. 5A). Furthermore, half of these TEs are more abundant on YL than on YS, suggesting that the TEs that have accumulated on YL are responsible for increased TE expression (Fig. 5B). Notably, this result is consistent irrespective of age, suggesting that differential regulation of TEs starts early in adult males (Supplemental Fig. S17).
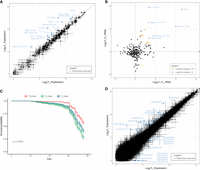
Differential transposon and gene regulation on the Y Chromosome. (A) Differential TE expression between YS and YL males irrespective of age. Data represent the mean of four replicates with standard error bars (50% higher or lower expression, P < 0.05, Wald test). (B) Differential TE expression between YS and YL plotted against their differential TE abundance from Illumina mappings. (C) Lifespan curves of YS and YL males and backcross females. (D) Differential gene expression between YS and YL irrespective of age. Data represent the mean of four replicates with standard error bars (50% higher or lower expression, P < 0.05, Wald test).
Old YL males had slightly higher TE expression compared with that of young YL males, but this was not true in YS males (Supplemental Fig. S18). Consistent with previous studies, we find that males live shorter than females (P-value < 0.0001). However, we find no significant difference in longevity between males carrying the differently sized Y Chromosomes (Fig. 5C; Supplemental Fig. S19); if anything, YL males live slightly longer than their counterpart YS males (median survival time, 77 vs. 74 d, respectively). Thus, although males with a larger Y Chromosome show slightly increased levels of TE expression, and more so in old males, these differences are relatively minor and do not result in faster aging. Small differences in TE expression and lifespan are consistent with previously published results using D. melanogaster Y-replacement lines (Griffin et al. 2015).
Previous studies in D. melanogaster have shown that different Y Chromosomes can influence the expression of hundreds of genes located on the X and autosomes (Lemos et al. 2010). We find that 930 genes on the autosomes and X show differential expression between YS and YL Y-replacement lines, 20 of which are significant (>50% fold change, Wald test, P-value < 0.05) (Fig. 5D; Supplemental Table S16). Many differentially expressed genes are involved in the regulation of chromatin and transcription such as aub, Ulp1, and mxc (Supplemental Table S16). Taken together, our results underline the impact Y Chromosomes can have on genome-wide expression regulation.
Repeated TE mobilization causes Y expansion in other Drosophila species
Our data suggest that modest changes in Y Chromosome size are caused by copy number changes of a large number of repeats (YM), but large size differences are due to mobilization of a few elements (YL). We used flow cytometry to study Y Chromosome size polymorphism in five different Y-replacement lines in Drosophila affinis (kindly provided by Rob Unckless), another member of the obscura species group and a distant relative of D. pseudoobscura. We found three Y Chromosomes that strongly differed in size (Fig. 6). Compared with the smallest Y Chromosome of D. affinis (YS,aff; strain YAF41), the medium-sized Y (YM, aff; strain YAF159) has an additional 12.7 Mb of DNA, and one of the largest Y Chromosomes (YL, aff; strain YAF79) has an extra 19.0 Mb of DNA (Fig. 6A). We used Illumina sequencing of males with the differently sized Y Chromosomes, and mapped reads to the same repeat library used for D. pseudoobscura to infer which repetitive elements are responsible for the size increase of the Y in D. affinis. Overall patterns of repeat accumulation on the large Y Chromosomes are similar between D. affinis and D. pseudoobscura, with only a small number of TEs accounting for most of the size increase of the large Y Chromosome (Fig. 6B; Supplemental Table S17). We find that the same TE (Helitron-1_DPe) that expanded on YL in D. pseudoobscura also is the main contributor to increased DNA content of YL, aff in D. affinis; 8 Mb of additional DNA (42%) on the large Y of D. affinis is due to additional copies of the Helitron-1_DPe element. The second most common repeat on YL, aff of D. affinis is Daff_Jockey_18 (3.4 Mb), followed by Polinton-1_DPe (2.2 Mb), the same element that also amplified on YL in D. pseudoobscura. Congruent with our findings in D. pseudoobscura, TEs on YL, aff and YM, aff also show low levels of sequence divergence (Fig. 6C). Thus, recent amplification of a small number of TEs is also responsible for Y expansion in other species.
