
Genome biology of the darkedged splitfin, Girardinichthys multiradiatus, and the evolution of sex chromosomes and placentation
- Kang Du1,
- Martin Pippel2,
- Susanne Kneitz3,
- Romain Feron4,5,
- Irene da Cruz6,
- Sylke Winkler2,
- Brigitta Wilde3,
- Edgar G. Avila Luna7,
- Gene Myers2,8,
- Yann Guiguen5,8,
- Constantino Macias Garcia7,8 and
- Manfred Schartl1,6
- 1The Xiphophorus Genetic Stock Center, Department of Chemistry and Biochemistry, Texas State University, San Marcos, Texas 78666, USA;
- 2Max-Planck Institute of Molecular Cell Biology and Genetics, 10307 Dresden, Germany;
- 3Biochemistry and Cell Biology, Biocenter, University of Wuerzburg, 97074 Wuerzburg, Germany;
- 4Department of Ecology and Evolution, University of Lausanne, and Swiss Institute of Bioinformatics, 1015 Lausanne, Switzerland;
- 5INRAE, LPGP, 35000 Rennes, France;
- 6Developmental Biochemistry, Biocenter, University of Wuerzburg, 97074 Wuerzburg, Germany;
- 7Instituto de Ecologia, Universidad Nacional Autónoma de México, C.P. 04510, Mexico City D.F., Mexico
-
↵8 These authors are co-senior authors and contributed equally to this work.
Abstract
Viviparity evolved independently about 150 times in vertebrates and more than 20 times in fish. Several lineages added to the protection of the embryo inside the body of the mother, the provisioning of nutrients, and physiological exchange. This often led to the evolution of a placenta. Among fish, one of the most complex systems serving the function of the placenta is the embryonal trophotaenia/ovarian luminal epithelium of the goodeid fishes. For a better understanding of this feature and others of this group of fishes, high-quality genomic resources are essential. We have sequenced the genome of the darkedged splitfin, Girardinichthys multiradiatus. The assembly is chromosome level and includes the X and Y Chromosomes. A large male-specific region on the Y was identified covering 80% of Chromosome 20, allowing some first inferences on the recent origin and a candidate male sex determining gene. Genome-wide transcriptomics uncovered sex-specific differences in brain gene expression with an enrichment for neurosteroidogenesis and testis genes in males. The expression signatures of the splitfin embryonal and maternal placenta showed overlap with homologous tissues including human placenta, the ovarian follicle epithelium of matrotrophic poeciliid fish species and the brood pouch epithelium of the seahorse. Our comparative analyses on the evolution of embryonal and maternal placenta indicate that the evolutionary novelty of maternal provisioning development repeatedly made use of genes that already had the same function in other tissues. In this way, preexisting modules are assembled and repurposed to provide the molecular changes for this novel trait.
Viviparity, a mode of reproduction that can exacerbate the conflict between sexes over the allocation of resources to offspring, has evolved independently among all classes of jawed vertebrates (except birds) about 150 times, 21 of which occurred in fish (for review, see Blackburn 2015). Viviparous species can be arranged along a continuum from lecithotrophic to matrotrophic based on whether the nutrients required for embryonic development are present in the egg (lecithotrophic development) or whether they are gradually provided by the mother through gestation (matrotrophic development), for instance, via a placenta (placentotrophy) (Blackburn 1999; Ghalambor et al. 2004; Pollux et al. 2009). Despite the large number of independent origins, our understanding of the evolutionary and functional development of matrotrophic viviparity, especially of that involving a placenta, comes mostly from the study of mammalian matrotrophy, and although vast, our knowledge of its genomic underpinning is limited to only one transition from oviparity to matrotrophic viviparity and a few recent studies on fish (see below) (Lynch et al. 2008; Kin et al. 2016). There is hence a need to incorporate more taxa to the study of the evolution of viviparity, ideally taxa that include both oviparous and viviparous (matrotrophic) species. Fish in the Order Cyprinodontiformes seem appropriate to conduct such comparisons, because different forms of viviparity have evolved independently in several families: Poeciliidae, Jenynsidae, Anablepidae, and Goodeidae (Meyer and Lydeard 1993; Blackburn 2015). Accordingly, comparative analyses revealed that placentation drives a shift from a reliance on pre-zygotic mate choice to increasing levels of polyandry, in conjunction with post-zygotic mechanisms of mate selection (Pollux et al. 2014; Furness and Capellini 2019). More recently, comparative transcriptomic analyses have found parallels between poecilid and mammalian placental viviparity (Guernsey et al. 2020). By focusing on the speciose Poeciliidae, this study explored the consequences of several transitions between lecithotrophy and matrotrophy, although still only covering one transition between oviparity and viviparity. It is likely a result of the lack of genomic resources that the family Goodeidae has not been used to investigate the genomic background of the transition from oviparity to placental viviparity. The Goodeidae is a small, yet diverse family of Cyprinodontiformes native to North America (Parenti 1981). The subfamily Empetrichthynae, from Nevada in the United States, includes two genera of oviparous fish, and the subfamily Goodeinae from Central Mexico includes ∼40 viviparous species distributed in 16 genera (Webb et al. 2004). This high ratio of genera to species is indicative of a burst of diversification (Mayr 1942), which has been identified as an adaptive radiation following the evolution of viviparity (Ritchie et al. 2005). There are at present no genomic resources that may allow the use of the Goodeidae to explore the genomic transitions that accompanied the evolution of matrotrophic viviparity from oviparity.
