
Early embryonic mutations reveal dynamics of somatic and germ cell lineages in mice
- Arikuni Uchimura1,2,
- Hirotaka Matsumoto3,4,
- Yasunari Satoh1,
- Yohei Minakuchi5,
- Sayaka Wakayama6,
- Teruhiko Wakayama6,7,
- Mayumi Higuchi2,
- Masakazu Hashimoto8,
- Ryutaro Fukumura9,
- Atsushi Toyoda5,
- Yoichi Gondo9 and
- Takeshi Yagi2
- 1Department of Molecular Biosciences, Radiation Effects Research Foundation, Hiroshima, Hiroshima, 732-0815, Japan;
- 2KOKORO-Biology Group, Laboratories for Integrated Biology, Graduate School of Frontier Biosciences, Osaka University, Suita, Osaka, 565-0871, Japan;
- 3School of Information and Data Sciences, Nagasaki University, Nagasaki, Nagasaki, 852-8521, Japan;
- 4Laboratory for Bioinformatics Research, RIKEN Center for Biosystems and Dynamics Research, Wako, Saitama, 351-0198, Japan;
- 5Comparative Genomics Laboratory, National Institute of Genetics, Mishima, Shizuoka, 411-8540, Japan;
- 6Advanced Biotechnology Centre, University of Yamanashi, Kofu, Yamanashi, 400-8510, Japan;
- 7Faculty of Life and Environmental Sciences, University of Yamanashi, Kofu, Yamanashi, 400-8510, Japan;
- 8Laboratory for Embryogenesis, Graduate School of Frontier Biosciences, Osaka University, Suita, Osaka, 565-0871, Japan;
- 9Department of Molecular Life Sciences, Tokai University School of Medicine, Isehara, Kanagawa, 259-1193, Japan
Abstract
De novo mutations accumulate with zygotic cell divisions. However, the occurrence of these mutations and the way they are inherited by somatic cells and germ cells remain unclear. Here, we present a novel method to reconstruct cell lineages. We identified mosaic mutations in mice using deep whole-genome sequencing and reconstructed embryonic cell lineages based on the variant allele frequencies of the mutations. The reconstructed trees were confirmed using nuclear transfer experiments and the genotyping of approximately 50 offspring of each tree. The most detailed tree had 32 terminal nodes and showed cell divisions from the fertilized egg to germ cell– and somatic cell–specific lineages, indicating at least five independent cell lineages that would be selected as founders of the primordial germ cells. The contributions of each lineage to germ cells and offspring varied widely. At the emergence of the germ cell–specific lineages, 10–15 embryonic mutations had accumulated, suggesting that the pregastrulation mutation rate is 1.0 mutation per mitosis. Subsequent mutation rates were 0.7 for germ cells and 13.2 for tail fibroblasts. Our results show a new framework to assess embryonic lineages; further, we suggest an evolutionary strategy for preserving heterogeneity owing to postzygotic mutations in offspring.
Newly occurring mutations produce genetic variations within cell populations, and they can be used as DNA barcodes for tracking cell lineages during development (Baron and van Oudenaarden 2019). Mutations occurring postfertilization are often detected as mosaic mutations, which exist at lower variant allele frequencies (VAFs) than inherited mutations that account for 50% in the case of heterozygous mutations. Researchers have reconstructed early embryonic cell lineages based on such mosaic mutations and tracked each lineage to several adult tissues (Bizzotto et al. 2021; Fasching et al. 2021), such as the gastrointestinal tract (Behjati et al. 2014), blood (Lee-Six et al. 2018), and brain (Evrony et al. 2015; Lodato et al. 2015; Bae et al. 2018). In mammals, germ cells are generated by induction from epiblasts—the main contributors to somatic tissues (Tang et al. 2016; Nguyen et al. 2019). Therefore, it is particularly important to understand how germ cell lineages emerge in embryos. However, reconstructing germ cell lineages has been limited to partial tracking (Ueno et al. 2009; Reizel et al. 2012; Lindsay et al. 2019; Yang et al. 2021), and the relationship between entire embryonic lineages and germ cell lineages remains unclear.
Here, we present a new direct method to reconstruct and track embryonic cell lineages based on mosaic mutations. Thus far, whole-genome sequencing (WGS) of multiple single cells with whole-genome amplification (Evrony et al. 2015; Lodato et al. 2015) or of multiple single-cell clones expanded in vitro (Behjati et al. 2014; Bae et al. 2018; Lee-Six et al. 2018; Bizzotto et al. 2021; Fasching et al. 2021) has been required for detailed lineage reconstruction. In single-cell-based lineage construction, researchers compare the accumulated somatic mutations in each clone and reconstruct the lineages. However, conducting WGS on many single-cell clones to cover the entire early lineages incurs massive sequencing cost, labor, and time, making it a difficult process. It would be desirable to develop an easier method for reconstructing cell lineages. In this study, we noted that the VAFs of mosaic mutations in adult tissues reflect the order in which the mutations occurred during embryogenesis; thus, we reconstructed the cell lineages according to the arithmetical law of VAFs (Fig. 1A): Rule 1 is if cells B and C are the two descendants of cell A and if mutations b, c, and a arose de novo in these three cells, respectively, then the allele frequency of a in any adult tissue equals the sum of the frequencies of b and c. Rule 2 is if cell E is part of the ancestral lineage of cell F and if mutations e and f arose de novo in these cells, respectively, then in any adult tissue, the allele frequency of e will be greater than or equal to the frequency of allele f.