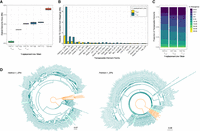
Y Chromosome expansions in D. affinis by the same TEs as in D. pseudoobscura. (A) Diploid genome size estimations from D. affinis Y-replacement lines. Rightmost boxplot (red) is the backcross female strain. (B) TE abundance in YM, aff and YL, aff relative to YS, aff from paired-end mappings to the TE library, made in a similar manner as in Figure 3B. TEs for which the absolute difference is >50 kb are shown. (C) Divergence in TE copies from whole-genome Illumina sequencing across the Y's estimated by dnaPipeTE, made in a similar manner as in Figure 3C. (D) Phylogenetic trees of Helitron-1_DPe (left) and Polinton-1_DPe (right) found in D. pseudoobscura (green), D. affinis (orange), and D. pseudoobscura YL (teal). Repbase sequences are highlighted in blue.
TEs can transfer horizontally between species, and this has previously been reported in the D. pseudoobscura group (Hill and Betancourt 2018). To evaluate whether horizontal transfer between species could have contributed to repeat accumulation on the large Y's, we examined patterns of sequence evolution of the amplified Helitron and Polinton elements in D. affinis and D. pseudoobscura. We extracted Helitron-1_DPe and Polinton-1_DPe copies found on the autosomes/X of D. pseudoobscura and D. affinis and our YL assembly from D. pseudoobscura, and we found that elements cluster by species (Fig. 6D; Supplemental Fig. S20). Thus, this suggests that these elements evolved independently in the two species (i.e., not through horizontal transfer), as found previously (Hill and Betancourt 2018). Because we lack a high-quality assembly of the large Y Chromosome in D. affinis, we cannot reconstruct the sequence of individual Y-linked TEs in this species, but our coverage analysis implies that a large majority of the Helitron and Polinton reads are derived from YL and YL,aff. We therefore constructed consensus sequences for these two TEs for both species and mapped genomic reads from D. affinis and D. pseudoobscura males with the large Y's to the consensus sequences to estimate the rate of species-specific mapping. As expected if TEs amplified independently in the two species, most YL reads map to the D. pseudoobscura consensus and most YL,aff reads map to the D. affinis consensus (Table 4). Thus, we find no signatures of horizontal transfer at these elements, which suggests that rampant amplification of autonomous TEs occurred independently on the two different species’ Y Chromosomes.
YL and YL, aff Helitron/Polinton mappings to the D. pseudoobscura and D. affinis consensus sequences
Discussion
In a series of papers, Dobzhansky cataloged variation in size and shape of the D. pseudoobscura Y Chromosome across its geographic range (Dobzhansky 1935, 1937). He surveyed roughly 100 naturally derived strains and identified five different Y Chromosomes based on size and shape: one large metacentric Y (called Type I in Dobzhansky's papers), two smaller acrocentric/submetacentric Y Chromosomes (Type IV and Type V), and two small metacentric Y's (Type VI and Type VII). Type I (found in nine lines) was restricted to Mexico and southern Arizona, where it exists together with Type IV (Dobzhansky 1937). Type IV and Type V are smaller acrocentric/submetacentric chromosomes that differ slightly in size of their longer arm (Dobzhansky 1935, 1937). These two types are widely distributed across the United States (found in 33 and 44 lines) (Dobzhansky 1937) with Type IV being more common along the West Coast, and Type V more common further inland along the Rockies, yet both types are found in many locations (Dobzhansky 1935, 1937). Type VI and Type VII, two metacentric small Y's that differ in their size, were found only inland in a few strains (in five and three lines) (Dobzhansky 1937). Our large metacentric Y Chromosome (YL) was initially collected in Sinola, Mexico, and its size, shape, and geographic location suggest that it may correspond to Type I of Dobzhansky's classification. We did not detect any other metacentric chromosomes in our Y-replacement lines, suggesting that we did not sample the rare Type VI and Type VII Y Chromosomes. Their relative sizes suggest that YM may correspond to Type IV of Dobzhansky's classification, and YS and all other Y Chromosomes could correspond to Type V. This would imply that the distribution and relative abundance of the different Y types has shifted since Dobzhansky's initial study, and it would be of great interest to resample some of the initial sites to study geographic shifts in Y type over time.