Matrotrophy in goodeids is linked to the possession of a dynamic placenta sensu Mossman (Mossman 1991) made up of a maternal part, the internal ovarian epithelium, and an embryonic component, the trophotaenia (Fig. 1; Lombardi and Wourms 1985; Uribe et al. 2005; Schindler 2015). The latter are extensions of the embryonic hindgut mostly shaped in the form of ribbons or rosettes (Turner 1937; Lombardi and Wourms 1985) made of an absorptive epithelium around a well-vascularized core (Mendoza 1972; Lombardi and Wourms 1985). The maternal part of the placenta is represented by the follicular epithelium of the ovary, which is closely associated with the trophotaenia. This is a dynamic system because early embryos rely on an initial supply of yolk, then develop the trophotaenia that reaches out to the modified ovarian lumen epithelium, but starts to regress, possibly through apoptosis, as gestation draws to an end (Iida et al. 2015). Maternally provided proteins, which can include vitellogenin (Vega-López et al. 2007) and also residues of dead siblings (Greven and Grossherr 1992), are taken up by the trophotaenial epithelium via receptor-mediated endocytosis and subjected to lysosomal degradation (Schindler and De Vries 1987; Schindler and Kujat 1990; Schindler 2003).
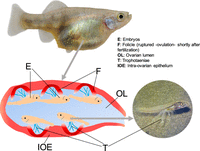
Images of a pregnant female adult darkedged splitfin, one laid embryo with the trophotaeniae, and a schematic drawing of the ovary/placental organ. For detail of the organ structure, see Uribe et al. (2010), Figures 31–33. For comparison of the placental organs in mammals, seahorse, and poeciliid fishes, see Griffith and Wagner (2017), Figure 3.
Unlike other viviparous fish such as poeciliids (Constantz 1984), splitfin females do not store sperm (Bisazza 1997), and they are only receptive for about 1 wk after giving birth, which takes place every 2 mo (Macías-Garcia and Saborío 2004). Additionally, males lack an intromittent organ, thus sperm is deposited in the outer part of the gonopore (Bisazza 1997; Garcia and Valero 2010; Greven and Brenner 2010). These particularities of splitfin reproductive biology may promote that males adjust their sperm production and mating effort to the probability of finding mates and of facing sperm competition (Simmons and Fitzpatrick 2012), enforcing novel conditions for sperm to reach the ova. Thus in addition to the consequences for females, it is likely that viviparity has also left a genomic footprint on males.
Splitfin males are heavily ornamented (Ritchie et al. 2005; Méndez-Janovitz et al. 2019) and, because they cannot force copulations (see previous paragraph), they perform complex courtship displays to secure mating (Macías-Garcia and Saborío 2004). This preponderance of sexual selection may be related to high rates of differentiation, as previously proposed (Darwin 1871; Panhuis et al. 2001; Ritchie 2007).
Mexican goodeids face a variety of environmental challenges (De La Vega-Salazar et al. 2003; Domínguez-Domínguez et al. 2006), including chemical pollution (De La Vega Salazar et al. 1997), which has made splitfins a model for ecotoxicological research (Arellano-Aguilar and Macías Garcia 2008a,b; Rueda-Jasso et al. 2014; Guerrero-Estévez and López-López 2016).
Here, we report the first high-quality reference genome of a goodeid, the darkedged splitfin (also known as Amarillo), Girardinichthys multiradiatus, assembled at full chromosome level including the X and Y Chromosomes. Using this genomic resource, we uncovered a pronounced sex difference in brain transcriptomes and established the expression signature of the trophotaenial placenta showing considerable overlap with human placenta and RNA-seq data from another live-bearing fish and seahorse.
Results
Genome sequencing, chromosome assembly, and annotation
To obtain a genome assembly of high contiguity and completeness, we used the pipelines of the international Vertebrate Genome Project that incorporate state-of-the-art sequencing technologies and assembly algorithms (Rhie et al. 2021). Pacific Biosciences (PacBio) continuous long reads (71-fold coverage), 10x Genomics Illumina read clouds (52-fold coverage), Bionano optical maps (1870-fold coverage), and chromosome conformation capture (Hi-C) Illumina read pairs (94-fold coverage) from three male individuals were generated (Supplemental Tables S1, S2). The PacBio reads were assembled into contigs using the customized assembler DAmar (https://github.com/MartinPippel/DAmar; Jebb et al. 2020). Next, we used the 10x Illumina read-cloud data to correct base errors and phase haplotypes, arbitrarily picking one haplotype in a phased block. Finally, we used Bionano optical maps and then Hi-C data to produce long-range scaffolds. This resulted in a final assembly of 1.1 Gb with a contig N50 of 17 Mb, arranged in 71 scaffolds. Of these, 24 are chromosome-size (Fig. 2) in agreement with the karyotype of G. multiradiatus (Uyeno et al. 1983), making up 99.7% of the genome assembly (Supplemental Table S3).