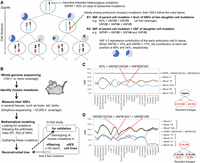
Principle of cell lineage reconstruction. (A) Inferred tree of lineage relationships and genotypes of the first two postzygotic divisions, as well as the arithmetical rules of variant allele frequencies (VAFs) of mosaic mutations used for the lineage reconstruction. The red and black numbers represent independently arising mutations that were first fixed and subsequently inherited, respectively. Pie charts represent example contributions of the early embryonic cell to the adult samples. (B) Overall experimental procedure for reconstruction of lineages. (C,D) Examples of the arithmetical relationship of VAFs of mosaic mutations in individual tissues of a mouse (mouse ID: ConC31). Mutations that show the same frequency in all tissues are considered co-occurring. The sum of the VAFs of the two mutation groups is indicated by the black line. The sum of the average VAF of mutations #2, #19, and #33A (VAF#2/19/33A) and the average VAF of mutations #28 and #33C (VAF#28/33C) equals 50%, indicating the first cell cleavage after a fertilized egg (B). The sum of VAF#14/41 and VAF#51/80 is equal to the VAF#2/19/33A, indicating the subsequent cell division after the first cell cleavage (C). Other examples are shown in Supplemental Figure S1.
To date, several studies have investigated clonal mosaicism by detecting low-VAF mutations with deep-coverage WGS (Rodin et al. 2021; Yang et al. 2021). However, some challenges still exist in reconstructing the detailed lineages directly based on the VAF of mosaic mutations. Here, we have improved several steps through measurement accuracy, using multiple samples, using appropriate mathematical modeling, and properly filtering false-positive variants. As a result, we have developed a new method that enables the reconstruction of reliable lineages simply by measuring the VAFs of mosaic mutations (Fig. 1B). This method is easily applicable to any tissue or species, and a more comprehensive analysis can be achieved using mosaic mutations detected in multiple tissues such as germ cells and somatic tissues. In addition, the lineages are reconstructed according to the magnitude of the VAF; therefore, the difference in VAFs between a parent cell and a daughter cell can be used to trace the assumed lineages that do not have unique mutations.
In this study, we applied this method to a mouse model and reconstructed the early embryonic lineages leading to somatic and germ cells. We also examined the offspring of the individuals for whom embryonic lineages had been reconstructed to assess the inheritance of mutations in the next generation. In particular, our analysis of both testis and tail samples to detect mosaic mutations revealed detailed cell lineages, which showed the cell division process from the zygote to germ cell– and somatic cell–specific lineages.
Results
Reconstructing early embryonic lineages
First, we conducted WGS with approximately 100× coverage depth to detect mosaic mutations using tail DNA (Fig. 1B). Then, we measured the VAFs of each mosaic mutation in various tissues (e.g., the cerebrum, liver, and testis) through amplicon-based targeted sequencing (approximately 10,000× coverage) with our optimized conditions, such as performing at least three independent initial reactions. We opted to analyze the VAFs in multiple samples to distinguish between mutations with coincidentally similar VAF values in one sample. We analyzed five mice and obtained 27–40 verified mosaic mutations (tail VAF range of 3.2%–39.1%) per individual. As expected, the VAFs fluctuated among tissue samples (Supplemental Fig. S1).
We first classified mosaic mutations with substantially similar VAFs across all tissues in the same group to reconstruct the lineages, indicating that these mutations occurred at the same developmental point. Next, we looked for mutation sets satisfying the arithmetic relationship (Fig. 1A). Using one mouse (mouse ID: ConC31) as an example, the sum of the average VAF of mutations #2, #19, and #33A (VAF#2/19/33A) and the average VAF of mutations #28 and #33C (VAF#28/33C) corresponded to ∼50% (Fig. 1C). Similarly, VAF#2/19/33A was equal to the sum of VAF#14/41 and VAF#51/80 (Fig. 1D). These relationships indicate critical single-cell division events from one parent to two daughter cells when the mutations were fixed. Cell lineages were reconstructed by gathering these mutation sets, and we placed mosaic mutations (those not part of any summation relationship) on an appropriate branch of the tree according to their relationship magnitude (example in Supplemental Fig. S1) to complete phylogenetic trees. We further developed a partial and top-down daughter cell–joining algorithm and reconstructed the cell lineages (Fig. 2A–D; Supplemental Fig. S1C).
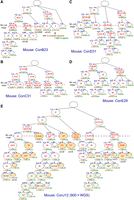
Reconstructed lineage trees based on de novo mutations occurring during the first several postzygotic divisions. Reconstructed lineage trees of five animals based on the mosaic mutations detected through whole-genome sequencing (WGS) with 100× (A–D) and 900× (E) coverages. The red numbers represent newly arising mutations. The underlined numbers represent mutations also observed in the offspring. Numbers with an asterisk represent mutations added using the genotyping results of the offspring. The numbers with a hash mark represent mutations with a GERP score of more than three (expected to have a harmful effect). The nuclear transfer embryonic stem (ntES) below the cell positions represents the lineages leading to each ntES cell line established in the mice ConB23 (A) and ConJ12 (E). The brown and green numbers (%) below each cell position represent the VAFs of labeled cells’ autosomal mutations (or the estimated values if there are no mutations) in somatic tissue averages and in a sperm sample, respectively. In the ConJ12 tree (E), in sperm or all somatic tissue samples, cells with VAF values comparable to the background were colored orange and light green as somatic cell–specific lineages and germ cell–specific lineages, respectively.
We performed two experiments to confirm mutation lineages. One was a nuclear transfer–based single-cell approach (Wakayama et al. 1998). Focusing on two mice (ID: ConJ12 and ConB23), we selected nuclei from tail fibroblasts to produce nuclear-transferred embryos, establishing 11 nuclear transfer embryonic stem (ntES) cell lines from ConJ12 and four lines from ConB23. We checked all mosaic mutations in each line using amplicon-based targeted sequencing (less than 1000× coverage). The genotypes of individual ntES cell lines were consistent with the possession of mutations at a unique terminal node of the lineage trees (Fig. 2; Supplemental Data 1). For example, in ntES cell line #1 for ConB23 (Fig. 2A), mutations #5, #11, #14, #22, and #43 were found to be heterozygous variants, but no other mutations were detected in this cell line. This finding supports the position that our reconstructed lineages are reliable. The other approach involved assessing the offspring. We produced about 50 F1 mice from each of the five mice and investigated their mosaic mutations using targeted sequencing. Genotyping of 287 F1 mice revealed complete consistency with reconstructed lineages: Mutations found together in the same F1 individual are always found on a common reconstructed lineage (Supplemental Data 1). Specifically, 71% (100/141) of the lineage-assigned mosaic mutations were detected in offspring. Moreover, inherited mutations (Fig. 2, underscored) were found in the genealogy, suggesting lineages that led to germ cells. These two experiments supported our lineage tree conclusions. We also assessed lineage-unassigned mosaic mutations in the ntES cell lines and offspring, identifying four mosaic mutation positions (Fig. 2, asterisks) and adding four branches to the trees. Consequently, our reconstructed trees comprised eight to 12 cell lineages with 19–35 mosaic mutations.