Although Dobzhansky first categorized Y polymorphism, the molecular basis or the mechanism of the origin of variation of Y size was unknown, and he speculated that the polymorphic Y's may have been derived by losses of sections from the largest Y Chromosome (Dobzhansky 1935, 1937). Here, we use a combination of classical cytogenetic methods and long-read sequencing to identify the molecular nature of Y size variation. We show that in contrast to Dobzhansky's hypothesis, most of the size differences between morphologically distinct Y Chromosomes are due to the recent expansion of a few TEs on the largest Y Chromosome. Thus, rather than being the ancestral form, this indicates that the large Y Chromosome is instead derived from a smaller ancestral Y. We find that the same TE families can contribute to Y expansion independently in different species of Drosophila. Why a few TEs amplified on some Y Chromosomes but not others is unclear. Repeat expansion has been found to drive genome size increases in other species (Ågren and Wright 2011; Rutkowska et al. 2012; Wong et al. 2019), with different dynamics of repeat expansions among taxa. In larvaceans (Naville et al. 2019) and rotifers (Blommaert et al. 2019), genome expansion is due to a combination of various repeat element classes, whereas a single TE family expanded in brown hydra lineages (Wong et al. 2019). Independent amplification of the same TE families in two different species suggests that some TEs, for unknown reasons, are inherently more prone to become deregulated on Y Chromosomes.
Why these differences in repeat content among Y Chromosomes exist is unclear. Dobzhansky tried to find a correlation between the type of Y Chromosome present in a given strain and other properties of that strain, including hybrid sterility (Dobzhansky 1933, 1935, 1937; Dobzhansky and Boche 1933), but concluded that the morphological and physiological characteristics of a particular male are not affected by the type of its Y Chromosome. By homogenizing the genetic background of the different Y types, we were able to detect subtle differences among Y Chromosomes. We show that overall heterochromatin enrichment is reduced on the Y of males carrying a larger Y Chromosome, and expression of a subset of TEs, especially those that have accumulated on YL, is increased. Additionally, expression of a few dozen of X-linked and autosomal genes is influenced by Y-type. Thus, Y variation can potentially influence male-related fitness traits. Thus, although differently sized Y Chromosomes can contribute to male fitness, we do not know the evolutionary forces driving Y size differences. Repeat accumulation on the Y is typically interpreted to reflect the reduced efficacy of natural selection (Bachtrog 2003; Bachtrog et al. 2008), but repeats can also contribute to diversification of gene families involved in meiotic drive on sex chromosomes (Bachtrog et al. 2019; Vogan 2021).
Morphological variation in Y Chromosome size and shape has been identified in many organisms, including humans (Chakraborty and Chakraborty 1984), but has not yet been studied at the DNA sequence level. Our study shows that novel sequencing approaches allow the assembly of even highly repetitive Y Chromosomes, and presents novel insights into the repetitive landscapes of variable Y Chromosomes.
Methods
Y-Replacement line backcrosses
A total of 26 different D. pseudoobscura males were backcrossed to the sequenced D. pseudoobscura stock (MVZ 25, courtesy of Stephen Schaeffer). The list of fly strains used for Y-replacement lines is given in Supplemental Table S1, and Supplemental Figure S1 shows the geographic origin of different lines. We backcrossed males to virgin females for nine generations, after which the Y-replacement lines were maintained on standard molasses culture. We verified homozygosity on the autosomes and X Chromosome by mapping Illumina data to the masked genome, calling sites with SAMtools mpileup (Li et al. 2009), and then dividing the number of sites that are homozygous across all strains by the total number of variant sites (Supplemental Table S18).
Y Chromosome karyotyping
We dissected brains from third instar larvae in 0.9% NaCl. First, we incubated brains in 0.9% NaCl with two drops of 0.1% colchicine for 10 min at room temperature, followed by another 10-min incubation in 0.1 M KCl solution. Then, we fixed brains in 3:1 methanol and acetic acid for 1 h. We moved the fixed tissues to 50 µL of 60% acetic acid solution and vigorously disassociated tissues via pipetting for 1 min. We dispensed the solution onto preheated 55°C microscope slides and stained them in 7% Giemsa solution (1× PBS at pH 6.5) for 40 min. Slides were imaged on a Leica DM5000B microscope with SPOT imaging software. We then measured chromosome lengths with KaryoType (Altınordu et al. 2016).