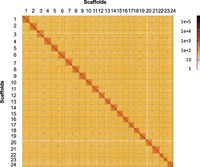
Hi-C contact heat map of the scaffolded portion of the splitfin genome assembly.
A comparison with other teleost chromosome assemblies disclosed a high degree of conserved synteny not only to other Cyprinodontiformes (Fundulus, Xiphophorus, and medaka), but also with the more distant Perciformes and to a lower extent with mostly small translocations and rearrangements to herring, cavefish, and zebrafish (Fig. 3). To annotate protein-coding genes, gene evidence from protein homology of other species, RNA-seq transcriptomes from darkedged splitfin and ab initio predictions were integrated. A total of 23,770 protein-coding genes were annotated. The BUSCO completeness based on the actinopterygii_odb9 data set was improved from 96.4% to 99.3% by the annotation process (Supplemental Table S4). Of the 23,770 genes, 23,463 (98.7%) have a BLAST hit to the Swiss-Prot/RefSeq database. Of the protein-coding genes, 2113 (8.9%) are single exon genes. Additionally, 1900 tRNA, 21 rRNA, 279 miRNA, and 355 other noncoding RNA genes were annotated (Supplemental Table S5).
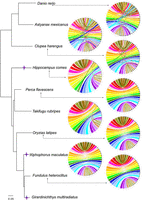
Species tree and chord diagrams showing orthology between Girardinichthys multiradiatus and other teleost species. For each species, chromosome orthology to G. multiradiatus is shown in a chord diagram, where blocks on the circumference represent chromosomes, and colored threads link orthologous genes with conserved synteny. Chromosomes of G. multiradiatus are represented by colored blocks, and those of other species are in black. Bootstrap support values are labeled on each node. Scale bar indicates average substitutions per site. Branches leading to live-bearing species are marked with purple four-pointed stars.
Genome evolution
The genome of G. multiradiatus consists of 37.3% of repetitive sequences according to RepeatMasker (Supplemental Table S6). This is in the range of many other teleost fish genomes (Shao et al. 2018), including in the closely related Fundulus and medaka species but considerably higher than the live-bearing platyfish. Of note, there is a strong expansion of ERV class I elements, which is not found in other genomes of the order Cyprinodontiformes (2.5% vs. 0.01%–0.4%). The transposon landscape history (Supplemental Fig. S1) indicates that darkedged splitfin experienced a strong wave of TE expansion rather recently, in contrast to other teleost genomes analyzed so far.
A phylogenomics reconstruction of the evolutionary relationships of splitfin to other teleosts using a gene set of 1425 high-quality orthologs confirmed previous groupings. The tree revealed a split of the goodeid branch from egg-laying cyprinodonts around 52 million years ago (MYA), about twice the age estimated from partial mitochondrial sequences (Webb et al. 2004). The last common ancestor of goodeids with the live-bearing poeciliids dated back almost to the same period, around 64 MYA (Fig. 3; Supplemental Fig. S2).
Gene family analysis revealed that 42 gene families expanded and 128 contracted in the splitfin genome. However, none of these families show a dynamic exclusive to the lineage of G. multiradiatus (Supplemental Fig. S2).
Sex-biased gene expression in the brain
The availability of an annotated genome allowed us to look for sexual dimorphism of the brain on a molecular level. Differential gene expression analyses of female and male brain transcriptomes revealed that 324 genes, with a log2 fold change (FC) > 1 and adjusted P-values < 0.05, were more expressed in males, whereas only 42 genes showed a biased expression toward females (Supplemental Table S7). In the female brain foxl2a, which is a main transcription factor in female gonad development, was higher expressed than in males. Enrichment for Gene Ontology (GO) terms revealed functions related to membrane structure and function including transporter activities (Supplemental Fig. S3A–D). The male biased genes were enriched for several categories related to germ cell development, male meiosis, and spermatogenesis (Supplemental Fig. S3E–G). Among these are dmrt1, an important regulator of male sexual development in fish, and genes that function in the early stage of meiosis (e.g., spo11, sycp1, sycp2, sycp3, hormad1) or sperm development (e.g., spata4, spata7, spata17, spata22, spef1, spag6, spag8). Of note, also genes from the piRNA pathway (exd1, piwil1, tdrd5, tdrd6, tdrd9), which is particularly active in testes, are predominantly expressed in male brain. Several genes of steroidogenesis pathways were also up-regulated in male (dhrs11b, akr1a1b, cyp11c1).