To assess the feasibility of expanding the application of our lineage reconstruction method, we conducted additional WGS using tail and testis samples of mouse ConJ12 with more than 900× total coverage (454× and 456× coverage for tail and testis DNA, respectively). We identified 53 additional mosaic mutations, including lower-VAF variants, and measured their VAFs. Using the same algorithm, we reconstructed a new cell lineage tree (Fig. 2E) consistent with that of 100× coverage data (Supplemental Fig. S1C). Genotyping of the ntES cell lines and offspring was also consistent with this tree. Here, we were able to add germ cell–specific lineages to the tree by detecting mosaic mutations in the testis (Fig. 2E, cell positions are colored light green).
Mutations reveal cellular dynamics
VAFs of mosaic mutations reflect the contributions of each lineage to tissues (Fig. 3A; Supplemental Figs. S2, S3). As expected, substantial VAF differences existed between somatic and germ cell samples. The inheritance frequency by offspring was proportional to the VAFs of mutations in germ cells, and the relationship was weak in somatic tissues (Fig. 3B). During development, the endoderm, mesoderm, and ectoderm are thought to originate from different cell populations, but within the cell lineages traced here, there were no apparent differences between each germ layer. Most of the cell lineages reconstructed here supposedly exist before gastrulation. Analyses tracing deeper lineages (e.g., adding mosaic mutations present in endoderm tissues because tail samples would not contain endoderm-derived cells) may reveal such differences between germ layers; for example, the testicular mosaic mutation analysis revealed indepth insights into germ cell lineages (Fig. 2E).
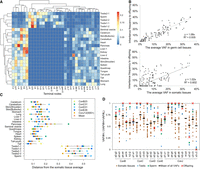
Contributions of each phylogeny to tissues. (A) A heatmap representing the contribution of the terminal nodes in the tree of mouse ConJ12 (Fig. 2E) for each tissue sample. The dendrogram represents the results of the hierarchical clustering. (B) The correlations of the average VAFs in tissues (somatic tissues or germ cell tissues) and the frequency of mutations inherited by the offspring. (C) The distance of the contribution of the terminal nodes between the values of each tissue and the average of all somatic tissues. (D) The contribution ratios of the two daughter cells after a single-cell division. VAF#H means the daughter cell had a higher average VAF relative to that of all samples, and VAF#L denotes the opposite condition. For comparison, the ratios of inheritance frequency of the mutations for the offspring are also shown.
Several tissues (e.g., from the liver and pancreas) had a greater distance in the contributions of the individual terminal nodes from the somatic tissue average (which reflects the expected average composition ratio in epiblast) than tail and heart tissues (Fig. 3C; Supplemental Fig. S2). In the pancreas, some terminal nodes often showed a specifically high contribution (Fig. 3A; Supplemental Fig. S3), indicating frequent clonal expansions. Thus, a developmental bottleneck or intercellular competition might function more strongly in these tissues than in the others.
We then investigated 24 critical cell divisions resulting in two distinguishable daughter cells. Consistent with previous reports (Ju et al. 2017; Bizzotto et al. 2021; Fasching et al. 2021), their contribution ratios to descendant cell populations were not equal but varied greatly (Fig. 3D). The overall contribution ratio based on the average VAFs in all tissue samples indicated asymmetric cell division, at a ratio of 65:35. When calculated based on the contribution to each tissue, the ratio was 68:32 on average in somatic tissues. In the testes and spermatozoa, the contribution showed more bias (on average 83:17). The frequency of the transmission to the F1 mice was also asymmetric. Of 22 divisions with observed mutations in offspring, only one daughter cell in 11 divisions was inherited by the F1 mice.
Our reconstructed cell lineage trees only showed lineages that contributed to adult tissues, omitting cells that became extraembryonic tissues or underwent apoptosis (Hashimoto and Sasaki 2019). The number of embryonic cells contributing to adult tissues is known to be limited (Morris et al. 2010); as such, there were only 7.9 cells at approximately the 100-cell stage. The bottleneck resulting from eliminated cells might affect the asymmetric contribution to adult tissues. A simple stochastic model predicted an asymmetric (64:36) contribution from the two daughter cells in one-cell division (Supplemental Fig. S4). In one- to two-cell divisions after fertilization, one daughter cell and its descendant were suggested to have intrinsically different potentials to contribute to adult tissues (White et al. 2016; Wang et al. 2018). Therefore, both stochastic and intrinsic factors could be involved in asymmetric cell divisions.
Primordial germ cell (PGC) specification has a significant role in germ cell development (Tang et al. 2016; Nguyen et al. 2019), which would explain the biased asymmetry in germ cells (Fig. 3D). Ohinata et al. (2005) detected approximately 16 PGCs on day 6.5 of embryonic development (immediately before gastrulation), which contains approximately 500 epiblast cells (Supplemental Fig. S5), but it remained unclear how many epiblast cells were initially selected as founder cells. The ConJ12 tree of 900× WGS (Fig. 2E) showed that the germ cell lineages appeared independently five or more times. The 100× trees with fewer lineages (Fig. 2A–D) showed that at least five (ConB), two (ConC), four (ConD), and three (ConE) independent lineages contributed to the offspring, indicating that the number of founder PGCs is small, but heterogeneity is maintained through selection from the heterogeneous early lineage population. However, contributions of the five germ cell positions (a-8, h-6, o-6, u-3, x-3) to the ConJ12 tree varied from 4.2% to 42.2%, suggesting that different founder cells might be selected for each lineage. High variance was also seen at the local testis tissue particle level (Fig. 3A; Supplemental Fig. S3). In contrast, the difference between left and right testes was unremarkable, suggesting that intercellular competition would function at least within local testis regions.