Flow cytometry
We estimated the sizes of Y Chromosomes using flow cytometry as previously described (Ellis et al. 2014). Briefly, we dissected and snap-froze heads from flies aged 3–7 d old. For an internal standard, we used heads from a D. virilis strain provided by Spencer Johnston (1C = 328 Mb). To prepare samples, we dounce homogenized one D. pseudoobscura head with one D. virilis female head in a 2-mL Kontes dounce homogenizer with 1 mL Galbraith buffer (45 mM MgCl2, 30 mM sodium citrate, 20 mM MOPS, 0.1% Triton-X 100 [v/v] at pH 7.2) and filtered the nuclei-containing solution through a 100-µM cell strainer. We then added 25 µL propidium iodide (1 mg/mL) to stain nuclei for 30 min at 4°C and ran the stained nuclei on a BD LSR II cytometer at 30–60 events/sec.
We exported raw fluorescence data on FlowJo (v.10.1) and used R (R Core Team 2020) to calculate the genome sizes. In brief, we used the base R density function to estimate the fluorescence of sample and
D. virilis standard peaks. Then, to calculate the genome size of the unknown Y-replacement line, we assumed fluorescent linearity and
used the following equation:
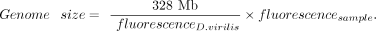 To estimate %AT content relative to D. virilis, we followed the method of Ellis et al. (2014) and used 1 µL of DAPI (1 mg/1 mL) in place of propidium iodide. We obtained heterochromatin estimates following the same
protocol as above. Note that we do not have a heterochromatin estimate for the D. virilis standard, only a whole-genome size estimate, thus the measurements are recorded as percentages. Regardless, all samples were
normalized by the same D. virilis standard so any differences reflect relative differences in %AT content among Y-replacement lines.
To estimate %AT content relative to D. virilis, we followed the method of Ellis et al. (2014) and used 1 µL of DAPI (1 mg/1 mL) in place of propidium iodide. We obtained heterochromatin estimates following the same
protocol as above. Note that we do not have a heterochromatin estimate for the D. virilis standard, only a whole-genome size estimate, thus the measurements are recorded as percentages. Regardless, all samples were
normalized by the same D. virilis standard so any differences reflect relative differences in %AT content among Y-replacement lines.
We estimated the proportion of underreplicated DNA in polytene tissue following the protocol previously described (Johnston et al. 2013). Briefly, we dissected thoracic tissue from flies and followed the protocol described above without the D. virilis standard. We ran the same protocol with head tissue as a positive control. We then used the base R density function to estimate the 4C and 2C peak fluorescences. All flow cytometry work was performed at the University of California Berkeley Cancer Research Laboratory Flow Cytometry Facility.
Lifespan assays
We conducted lifespan assays following the method of Linford et al. (2013) with the following rearing conditions: 18°C, 60% relative humidity, 12-h light, and standard Bloomington food. Briefly, we collected synchronized embryos on a molasses plate with yeast paste for 16–20 h. We washed embryos with 1× PBS (pH 7.2) three times and dispensed 10 µL of embryos per culture vial by pipette. To obtain synchronized adults, we collected emerging adults over a 3-d window and aged the adults for 3 d to mate and copulate. Male and female flies were separated into separate vials, placing 30 flies per vial. We moved flies to new vials every 2–3 d without CO2 and recorded the number of deaths. Flies that escaped were censored. In total, 1196 sequenced strain female flies, 722 sequenced strain YS male flies, and 987 YL male flies were tracked for the lifespan assays recorded here.
Tissue collection
Tissue for gDNA-Seq was collected by freezing male flies and popping off heads via vortexing. To collect tissue for the RNA-seq experiment, we censored the entire experiment once it reached 50% survivorship for one line. We collected head tissue for RNA-seq in a similar manner to the gDNA-Seq. For the ChIP-seq experiment, we anesthetized young (5–7 d old) flies using CO2, dissected the brains in 1× PBS (pH 7.2), and immediately froze two brains per sample using dry ice.