Placental development
The placenta is an organ formed by the sustained apposition or fusion of fetal membranes and parental tissue for physiological exchange (Mossman 1991). Goodeids have been described as having the most complex placental organ among live-bearing fishes to support the developing embryos, given its large array of histological and cytological modifications of the intra-ovarian epithelium and the trophotaenial absorptive epithelium, which are apposed to each other during pregnancy (Uribe et al. 2010; Schindler 2015). To contribute to the understanding of the evolution, function, and development of this outstanding trait, we analyzed transcriptomes of the trophotaenia (embryonal part of placenta) and the ovary after dissecting out the embryos (maternal part of the placenta). Both data sets were then compared to all other transcriptomes (male and female brains, testis, organ mix, early and late embryos, and trophotaeniae or ovary, respectively). Trophotaenia and ovary predominant expression signatures were defined by a log2 fold change > 2 compared to all other transcriptomes in accordance with the definition of “genes preferentially expressed in human placenta” (https://www.proteinatlas.org/humanproteome/tissue/placenta). This revealed 1653 trophotaenia and 1132 ovary enhanced genes (Supplemental Tables S8, S9).
The ovarian transcriptome is enriched for genes with functions for formation of the zona pellucida and extracellular matrix, fertilization, immune response, and steroidogenesis (Supplemental Fig. S4). The most up-regulated gene in the ovary is cyp19a1a (log2FC = 7.46), the ovarian isoform of the estrogen producing enzyme aromatase.
The genes that constitute the expression signature of the trophotaenia are highly enriched for genes with a function in brush border membranes, transport of ions and metabolites, and metabolic pathways (Supplemental Fig. S4) in agreement with the role of the embryonal part of a placenta. Of note, the telomerase reverse transcriptase is highly up-regulated (log2FC = 5.82 compared to all other organs), as well as the gene encoding the prolactin receptor (log2FC = 4.41).
Insulin-like growth factor 2 (IGF2) signaling has an important role in regulating placental supply of nutrients in mammals and is a key gene undergoing paternal imprinting (DeChiara et al. 1990, 1991). In the darkedged splitfin, the insulin-like growth factor-binding protein1-like and insulin-like growth factor-binding protein4-like genes are predominantly expressed in the trophotaenia, and insulin-like growth factor-binding protein 1 and 3, insulin-like growth factor 1, and IGF-like family receptor 1 are predominantly expressed in the ovary. However, igf2 is not expressed in the ovary and also is not part of the trophotaenia transcriptome.
We then checked the trophotaenia and ovary transcriptomes for expression of other genes imprinted in mammals (N = 255; https://www.geneimprint.com/site/genes-by-status) and found that 16 of these are expressed in trophotaenia and 10 in ovary (Supplemental Table S10).
In another family of live-bearing cyprinodontiform fishes, the Poeciliidae, several species have evolved maternal provisioning of nutrients to the developing embryo throughout pregnancy. Here, the follicular epithelium like in splitfins is the maternal part of the placenta supplying support. We compared the transcriptomes of follicles of the matrotrophic species Poeciliopsis retropinna (Guernsey et al. 2020) with our data set. In total, 1640 genes were found to be overexpressed in follicles of P. retropinna compared to follicles of the lecithotrophic species P. turrubarensis, and among those 59 overlapped with the ovary-specific expression pattern of G. multiradiatus (Supplemental Fig. S5; Supplemental Table S11). The shared genes are enriched for extracellular matrix and cytokine signaling related GO terms (Supplemental Fig. S6A).
The lineage of seahorses has also developed live-bearing, convergently. In addition, seahorses have evolved provisioning of nutrients to the embryo by a specialization of the ventral skin integument, the male brood pouch epithelium, representing the parental part of the placenta (Griffith and Wagner 2017). A transcriptomic study identified 147 significantly up-regulated genes in the pregnant (compared with the nonpregnant) and post-parturition seahorse brood pouch (compared with the late-pregnant) (Whittington et al. 2015). Nine of these are also up-regulated in the ovary of splitfin (Supplemental Fig. S7), with functions in calcium and lipid transport and tissue remodeling.
The human placenta is formed from the endometrium as the maternal part and the trophoblast, which develops into the embryonal contribution of the placenta. The available human placenta transcriptome combines the embryonal and maternal part. Thus, we compared it to both parts of the splitfin placenta. The trophotaenia and ovary transcriptomes of the splitfin overlap with 89 of the 493 genes of the human placenta transcriptome (https://www.proteinatlas.org/) (Supplemental Fig. S8A; Supplemental Table S12). These include the vascular endothelial growth factor family member placental growth factor (PGF), the prolactin receptor and many components of the extracellular matrix, and genes enriched for GO terms angiogenesis, growth, and morphogenesis (Supplemental Fig. S6B,C). A comparison of trophotaenia with the human trophoblast, which gives rise to the embryonal part of the placenta, revealed 97 genes in common between both tissues (Supplemental Table S12; Supplemental Fig. S8B), including many transporters and transcription factors enriched again for morphogenesis, extracellular matrix, and angiogenesis GO terms (Supplemental Fig. S6D).
The evolution of a highly sophisticated placental organ mediating matrotrophy in goodeids may have required changes in protein structure and function that should be visible as signatures of positive selection. To uncover such genomic traces of natural selection we collected 7102 one-to-one orthologs from G. multiradiatus and 11 other teleost species (Supplemental Fig. S2), including the live-bearing species from the Poecilidae family and the more distantly related seahorse, and we tested these genes for positive selection in G. multiradiatus. With site class 2a (positive selection in marked branch and conserved in rest) we identified 48 genes, and with site class 2b (positive selection in marked branch and relaxed in rest) we identified 122 genes under positive selection specifically in the splitfin lineage (Supplemental Table S13). These genes include 16 from the trophotaenia and six from the ovary-specific transcriptomes (Supplemental Table S14).