The ConJ12 tree indicated that the epiblasts immediately before PGC specification did not strictly commit to a specific germ layer (Supplemental Fig. S6). For example, considering the cell position immediately before the germ cell–specific position, cell position o-5 (mutation #75) existed only in the small intestine. However, positions a-7 and h-6 (mutations #67 and #83, respectively) were found in various tissues such as the cerebrum, seminal vesicle, and tongue.
Mutation rates and characteristics
We estimated the mutation rates from the mutation accumulation (MA) in the lineage trees. From the five trees of 100× WGS data (Supplemental Table S1), we estimated 2.2 mutations per branch, resembling previous estimates for humans (Ju et al. 2017; Rodin et al. 2021). An in vitro observation (Morris et al. 2010) of mouse embryonic lineages indicated that one branch in our reconstructed trees should correspond to 2.2 actual cell divisions (for details, see Methods), suggesting that the per-mitosis mutation rate was 1.0. Furthermore, we found that 10–15 mutations (mean 12.2) accumulated in each of the most upstream germ cell–specific positions of the ConJ12 tree (Fig. 2E). Considering that 12 cell divisions occur from zygote to PGC specification (Drost and Lee 1995), the mutation rate of 1.0 mutation per mitosis is suggested to be consistent up to the PGC specification (Fig. 4A).
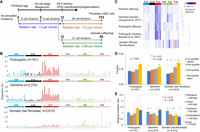
Mutation rates and characteristics of the postzygotic mutations. (A) Schematic of the number of spontaneous mutations along with the developmental period. (B) The trinucleotide context of postzygotic mutations (validated mosaic mutations detected during deep-coverage WGS on five mice), germline mutations (de novo mutations accumulated in long-term breeding mouse lines) (see Supplemental Fig. S8), and somatic mutations (de novo mutations accumulated in ntES cell lines derived from mouse tail fibroblasts) (see Supplemental Fig. S7). (C) Hierarchical clustering of the mutation signatures. Human germline and postzygotic mutations were obtained by Jónsson et al. (2017) and Bizzotto et al. (2021), respectively. (D) Comparison of replication timing at the genome position where the mutation occurred. Significant differences were calculated using a chi-square test. Error bars indicate binomial 95% CIs. (E) Mutation occurrence frequency in genomic regions. Each nucleotide in the effective whole-genome coverage (EWC) regions was annotated with SnpEff software. “Upstream” and “Downstream” indicate 5000-bp regions of each gene. Significant differences were calculated using a chi-square test. Error bars indicate 95% CIs for the Poisson means.
To characterize postzygotic mutations, we compared them to somatic and germline mutations. For somatic mutations, we conducted WGS on four ntES cell lines derived from tail fibroblast clones, finding 517–886 mutations per cell line (Supplemental Fig. S7). The mutation rate was estimated as 13.2 mutations per mitosis. For germline mutations, we investigated long-term breeding mouse lines (Uchimura et al. 2015) and identified approximately 430 mutations that accumulated in 30 generations of inbreeding (Supplemental Fig. S8). We estimated 26.3 mutations per generation and 0.68 mutations per mitosis, after the PGC specification. This rate was confirmed by the WGS results of four offspring born to mouse ConJ12 (Supplemental Fig. S7). The pregastrulation mutation burden might correspond to ∼1.6% of accumulated mutations in a single fibroblast cell and ∼46% of the per-generation germline mutations (Fig. 4A).
The signature (Alexandrov et al. 2020) of mouse postzygotic mutations (validated mosaic mutations detected using deep-coverage WGS on five mice) resembled human postzygotic (Bizzotto et al. 2021) and mouse germline (de novo germline mutations accumulated in the long-term mouse inbreeding) mutations, whereas it differed from mouse fibroblast mutations identified in the ntES cell lines (Fig. 4B,C). C > T transitions (46%) were predominantly found in postzygotic mosaic mutations. Particularly, transitions at CpG sites, possibly derived from the spontaneous deamination of 5-methylcytosine, were observed (Supplemental Fig. S9). All mutations tended to occur more in the late replication region (Fig. 4D) and were observed in all regions (exons, introns, and intergenic regions) (Fig. 4E). A significant difference in postzygotic mutation occurrence was not observed among regions (P = 0.60, chi-square test).
Finally, we found a peculiar pair of postzygotic mutations, namely, mutations #33A and #33C, at the first cleavage in the ConC31 mouse (Fig. 2B). The “T-to-A” substitution was inherited by one daughter cell, and the “T-to-C” substitution was inherited by another at the same position. As the parents of ConC31 did not carry either mutation, these mutations may have occurred simultaneously during the first cleavage.
Discussion
We report that reliable cell lineages can be reconstructed according to the arithmetic relationships between VAF values obtained from multiple tissue samples. This method allows for tracing cell lineages with a single-cell division resolution if one or more mutations occur per mitosis. In early embryos, the mutation rate is estimated to be 1.0 event per mitosis on average, and bottlenecks in the cell population owing to epiblast formation increase the number of mutations per branch, which makes it easy to reconstruct the detailed lineages. This method is applicable to multiple cell types if an average of one or more mutations occurs per mitosis in them.
Our method directly uses the mosaic mutations detected in bulk tissue samples and is therefore easier to conduct than lineage reconstruction conducting WGS for many single-cell clones. However, the developed method has some technical requirements. First, it requires efficiently detected true mosaic mutations from high-coverage WGS data. Second, it is necessary to obtain accurate VAF values to effectively reconstruct reliable lineages; therefore, mutations for which accurately measuring VAFs was difficult, such as indels occurring in the tandem repeat regions, were excluded in this study. Third, multiple VAF values measured in several tissue samples are required. The lineages were reconstructed based on the VAF values obtained from 18 different tissues; however, comparable lineages can be reconstructed using fewer tissue samples. For example, in the ConJ12 tree, germ cell–specific lineages were substantially reconstructed using only testis and sperm samples. This lineage reconstruction method can be applied directly to living individuals using only noninvasive tissue samples. Once the lineages are reconstructed, the cell populations derived from the early lineages can be tracked over the entire lifespan of an individual. Naturally occurring mutations are the ultimate DNA barcodes for high-resolution cell lineage tracking, even when compared with CRISPR-Cas9 and other genetically engineered DNA barcodes (Baron and van Oudenaarden 2019; Chan et al. 2019). Phylogenetic tracking methodologies based on mosaic mutations and their VAF values can bring new benefits to a wide range of life sciences.