DNA, RNA, and ChIP library preparation
We extracted DNA from pooled heads of each Y-replacement male using the DNeasy Qiagen extraction kit. We then prepared gDNA libraries using the Illumina TruSeq Nano DNA library kit (20015965). For the cDNA libraries, we first extracted RNA from 20 pooled heads for Y-replacement line males using TRIzol (Invitrogen 15596026). We then made libraries using the Illumina TruSeq stranded total RNA Ribo-Zero Gold kit (Illumina RS-122-2201).
For the ChIP-seq libraries, we started with brain tissue and prepared ChIP pull-downs as previously described (Nguyen and Bachtrog 2021). Briefly, we modified the native and ultra-low input ChIP protocol (Brind'Amour et al. 2015) for double-brain ChIP-seq. First, we added 40 µL EZ nuclei lysis buffer to frozen tissues and homogenized them with a pestle grinder on ice. We spun cells down at 1000g for ∼10 min and decanted 20 µL of the supernatant. We froze the cell pellet in the lysis buffer at −80°C. We then followed the 100,000 cell count digestion protocol from Brind'Amour et al. (2015) with a few modifications. For each D. pseudoobscura sample and D. melanogaster spike-in sample, we added 4.4 µL of 1% DOC and 1% Triton-X 100 to resuspended cells and mixed thoroughly. Briefly, we digested each sample at 37°C for 04:58 (mm:ss) with MNase and quenched the reaction with 4.9 µL of 100 mM EDTA (pH 8.0). We then added the fragmented spike-in samples to each D. pseudoobscura sample to account for ∼20% of the final pooled sample volume (i.e., final sample consisted of 80% D. pseudoobscura, 20% D. melanogaster). We reserved 10% of the pooled fragmented sample for the input (fragment control) and used the remaining 90% to perform the chromatin pull-down (ChIP sample, target antibody: H3K9me3 polyclonal classic Diagenode C15410056) according to the 100,000 cell specifications. We used the Rubicon Genomics ThruPlex kit to prepare ChIP-seq libraries for sequencing with 10 PCR amplification cycles for the input samples and 12 cycles for the ChIP pull-down samples. We sequenced libraries on the Illumina HiSeq 4000 platform at the Vincent J. Coates Genomics Sequencing Center.
Repeat content estimations by reference library
We used the D. obscura group consensus TE library from Hill and Betancourt (2018) and included a Y Chromosome–specific 267-bp repeat sequence that was later confirmed as the 267-bp intergenic spacer (Larracuente et al. 2010). We masked the D. pseudoobscura or D. affinis genome using this repeat library with RepeatMasker (Smit et al. 2013–2015) with the following parameters: -no_is -no_low -norna, v4.1.0. Then, we mapped reads with BWA-MEM (Li and Durbin 2009) to the repeat-masked genome and the TE library. In this way, all reads derived from TEs will map to the consensus library to decrease reference bias for the small (reference) Y. We used BEDTools (Quinlan and Hall 2010) coverage to calculate coverage per TE and normalized coverage by the median autosomal coverage. The autosomes were confirmed to have approximately two times coverage (i.e., diploid genome).
We summed the copy numbers of all TEs multiplied by their respective lengths to estimate the difference in TE base pair contribution between Y-replacement lines. Note that this sum includes TEs derived from autosomes; however, these contributions should be negligible because the strains have isogenic backgrounds.
Repeat content estimation and divergence by dnaPipeTE
We ran dnaPipeTE (Goubert et al. 2015) on the first strands of samples with the following parameters: -coverage 0.5 -genome-size 175000000 -sample-number 2. Note
that we used a genome size of 175 Mb for all three Y-replacement lines even though the total genome size differs among strains.
Larger, repeat-rich genomes should have a smaller fraction of their genome composed of single-copy elements, and we used differences
in the single-copy elements to estimate relative differences in repeat content between Y's. We used the following equation
to estimate this for each male strain:
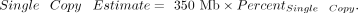 We also used the estimated divergence to identify recently active TEs on a genome-wide basis.
We also used the estimated divergence to identify recently active TEs on a genome-wide basis.