The positive selection analysis revealed 440 genes (site class 2a, 128 genes; site class 2b, 312 genes) in seahorse (Supplemental Table S15). In Poeciliopsis, because of the unavailability of an annotated genome, the orthologous genes could only be identified by a BLAST to the genome assembly. This restricted the number of one-to-one orthologs for comparison with splitfin to about one-quarter (n = 1521), of which 65 genes (site class 2a, 35 genes; site class 2b, 30 genes) are under positive selection (Supplemental Table S16). Between all three species, there are two genes commonly under positive selection in Poeciliopsis and splitfin, eight genes shared between seahorse and splitfin, and one gene, sterol-O-acyltransferase 1, which is under positive selection in all three species (Supplemental Fig. S9).
Sex determination and sex chromosomes
To characterize the sex-determination system of the darkedged splitfin we first searched for sex-linked markers using restriction site associated DNA sequencing (RAD-seq) (Feron et al. 2021) in 29 females and 27 males. A total of 6,359,801 markers was found, of which 892 were significantly associated with male phenotype (P < 0.05, χ2 test with Bonferroni correction) and none were associated with female phenotype (Fig. 4A; Supplemental Table S17). These results suggested a stable male heterogametic genetic sex-determination system. The absence of outliers most likely indicates the absence of relevant environmental factors or autosomal modifiers influencing the XY sex chromosomal system.
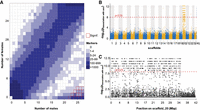
Identification of the sex chromosomes by RAD-tag sequencing. (A) Distribution of RADSex markers between males (horizontal axis) and females (vertical axis) using a minimum depth of 1 to consider a marker present in an individual. There were 892 markers significantly associated with male phenotypes in the tiles highlighted in red. (B) Manhattan plot showing the negative log of P-value of association with sex for all RADSex markers aligned to the genome. The vast majority of aligned markers significantly associated with sex (97%) was aligned to scaffold_20. (C) Negative log of P-value of association with sex for all RADSex markers aligned to scaffold_20. Markers significantly associated with sex aligned to a continuous region spanning from the start of the chromosome to ∼32.5 Mb.
We then aligned these RAD-seq markers to the genome assembly of darkedged splitfin. Among the 892 markers significantly associated with male phenotype, 675 (76%) were aligned to a unique position with mapping quality higher than 20, and of these good quality markers, 654 (97%) were aligned to scaffold 20 in a region ranging from 22 kb to 34.5 Mb (Fig. 4B,C). These results indicate that scaffold 20 is the sex chromosome in darkedged splitfin, and that a large region spanning 80% of the sex chromosome is differentiated between the X and Y Chromosomes.
To identify the male-specific region and to elucidate the evolution of the sex chromosomes of splitfin, a haplotype-resolved assembly of scaffold 20 was performed. Mapping of the male-specific RAD-tags identified the putative Y Chromosome (Fig. 5A), where 645 (71%) had a significant hit.
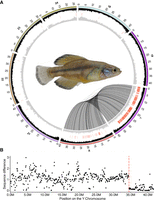
Circos and dot plots showing synteny and sequence difference between Chromosomes X and Y. (A) Circos plot of Chromosomes 18 to 24 and both haplotypes of scaffold 20 (X and Y) showing repeat content (gray histogram), GC value (black histogram), male-specific RAD-tag hits (red bars), and synteny (linking ribbon) on chromosomes; (inset) image of a male darkedged splitfin adult. (B) Dot plots showing the sequence difference (percentage of SNPs and indels in windows of size 100 kb) between X and Y.
Sequence difference between X and Y was determined in 10-kb sliding windows as the percentage of SNP and indels. It uncovered a large region from 0 to 34.5 Mb on the Y, where the nucleotide difference was uniformly in the range of 1.4% (Fig. 5B). Beyond this point the overall sequence difference between X and Y was close to zero. The dS values were high in the XY-differential region (median 0.01) and low in the terminal segment (Supplemental Fig. S10A). The dN/dS values in the terminal region are predominantly around 0.2, indicating purifying selection. Most genes in the XY-differential region have on average twice as high values and many close to 1 or even higher, indicating relaxation from purifying selection (Supplemental Fig. S10B). Together this identifies a possible terminal pseudoautosomal region of 7 Mb with about 200 genes and a large region on the Y of about 34.5 Mb containing around 900 genes, where it is different from the X (Supplemental Fig. S10C).
To estimate the age of the Y, we used the dS distance between the X and Y in the differential region and calibrated it to the dS based divergence time estimate of splitfin to Fundulus (52.6 MYA) (see Supplemental Fig. S2). This revealed that the Y Chromosome of splitfin originated only about 2 MYA (Supplemental Fig. S11).