The accumulated mutations may indicate the number of experienced cell divisions. There was some variation between counts of new mutations at each cell position of the reconstructed lineage trees. Considering a single-cell division, more mutations occurred in one daughter cell that on average contributed less to the somatic tissues (P = 0.015, two-tailed paired t-test). This suggests that daughter cells with many mutations undergo more cell divisions, producing daughter cells that do not contribute to adult tissues. In addition, this assumption is supported by the result suggesting that the number of accumulated mutations at each of the most upstream germ cell–specific positions is similar, although the number of phylogenetic branches from the zygote to those positions varied (four to eight branches). Assuming a constant mutation rate, it may be possible to estimate the number of actual cell divisions corresponding to the reconstructed lineage tree.
Whether genetic heterogeneity owing to postzygotic mutations would affect the cell fate for PGC specification has long been debated (Nguyen et al. 2019). Our reconstructed trees showed no apparent difference in the number of accumulated mutations between somatic and germ cell lineages. However, assuming that mutations with a genomic evolutionary rate profiling (GERP) score (Cooper et al. 2005) of more than three are deleterious (deleterious mutations are shown with hash marks in Fig. 2), the ratios of harmful mutations differ as follows: 0.7% (1/137) of mutations that occurred in lineages contributing to offspring were harmful, whereas 6.0% (3/50) of the mutations were harmful in the side branches and did not contribute to the offspring. Although the difference was not significant (P = 0.059, Fisher's exact test), some deleterious mutations may be eliminated from germline lineages by PGC selection. A recent report suggested that cells with chromosomal aberrations might be eliminated from extraembryonic tissues (Coorens et al. 2021). Therefore, a more detailed analysis of lineage selection with genetic heterogeneity is required.
Germline mutation rate is an important parameter for organism evolution (Kimura 1983; Lynch 2010; Moorjani et al. 2016). Consistent with the study by Lindsay et al. (2019), our results clearly showed that the mutation rate of pre-PGC-stage cell division is higher than that of subsequent germ cell divisions and that postzygotic mutations make up a significant portion of de novo germline mutations. Multiple early lineage contributions to germ cells effectively reduce de novo mutations shared between siblings (Jónsson et al. 2018; Lindsay et al. 2019; Sasani et al. 2019) and may promote intercellular competition for genetic heterogeneity. Such developmental processes should function as an evolutionary strategy to increase the genetic variations in offspring while reducing the effects of harmful mutations. Elucidating the mutagenic mechanisms associated with cell lineages is important for a better understanding of the principles that create biodiversity.
Methods
Animals
All animal experiments were approved by the Animal Committee of Osaka University (Osaka, Japan; approval no. FBS-17-006) and by the Experimental Animal Care Committee of the Radiation Effects Research Foundation (RERF; Hiroshima, Japan; approval no. 2018-02). They were performed in accordance with the 1996 National Institutes of Health guidelines and with the Guidelines for Animal Experiments of the RERF. For our cell lineage analysis, we used five wild-type C57BL/6J mice (Supplemental Table S2), which were maintained as separate inbred lines (Supplemental Fig. S8). The mice were maintained at a constant temperature (22 ± 1°C) in a 12-h light/dark cycle, with ad libitum food and water access. The housing environment and diet were kept constant throughout the experiment.
WGS analysis
Originally, WGS was performed to analyze mutations accumulated in the MA lines (Supplemental Fig. S8), and many mosaic mutations were detected; thus, we applied this method. The five mice used to analyze the mosaic mutations were determined based on sample storage conditions (frozen sperm, etc.). The summary of WGS analysis is shown in Supplemental Table S3. Genomic DNA was extracted from the tail and testis using the DNeasy blood and tissue kit (Qiagen) or smart DNA prep (m) kit (Analytik Jena). The extracted DNA solutions were used to prepare paired-end libraries according to the user manual of the Illumina sample preparation kit. WGS was conducted using HiSeq or NovaSeq (Illumina).
Mapping and variant calling
First, we used BWA-MEM v0.7.12 (Li and Durbin 2009) with the “–M” option for Picard compatibility to map sequence reads to the mouse reference genome (UCSC mm10). Next, we performed deduplication and base-quality recalibration using Picard v1.110 and GATK v3.6 (McKenna et al. 2010), respectively. Finally, highly reliable (HR) reads that met the following conditions were extracted: (1) properly mapped according to the aligner, (2) mapped with a mapping quality of 60 or more, and (3) mapped to the reference without clipping. Only HR reads were used in variant calling.
De novo mutations can be identified accurately by limiting the analyzed regions (Uchimura et al. 2015; Satoh et al. 2020). In this study, the mutation call was basically limited to the effective whole-genome coverage (EWC) regions of each analysis. In the analyses of postzygotic mutations and variants in offspring, we called additional candidate variants outside the EWC regions by comparing VAFs and read depths among samples.