Nanopore sequencing and Y Chromosome de novo assembly
For the reference strain male (YS), we used reads from Bracewell et al. (2019). For YM males, we used one Nanopore MinION flow cell (v9.4.1 RevD) with the ligation sequencing kit (SQK-LSK109) and obtained 540,115 total filtered reads with a mean read length of 15,972 bp. We sequenced YL males using three Nanopore MinION flowcells (v9.4) with the ligation sequencing kit (SQK-LSK108 and SQK-LSK109). We combined the total output from these three runs and obtained 482,379 filtered reads with a mean read length of 19,650 bp.
We followed a heterochromatin-enriched assembly approach from Chang and Larracuente (2019). Briefly, we mapped reads to the D. pseudoobscura assembly (Bracewell et al. 2019) using minimap2 (v2.1) (Li 2018) with default parameters. Then we extracted reads that did not map to putative euchromatin in the D. pseudoobscura assembly (Bracewell et al. 2019) to make heterochromatin-only assemblies using Canu (v1.6) (Koren et al. 2017) and FALCON (v1.4.2) (Chin et al. 2016) independently of each other.
We then removed contigs containing bacterial DNA using BLASTN against the NCBI database and used QuickMerge to merge contigs between the Canu- and FALCON-based assemblies. We settled on assemblies that had a high N50 and generally fewer contigs, all of which resulted from merges between assemblies from a Canu to FALCON direction with assemblies from a FALCON to Canu direction. To identify Y contigs, we mapped Illumina male and female reads to the corresponding heterochromatin-based assemblies with BWA-MEM and obtained genomic coverages with BEDTools coverage. Y Chromosomes can contain repeats found elsewhere in the genome so we used differences in female and male coverages to identify male-bias/Y-linked sequences. Contigs whose Log2(female/male) coverage was below −1 and whose overall male coverage was at least five times were considered Y contigs. We then appended these Y contigs to the assembled autosomes and X scaffolds to generate full male genomes. Using our coverage analysis, we also split contigs whose coverage was partially male-biased for the medium and small Y assemblies. Additional information can be found in Supplemental Material 1.
Repeat content estimation by de novo assemblies
To calculate the estimated amount of DNA and repeats on each Y Chromosome variant, we normalized the coverage of the Y contigs
by the coverage of the autosomes. We used this normalized coverage as the corresponding copy number (estimated abundance)
of each Y contig with which we used to adjust the estimated total DNA length and total repeat abundance. Repeats and their
lengths were identified by RepeatMasker v4.1.0.
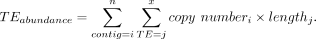
Bionano molecules and hybrid assembly
We used optical mapping (Bionano Genomics) to stitch Y contigs from the large Y Chromosome together. Briefly, DNA was extracted from 45 mg of frozen male larvae using the Bionano prep animal tissue DNA isolation kit (80002) with modifications from a protocol by Susan Brown at Kansas State University. Then, DNA was homogenized and embedded in agarose plugs, treated with Proteinase K and RNase A, and isolated from agarose and cleaned via drop dialysis. DNA was then left for several days at room temperature to homogenize before quantitation. Seven hundred fifty nanograms of DNA was labeled using the DLS labeling kit (80005) following the Bionano prep direct label and stain (DLS) protocol (document 30206).
Labeled DNA was run through the Bionano Saphyr platform. These reads were then assembled into full optical maps and then allowed for stitching of Y Chromosome sequences. In total, 334,498 molecules were generated with an N50 of 223.875 kb, and this was down-sampled to proceed with the hybrid assembly pipeline (i.e., 189,039 molecules and N50 of 273.571 kb). The Bionano hybrid assembly contained 32 hybrid scaffolds, six belonging to the autosomes and X Chromosome and the remaining 26 to YL Chromosome. We confirmed the six scaffolds were from the autosomes and the X Chromosome with NUCmer (v3.1) alignments to the D. pseudoobscura genome. Additional information can be found in Supplemental Material 2. The Bionano preparation and hybrid assembly were completed at the UC Davis DNA Technologies Core.
Y-linked TE divergence estimates
To obtain Kimura divergence estimates for each TE family and their copies on the Y's, we used parseRM_getLandscape.pl from Kapusta et al. (2017) with our RepeatMasked Y Chromosome assemblies.