The region on the Y that is different from the X does not have a higher content of repeats and TEs. Also, the transposon landscape is similar to the corresponding region on the X and all autosomes except for a slightly higher percentage of LINE elements (Supplemental Fig. S12). The region on the Y has a higher content of pseudogenes (20.4%) than the corresponding region on the X (13.7%) and all autosomes (16.1%) (Supplemental Methods, Comparison of the X and Y Chromosomes). Mapping the gene contents of the X and the Y to each other revealed that no gene from the X is missing from the Y, whereas there are 12 gene models annotated that are present only on the male sex chromosome. Manual inspection revealed all of these 12 genes are either corrupted, pseudogenes, or transposon-derived sequences (Supplemental Table S18).
New sex determining genes arise either from local gene duplications or allelic diversification (Herpin and Schartl 2008). Because we did not find any hint of duplicated Y-specific genes, we considered that the splitfin male determining gene would show allelic differentiation between the X and Y copies. We manually checked the 948 genes of the differentiated region on the Y for the recurrently appearing sex determining genes in vertebrates. From these (Tgfb signaling, dmrt1, and sox family members) only sox8a, sox9a, and sox10a are on scaffold 20. Although sox9a and sox10a have dN/dS values of 0.32 and 0.23, which indicate a purifying selection, sox8a has a value of 0.81. The X and Y sox8a copies differ by 12 (2.6%) amino acids. Overlaying the computer-modeled protein structures revealed that none of these differences would compromise the three-dimensional structure of Sox8a as a whole, suggesting that both copies encode functional transcription factors (Supplemental Fig. S13). The Y copy presents two amino acid changes at a highly conserved position (L364M, S360N). The S360N exchange adds a ß-sheet at this place to the secondary structure. Further functional studies are required to elucidate what could be the impact of these changes on Sox8a activity in splitfin. Of note, the Y allele is preferentially expressed in testis, however, in male brain, the X and Y alleles are equally expressed (Supplemental Table S19).
Discussion
As part of the Vertebrate Genomes Project (VGP), a project of the Genome 10K Consortium, which aims to generate near error-free reference genome assemblies of approximately 70,000 extant vertebrate species, the darkedged splitfin was selected to represent the group of goodeid fishes. The availability of a chromosome-level genome assembly of high completeness allowed us to perform genome-wide analyses to contribute to a better understanding of several highly interesting characteristics of goodeids and also benefit ecotoxicological research.
We identified a differentiated XY sex chromosome system in G. multiradiatus and succeeded in assembling X and Y separately. The comparison of these two sex chromosomes revealed a large MSY with uniform levels of sequence differentiation over its whole length. Such distribution of male-specific SNPs would be expected if recombination stopped simultaneously in the Y-specific part by a large chromosomal inversion. Manual inspection of the Hi-C maps did not support such a single-large inversion, however, several smaller inversions are possible. Further studies are needed to explain this pattern.
The differentiation of the Y occurred in less than 2 million years since its origin, about 6 million years later than the split between G. multiradiatus and its sister species G. viviparus (Webb et al. 2004). The two species are different in the expression of epigamic characters, with G. multiradiatus males being more ornamented (larger and colorful median fins) and having a large repertoire of courtship displays, whereas G. viviparus has smaller dark fins and very simple courtship behavior (González Zuarth and Macías Garcia 2006; Méndez-Janovitz and Garcia 2017). A full assembly of the genome of G. viviparus is needed to better understand sex chromosome evolution in the genus Girardinichthys. Comparison of the sex chromosome-specific region to the corresponding region of the G. multiradiatus genome can then be used to search for genes involved in male ornaments.
Our brain transcriptome analyses uncovered a large number of genes that were more expressed in males and are known to fulfill their functions in testis development and spermatogenesis. This is in agreement with RNA-seq data from human, mouse, and zebrafish (Guo et al. 2003, 2005; Sreenivasan et al. 2008). A closer relationship of female and male brains to their respective gonad was also reported (Sreenivasan et al. 2008); however, at least to our knowledge, such a strong bias as in splitfin of male brain toward testis has not been noted. The biological meaning of the abundance of testis gene expressions in the male brain is unknown as well as its molecular mechanisms. Shared characteristics between neurons and sperm, including the importance of the exocytotic process and the presence of similar receptors and signaling pathways have been hypothesized (Matos et al. 2021). In humans it has been shown that meiosis genes expressed in the brain are not translated. A possible reason for the absence of translation of such transcripts to proteins may be connected to the lack of Dazl, which is a key regulator of translation of spermatogenesis genes (Li et al. 2019). In splitfin brain, dazl is expressed only at low levels in male, not at all in females, and abundantly only in testis. In splitfin, we lack the tools to analyze protein expression, but it may well be that a large fraction of the testis genes is not translated owing to the lack of Dazl (Supplemental Fig. S14). Even if not, all expressed “testis” genes may have a function in brain they might reflect a coregulatory expression mechanism, which up-regulates genes that are of importance in both organs, for example, steroidogenesis genes. The reproductive biology of splitfins requires a complex male-specific behavior, including searching for receptive females, a whole suite of courtship behaviors (Garcia and Valero 2010), and constantly adjusting sperm production to suit the current availability of partners and of potential sperm competitors (Garcia et al. 1998; Simmons and Fitzpatrick 2012), which is orchestrated in the brain and may be regulated by neurosteroids. We found up-regulation of genes involved in steroidogenesis specifically in the brain of males. Further studies have to show whether these enzymes are indeed involved in the synthesis of neurosteroids.