To detect low-frequency variants and accumulated mutations in MA lines, the following were used to define the EWC regions: (1) a lower-bound coverage of 50% and upper-bound coverage of 3× peak coverage of the HR reads; (2) bases covered by both forward- and reverse-strand HR reads; (3) a minimum 80% ratio of HR reads to all mapped reads at each site; and (4) a minimum base quality of 20. The EWC region was defined for each sample, and the common region was used for analysis. In this study, we used the EWC regions as follows: 1,961,028,804 bp in autosomes and 111,278,170 bp in Chromosome X for mice ConA to ConH (Supplemental Fig. S8), as well as 1,971,853,032 bp in autosomes and 116,426,256 bp in Chromosome X for the ConJ12 and ConJ12b mice. To detect the accumulated mutations, we used GATK HaplotypeCaller with the “‐‐min_base_quality_score 20” option for HR reads as previously described (Satoh et al. 2020). Mutations were called for >33% VAFs for heterozygous alleles and >80% for homozygous alleles. De novo mutations were identified by filtering with the following inclusion criteria: a VAF of <10% in both the ancestor male and the ancestor female of the MA lines. To detect low-frequency variants, we used GATK MuTect2, which calls somatic variants. The “‐‐min_base_quality_ score 20” option was specified to avoid false-positive detection. For each sample, variants were called between the ancestor male and the ancestor female, respectively. We extracted the variants called in both cases that were supported by at least four reads as candidate de novo mutations in 100× WGS data. For analysis using 900× WGS data, we also used GATK MuTect2 with the same setting and called candidate de novo variants by stringent filtrations, such as (1) VAFs > 2% and close-to-zero VAFs in control WGS data (the ancestor male and female samples) (Supplemental Fig. S8), (2) an upper-bound coverage of 3× peak coverage of the HR reads, (3) a forward- and reverse-strand bias (P > 0.0001), (4) >35 bp in the median of variant locations on a single read, and (5) removing cluster mutations (when more than one alteration occurred within 100 bp).
To detect variants in “ntES cell lines” and “offspring,” we used GATK HaplotypeCaller with the “‐‐min_base_quality_score 20” option for HR reads. Mutations were called with a VAF > 33% (for Chromosome X in “ntES cells,” with VAF > 80%) in each sample. De novo mutations were identified by filtering according to several criteria, shown in Supplemental Data 2. As the EWC regions (here, defined except for the condition 2 previously mentioned herein), 1,964,032,971 bp for autosomes and 108,214,682 bp for Chromosome X were used for “ntES” analysis, and 1,888,954,114 bp in autosomes was used for “offspring” analysis.
Multisite mutations involving more than one alteration (single-base substitution or indel) that occurred within 100 bp in the same allele were counted separately as distinct mutations from single-base substitutions and indels. All multisite mutations were manually confirmed using the Integrative Genomics Viewer (IGV) (Robinson et al. 2011). For analysis except for the detection of mosaic mutations, we excluded indels that occurred in homopolymers (>3 bp) and di-, tri-, tetra-, penta-, or hexa-nucleotide repeats from this analysis.
Selection of candidate mosaic mutations from called variants
We picked candidate mosaic mutations with VAFs > 3.5% in 100× coverage WGS data (or VAFs > 2.0% in each 450× coverage WGS data set) and excluded the mutations for which the maximum VAF of the negative control samples was more than half the VAF of the target sample. Mutations, especially insertions and deletions, occurring on the repeat sequence were excluded from the analysis because of the difficulty in measuring VAFs with amplicon sequencing. All candidate mutations in the EWC region were subjected to amplicon sequencing validation experiments. For areas outside the EWC regions, we manually inspected all candidates using JBrowse (Buels et al. 2016) before performing amplicon sequencing. The breakdown of the validation results for these candidate mosaic mutations is shown in Supplemental Table S4.
Measuring VAFs of mosaic mutations
We measured the VAFs of mosaic mutations using amplicon sequencing. The amount of each tissue particle used for DNA extraction is shown in Supplemental Table S5. We designed PCR primers using the Primer3 (Untergasser et al. 2012)-based software Qprimer v1.6.0 (Amelieff) for each candidate and attached forward and reverse overhang adaptor sequences to the 5′ termini of each primer pair, in accordance with Illumina's instructions. All primers were purchased from Thermo Fisher Scientific. These primers were used for multiplex PCR of several PCR sites using KAPA fast multiplex mix (Kapa Biosystems). Because the multiplex PCR did not work well for several PCR sites, such PCRs were performed individually. The detailed PCR conditions are shown in Supplemental Data 3. To measure the VAF accurately, we conducted at least three initial PCRs per sample. PCR products were cleaned up using AMPure XP beads (Beckman Coulter). The second round of PCR was performed using a Nextera XT index kit v2 set A and D (Illumina) and KAPA HiFi HotStart ReadyMix (Kapa Biosystems). PCR products were pooled and sequenced on Illumina MiSeq or HiSeq platforms (around 10,000× coverage, ranging 3000–1,000,000× [for measuring accurate VAFs in tissues] and approximately 1000× coverage [for detecting heterozygous variants in offspring and ntES cell lines]). Reads were mapped to a mouse reference genome (UCSC mm10) using BWA. To distinguish true mosaic mutations and artifact mutations, we checked the variants using a quasi-dilution series made from mixtures with the DNA solutions of negative controls (samples of unrelated individuals). Each VAF was used for analysis after subtracting the VAF value of negative control samples. For data replication using the same DNA sample, the average measured VAF value was used as the final result (when unreliable data [read count below the standard] were included, such data were excluded). All lineage analyses were performed on male mice; therefore, for mutations on Chromosome X, the measured value divided by two was used as the VAF of the mutation. All VAF values are shown in Supplemental Data 1. Because the tissue particles used for DNA extraction were not very small (Supplemental Table S5), the variation in the VAF was not likely owing to clonal growth that occurred in a local area.