Tandemly repeated transposable element analysis
We adapted an approach from McGurk and Barbash (2018) to infer if transposons were tandemly repeated by identifying violations in paired-end alignments. Paired-end reads were first aligned with BWA-MEM in single-end mode to the masked D. pseudoobscura genome and the TE library. We then obtained read pairs in which both forward and reverse reads aligned to the same TE and then plotted their alignment coordinates.
To infer the number of tandemly repeated Helitron-1_DPe transposons in the YL assembly, we made artificial queries of full head-to-tail sequences as previously described (Pritham and Feschotte 2007). We used BLASTN (v2.6.0+) to BLAST this sequence to the Y assemblies and identified tandem junctions based on matches with >80% aligned.
Transposable element phylogenies
We used BLASTN (2.6.0+) to extract transposon sequences from our Y Chromosome assemblies. We kept alignments of at least 70% length with the exception of Gypsy22-I_Dpse, Copia-1_DPer-I, Polinton-1_DPe, and T150_X, where we kept alignments mapping to regions of elevated coverage. We used Clustal Omega (v1.2.4) using default parameters to generate multiple sequence alignments and then RAxML (v8.2.12) (Stamatakis 2014) using the following parameters: -f a -x 1255 -p 555 -# 100 -m GTRGAMMA. Phylogenies were visualized using ggtree (Yu 2020) and FigTree (http://tree.bio.ed.ac.uk/software/).
Gene expression analysis
Briefly, we mapped RNA-seq reads for each replicate to a repository of ribosomal DNA scaffolds from NCBI and removed all reads that mapped to this scaffold. Differences in rRNA abundance in sequenced samples are likely to be technical artifacts following RNA library preparation. We then mapped the remaining reads to the D. pseudoobscura genome and to the repeat library separately using HISAT2 (Kim et al. 2015) with default parameters. We then used Subread featureCounts (Liao et al. 2014) to calculate gene counts and repeat counts and used DESeq2 (Love et al. 2014) to normalize the libraries and perform differential expression analysis between male/female flies of different ages.
ChIP enrichment analysis
We followed the approach from Nguyen and Bachtrog (2021). We took ChIP mappings to the entire genome and called enrichment at repetitive elements through TECounts (part of TEtranscripts) (Jin et al. 2015). To call for repeat enrichment on a per chromosome basis, we split mappings by chromosome (autosome/X Chromosome and Y Chromosome) and ran TECounts separately for each split mapping. We then normalized counts at repeats in both the ChIP and input by mean autosomal coverage and calculated ChIP enrichment as ChIP/input.
Transposon cross-mapping analysis
We identified autosome/X Helitron and Polinton elements in both the D. pseudoobscura and D. affinis genomes using BLASTN, keeping alignments with at least 70% length. We generated consensus sequences using EMBOSS cons with default parameters (https://www.ebi.ac.uk/Tools/msa/emboss_cons; accessed April 22, 2021). For YL and YL, aff samples, we extracted reads mapped to Helitron-1_DPe/Polinton-1_DPe. Then, we used BWA-MEM to map reads to both consensus copies of the corresponding transposon simultaneously.
Data access
All the raw sequence data generated in this study along with the Y Chromosome assemblies have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA799077. The Bionano data can be accessed through Dryad (https://doi.org/10.6078/D17Q6Z).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank members of the Bachtrog laboratory, especially Kevin H.C. Wei and Ryan Bracewell, for help with data analysis. We thank Carl Hjelman and Hector Nolla for advice on flow cytometry. We thank Spencer Johnston, Stephen Schaeffer, Nitin Phadnis, Ryan Bracewell, Rob Unckless, and the Cornell Stock Center for providing fly strains. This research was funded by National Institutes of Health grants (R01 GM101255, R01 GM076007, and R56 AG057029) to D.B.
Author contributions: A.H.N. performed fly crosses, established cytological and phenotypic assays, and conducted all sequencing analyses. W.W. performed the karyotype and flow cytometry experiments and phenotypic assays and helped with sequencing analyses. E.C. assisted with fly crosses and flow cytometry experiments. K.C. optimized the DNA extraction method and generated long-read sequencing data. D.B. conceived the experiments and wrote the paper.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275996.121.
- Received July 13, 2021.
- Accepted April 15, 2022.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