So far, from the other placental fish, seahorse and Poeciliopsis, only the parental part of the placenta was analyzed. Here in splitfin, for the first time information also from the embryonal part could be generated. The placental organ of G. multiradiatus shows a high similarity with the expression signature of human placenta. This is indicative of a convergent evolution of organs that serve embryonic nutrition although they are phylogenetically quite distant and developmentally totally unrelated. The mammalian placenta descends from the maternal endometrium and the trophoblast, which have no counterpart in fish. The goodeid trophotaeniae, however, are outgrowths of the hindgut that interact with the maternal ovarian wall. The similarities to the human placenta transcriptome, which is derived from the whole composite organ, are equally shared between the maternal and embryonal part of the G. multiradiatus placenta.
The trophotaeniae show a high expression of the prolactin receptor indicating that this organ is responsive to prolactin stimulation. Prolactin, the lactation hormone of mammals, is a hormone that plays multiple roles in parental care in fish, including mouth breeding, nutrient provision by mucus or fluids, parental care behavior, and viviparity (Whittington and Wilson 2013; Dobolyi et al. 2020). Expression of the prolactin receptor gene has also been observed in the follicular epithelium of matrotrophic Poeciliopsis species (Guernsey et al. 2020). The prolactin gene of G. multiradiatus is not only expressed in brain but also at similar levels in trophotaenia and ovary, indicating not only endocrine but also juxtacrine hormone signaling.
The trophotaenia is an embryonic organ and thus high telomerase expression is within expectation (Wright et al. 1996; Anchelin et al. 2011); in splitfin, its level of expression achieve 15- to 50-fold higher than in the embryo proper. In the human placenta dysregulation of telomerase activity is connected to a variety of pregnancy complications (Fragkiadaki et al. 2016).
The absence of igf2 in the ovary and the trophotaenia of G. multiradiatus is intriguing (Lawton et al. 2005). Parent-of-origin specific expression of this growth factor gene constitutes an iconic example of imprinting linked to sexual conflict. In mammalian embryos, the paternal copy can be overexpressed, hence promoting embryo growth beyond the optimum for the mother, and the maternally inherited copy silenced through methylation, thus resisting the effects of the paternally inherited allele. Conversely, the maternal copy of the cation-independent mannose-6 phosphate receptor, which binds the growth factor and transports it to lysosomes, is overexpressed, and the paternal allele is silenced (DeChiara et al. 1990, 1991). Igf2 is expressed in fish embryos (Yuan et al. 2011), including those of G. multiradiatus, where there is evidence suggesting that, as in mammals, the paternal copy is expressed and the maternal allele is silenced through methylation (Saldivar Lemus et al. 2017). Thus, we would have predicted its absence in the ovary transcriptome but expected it to be expressed in the trophotaenia. At present we have no information of where in the embryo igf2 is being expressed or why it is not being expressed in the trophotaenia. It is still possible that its expression in the trophotaenia depends on the developmental stage of the embryo, yet we collected trophotaeniae from embryos of 3–4 and 5–6 wk of development, which were, at those stages, not producing this growth factor.
Live-bearing and maternal or paternal provisioning through placenta-like structures has evolved independently several times in fish. We asked the question if this physiological and morphological convergent evolution is connected to similar convergent changes on the molecular level. Proteins that may have adapted to new functional properties required for placentation should show signatures of positive selection in the respective lineage. Indeed, about one-fifth of the genes identified as positively selected in splitfin are specifically expressed in the placenta. An even higher fraction was noted in the matrotrophic Poeciliid species Heterandria formosa (van Kruistum et al. 2019). However, comparing positively selected genes of seahorse, Poeciliopsis, and splitfin revealed only a single gene in common, sterol-O-acyltransferase 1. This gene encodes a lipid metabolism enzyme, which has a key role as the rate-limiting enzyme for regulation of the transport of circulating cholesterol and fatty acids, their metabolism, and the turnover of lipoproteins. Perturbations of the placental lipid metabolism have been connected to pregnancy complications like preeclampsia (Hu et al. 2021). The low overlap between seahorse and the other two taxa could be a result of the different sexes contributing to the placenta.