Reconstruction of mutation occurrence history
First, we clustered the mosaic mutations, which showed essentially the same VAF values across all tissues. We calculated the mean VAF values for the mutations in the cluster for each tissue and used the averaged VAF values thereafter. For simplicity, we described the mutation cluster and the averaged VAF value as Mos#i and VAF#i, respectively. Second, we reconstructed partial lineage relationships based on the contribution ratio rules such as “R1” (Fig. 1A). We evaluated the difference between VAF#i and (VAF#j + VAF#k) for all i, j, and k combinations and extracted relationships between one parent and two daughter cells for which differences were under the threshold value. Setting an appropriate threshold value was necessary in this procedure because we estimated false-positive relationships with a large threshold and overlooked genuine relationships with a small threshold. If the sum of all relationships contains false-positive relationships, the relationships must be inconsistent when a mutation has more than three daughter cells or more than two parent cells. Therefore, we set a large threshold value initially and gradually decreased the value until the entire lineage tree contained no inconsistent relationships. Next, we added lineage relationships for the current reconstructed tree based on the contribution ratio rules such as “R2” (Fig. 1A). We investigated whether we could add an unassigned mutation Mos#i to the current tree. We compared VAF#i and VAF#j for all j mutations that corresponded to the terminal nodes of the current tree. If the magnitude of the relationship VAF#i < VAF#j was satisfied for only one terminal node j, we assigned Mos#i to one daughter cell of Mos#j. We also attached a virtual node corresponding to another daughter cell of Mos#j and defined the VAF values with (VAF#j − VAF#i). We ran this procedure for all unassigned mutations in decreasing order of averaged VAF values of the unassigned mutations. As a result, we reconstructed the entire tree computationally and made manual fine adjustments (we checked if the sum of the daughter cells matched that of the parent cells and if there were appropriate cell locations for the unassigned mutations) for the final cell lineage tree. The trees (in Fig. 2; Supplemental Fig. S1C) are displayed so that cell positions were horizontally placed when reaching six to seven branches, which corresponds approximately to the 64-cell-stage embryo. When there were multiple mutations in a node, the average VAF value was used as the representative value for that node (if there were mutations with unreliable VAF measurements [Supplemental Data 1], the former were excluded from average calculation).
We evaluated the reconstructed tree using the offspring's genotype data. We developed a probabilistic model that generated a genotype from the tree and confirmed the validity of the tree. This probabilistic model could estimate the lineage origin of each offspring. Therefore, we could also assign unassigned mutations if the source was uniquely specified. The detailed explanation for the algorithm and probabilistic model is described in Supplemental Materials.
Nuclear transfer and establishment of ntES cell lines
Nuclear transfer was performed as described previously (Wakayama et al. 1998). Tail tips derived from mouse ConJ12 (fresh sample) or ConB23 (frozen sample with cell cryopreservation medium “CELLBANKER 1 plus”; Zenoaq) were cultured on a Petri dish for 12–14 d. The cultured tail-tip fibroblasts were used as nuclear donors. After nuclear transfer, the reconstructed oocytes were activated using 5 mM SrCl2 in Ca-free CZB medium in the presence of 5 μM latrunculin A supplemented with trichostatin A (50 nM) for 9 h. After three washes in CZB, cloned embryos were cultured for 4 d in the same medium, and morula- or blastocyst-stage embryos were used to establish ntES cell lines as described previously (Wakayama et al. 2001; Wakayama and Wakayama 2010).
Estimation of mutation rates
To evaluate the mutation rate during postzygotic cell division, we first estimated the mutation rate per branch in the reconstructed lineage trees. Here, we focused on mosaic mutations with VAFs of ≥6% in tail samples that were subjected to 100× WGS, because such mutations were expected to have a low missing variant rate. To estimate the rate, it is necessary to consider the branches that do not have unique mutations. In cases of trichotomy (three-way split), we added one tentative daughter cell position with no unique de novo mutations for calculation. This means that the number of each branch of trichotomy was regarded as 1.33. For most downstream cell positions, depending on the VAF values at that position and VAF values at the observation limit (6% for 100× data), an expected number of virtual cell positions with no unique de novo mutations were added. Supplemental Table S1 shows the estimates for each phylogenetic tree. The mutation rate μ was calculated as μ = m/n, where m is the total number of mutations, and n is the number of branches. The frequency of mutations missed in our mutation-detection pipeline was evaluated by comparing the 100× and 900× results of the ConJ12 mouse. After the correction according to this frequency, an estimate of the mutation rate was obtained.
To evaluate the per-mitosis mutation rate, we used the relationship between the number of cell lineage branches and the number of actual cell divisions in mouse embryos, as suggested by a previous paper (Morris et al. 2010). The data showed that the number of cells leading to adult tissues (precisely, epiblast cells in approximately 100-cell-stage embryos) was limited as follows: 3.6 cells became epiblast cells in the 16-cell stage; 4.9 cells, in the 32-cell stage; and 6.7 cells, in the 64-cell stage. For example, each cell in 16-cell-stage embryos had actually experienced four cell divisions; however, in our reconstructed lineage trees, each corresponding cell position would be expected to have only 1.8 branches (3.6 = 21.8) from fertilized eggs. Therefore, we considered that each branch of the tree corresponded to 2.2 (=4/1.8) actual cell divisions. In this manner (3.6 = 21.8, for 16[ = 24]-cell stage), the relationships of 4.9 = 22.3 (for 32[ = 25]-cell stage) and 6.7 = 22.7 (for 64[ = 26]-cell stage) provided the consistent estimate that 2.2 actual cell divisions would correspond to one branch of the lineage tree on average. Using this value, we estimated the per-cell division mutation rate during this period.
To estimate the downstream mutation rate, we investigated the tree of mouse ConJ12 with 900× WGS data and counted accumulated mutations at the cell positions that approximately corresponded to the PGC specification stage. At six cell positions (Fig. 2E, a-8, h-7, k-7, o-6, x-4, and β-4), where VAFs became essentially zero except for germ cell tissues, an average of 12.3 mutations were accumulated. Because it is known to take 12 cell divisions to go from a fertilized egg to PGC induction (corresponding to embryonic day 6.5) (Drost and Lee 1995), the rate was estimated to be approximately one mutation per mitosis during that period.
To estimate the mutation rate in fibroblast cells, the number of cell divisions was estimated as previously described (Milholland et al. 2017). In our experiment, the tail specimen was collected when mouse ConJ12 was 11 mo old. We assumed that the weight of a 3-mo-old mouse is 30 g (corresponding to 39 cell divisions) and that two cell divisions occurred per month (referring to a previous study for mouse keratinocytes) (Busch et al. 2007) for 8 mo (3–11 mo old). In the primary culture period of fibroblasts (13 d), 13 cell divisions would have taken place. We used 68 cell divisions (=39 + 16 + 13) to estimate the per-cell division mutation rate. For germ cell divisions, we calculated the rate using 33 total cell divisions (an average of 41 times for males and 25 times for females) per generation in our experiment, as previously described (Drost and Lee 1995).
Mutation signatures and variant annotation
Mutational signature analysis and visualization of single-base substitutions that occurred in the EWC regions were performed using SigProfilerMatrixGenerator v1.2.2 (Bergstrom et al. 2019) and SigProfilerExtractor v1.1.4 using “cosmic_version = 3.2” specifying “reference_genome = “mm10”,” “opportunity_genome = “mm10”,” and “nmf_replicates = 100” (Alexandrov et al. 2020). Mutational signatures of each sample were classified by hierarchical clustering with the Ward's minimum variance method (Ward.D2) using the heatmap.2 program of the “gplots” package of R (R Core Team 2019). The replication timing profile of mutations was calculated using Repli-seq data measured in mouse ES cells (Rivera-Mulia et al. 2018). The Repli-seq data obtained from early and late S phase fractions, consisting of 50-kb bins found throughout the EWC regions of the mouse genome, were divided into four quartiles from early-to-late replicating regions. Fractions of mutation sites in each quartile were counted. Significance was assessed with a chi-square test using GraphPad Prism software version 7.0 (GraphPad Software). Each nucleotide in the EWC regions was annotated with SnpEff software (Cingolani et al. 2012) using GRCm38.86 information from the Ensembl reference genome to analyze the rate in genomic regions. Mutations were also annotated based on GERP scores (Cooper et al. 2005) using the mouse base-wise GERP score derived from mouse mm9 reference data and mammalian alignments available at UCSC in late 2010 (see the webpage of Dr. Sidow's laboratory: http://mendel.stanford.edu/SidowLab/downloads/gerp/).
Analysis of the contributions of each lineage
The contribution of a node is defined as double the VAF. We used averaged VAFs when the node had multiple mutations and used expected VAFs calculated with VAFs of the parent and counterpart nodes when the node had no mutation. When there were multiple co-occurring mutations that were not sufficiently accurate to measure the VAF, the average of the remaining mutations was used, excluding the inaccurate mutations. For the analysis of asymmetric cell divisions, the measured values with a VAF ≤ 2% in both daughter cells were excluded from the analysis owing to the uncertainty caused by VAF measurement errors.
Simulation analysis for asymmetric cell division
We evaluated the asymmetric cell division caused by the bottleneck effect at the 128-cell stage, from which n cells are randomly selected and contributed to the epiblast cells (n = 7–10). The probability that x and y cells in the lineages of two daughter cells of a kth-stage parent cell (k = 1–2) are selected for the n cells (epiblast cells) is defined as follows: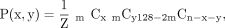 where m is 27−k and Z is the normalizing constant. Because we cannot observe the cell lineage when x = 0 or y = 0, we defined the domain of x and y as x ≥ 1 and y ≥ 1, in addition to x + y ≤ n. Based on the above probability, we evaluated the asymmetry with max (x/(x + y), y/(x + y)), and the distribution and expected values are shown in Supplemental Figure S4.
where m is 27−k and Z is the normalizing constant. Because we cannot observe the cell lineage when x = 0 or y = 0, we defined the domain of x and y as x ≥ 1 and y ≥ 1, in addition to x + y ≤ n. Based on the above probability, we evaluated the asymmetry with max (x/(x + y), y/(x + y)), and the distribution and expected values are shown in Supplemental Figure S4.
Data access
A list of the mosaic mutations and their VAFs in tissue samples, the offspring, and the ntES cell lines can be found in Supplemental Data 1. Information about de novo mutations identified in WGS analysis using ntES cell lines, the offspring, and trans-generational MA lines can be found in Supplemental Data 2. DNA sequencing data generated in this study have been submitted to the DDBJ Sequence Read Archive (DRA; https://www.ddbj.nig.ac.jp/dra/index-e.html) under the accession number DRA012698. The detailed procedure and algorithm for lineage reconstruction and lineage origin inference of offspring are described in the Supplemental Text, and all scripts and VAF data for lineage reconstruction can be found as Supplemental Code.
Competing interest statement
The authors declare the following competing interests: Osaka University and the Radiation Effects Research Foundation (inventors: A.U., H.M., Y.S., and T.Y.) have filed patent applications related to the work described here. The title of the patent application is “Method and program of construction of cell lineage.” The Japanese patent was filed on July 30, 2019, 2019-139833. The remaining authors declare no competing financial interests.
Acknowledgments
We thank T. Tsuji, M. Imanaka, M. Toshishige, S. Matsu-ura, and A. Miura for technical assistance. The computations were partially performed using the NIG supercomputer at the ROIS National Institute of Genetics. Part of the experiment was performed at the Natural Science Center for Basic Research and Development, Hiroshima University. The Radiation Effects Research Foundation (Hiroshima and Nagasaki, Japan) is a public-interest foundation funded by the Japanese Ministry of Health, Labor and Welfare and the U.S. Department of Energy. The views of the authors do not necessarily reflect those of the two governments. This research was supported by JSPS KAKENHI (Ministry of Education, Culture, Sports, Science and Technology, grant nos. 15K12205, 16H05890, 18K19340 to A.U.; 15K00553 to Y.S.; 16H06279 to A.T.; and 17H00789 to R.F. and Y.G.) and partly by RERF Research Protocols, RP 2-13.
Author contributions: A.U. designed the research and wrote the manuscript. A.U. and Y.S. performed the experiment and analyzed the data. H.M. contributed to the theoretical analysis and analyzed the data. S.W. and T.W. performed the nuclear transfer experiment. T.Y. and M. Higuchi contributed to the establishment of mutation accumulation mouse lines. Y.M. and A.T. were involved in the whole-genome sequencing and detection of genetic variants. R.F. and Y.G. performed some of the amplicon sequencing analysis. M. Hashimoto performed some of the experiments and provided useful comments with respect to embryogenesis. T.Y. and Y.G. supervised the project.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276363.121.
-
Freely available online through the Genome Research Open Access option.
- Received November 8, 2021.
- Accepted April 1, 2022.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