Changes in expression levels, for example, of proteins involved in trafficking molecular cargo between compartments, would be another common adaptation connected to placentation. Indeed, ∼20% of the genes preferentially expressed in human placenta are also found in the placental transcriptomes of splitfin. Nevertheless, all transcriptomes were enriched for similar pathways and gene categories, in particular those related to transport across membranes, extracellular matrix, cell interactions, and metabolism, but our data do not point to specific “placentation genes,” which are specifically recruited repeatedly. We hypothesize that the results may instead indicate that the independent development of parental provisioning repeatedly made use of genes that already had the same function in other tissues (e.g., in the transport of metabolites or interaction of between cells and organs) when it became necessary to make up a novel organ for embryo nutrition. In this way, preexisting modules could have been assembled to provide the molecular changes for the novel trait. This is consistent with the idea that regulatory evolution is crucial to major evolutionary innovation (Carroll 2008; Emerson and Li 2010). We have generated genomic tools for G. multiradiatus as a representative of the live-bearing placental goodeids and could identify molecular changes that are connected to this form of maternal care. Further analysis will benefit from the inclusion of genomes and transcriptomes of lecithotropic splitfin species and the basal oviparous Goodeidae; the Empetrichthynae, from which the viviparous splitfins (Goodeinae) diverged 16.8 MYA (Webb et al. 2004).
Methods
Experimental animals
Most samples were from laboratory-born descendants of fish collected in 2011 at Salazar Lagoon (19°18′26′′ N, 99°23′20′′ W, 3000 meters above sea level [MASL]) under license DGOPA/05332/071007.2437 from Dirección General de Gestión de Pesca y Acuicultura. We also used, for genome annotation, one sample from an aquarium stock originating from Zempoala (19°03′00′′ N, 99°18′42′′ W) and collected under permit DGOPA A/01584/110310.0785.
Live fish and preserved tissues were exported under permit B00.02.04.-1277.2019 from the Animal Health Direction (DGSA; SENASICA) of the National Ministry of Agriculture and Rural Development (SAGARPA).
Adult fish were sedated by placing them in ice-cold water, then quickly sacrificed by decapitation and immediately dissected. Embryos were classified as “early” (≤4 wk of development) or “late” (5–6 wk of development; gestation lasts around 8 wk in this species). Embryos were separated from the single ovarian lumen. The corresponding remaining ovary tissue and trophotaeniae were collected and processed separately; hence, we used whole ovaries from pregnant females, which were free of any embryonic tissue. For RNA-seq tissues were promptly placed in RNA-later (2019). For genome sequencing, dissected organs were flash frozen and stored at −80°C, and for RAD-tag sequencing tissues were stored in 100% ethanol.
For whole-genome sequencing and assembly, three individuals from the same family were used (Supplemental Table S2).
As transcript evidence for genome annotation, RNA-seq reads from a total RNA organ mix of one female (brain, eyes, gills, muscle, skin, kidney, liver, embryos) and ovary from a second female originating from Zempoala were used.
For expression analysis, pools of organs and embryos were used (Supplemental Table S20).
DNA extraction and genome sequencing
Ultralong genomic DNA of different organs of two splitfin individuals was extracted with the agarose plug based Bionano Prep Animal tissue kit following the manufacturer's instructions. Residual pigments were precipitated by adding Triton X-114, SDS, and NaCl to the genomic DNA (Ma et al. 2012). The fragment size of all genomic DNAs was controlled by pulse-field gel electrophoresis before library construction. Two size-selected PacBio continuous long-read libraries of 24 and 29 kb in size were run on the SEQUEL system with 10-h movie times. Linked Illumina reads were generated with the 10x Genomics Chromium genome protocol following the manufacturer's instructions. These libraries were sequenced on the Illumina NovaSeq with a 150-bp paired-end regime. For Bionano optical mapping, genomic DNA was labeled following the Bionano DLS protocol. Labeled genomic DNAs were run on the Bionano Saphyr instrument to 200× genome coverage. Hi-C conformation capture of a third individual was performed following Arima Genomics Hi-C protocol (Arima Hi-C user guide for Animal tissues, v01), an Illumina library of the enriched regions was prepared with the KAPA HyperPrep Kit, and sequencing was performed on an Illumina NovaSeq applying 150-bp paired-end conditions.
Details of all protocols are provided in Supplemental Methods (DNA extractions and genome sequencing).
Genome assembly
We assembled the PacBio long reads into contigs using a customized assembler we termed DAmar, a hybrid of the earlier Marvel (Nowoshilow et al. 2018), Dazzler, and Daccord (Tischler and Myers 2017) systems. Next, we used 10x Illumina read-cloud data to correct base errors and phase haplotypes, arbitrarily picking one haplotype in a phased block. Afterward we used Bionano optical maps and then Hi-C data to produce long-range scaffolds. A manual curation step was performed that uses the Hi-C reads. By inspecting the Hi-C interaction contact matrix with HiGlass (Kerpedjiev et al. 2018), remaining false joins were split and curated scaffolds were joined into chromosomes. Finally, the curated chromosomes were phased by applying an adapted version of the DipAsm pipeline (Garg et al. 2021).
Details of the procedures are provided in Supplemental Methods (Genome assembly).
Genome annotation
The genome was annotated using a pipeline adapted from our previous study (Du et al. 2019, 2020; Powell et al. 2020). The repeats of the genome were identified and masked using RepeatModeler, RepeatProteinMask, and RepeatMasker (http://www.repeatmasker.org). Then the protein-coding genes were annotated by collecting and synthesizing gene evidence from homology, transcriptome, and ab initio predictions. Annotated genes were blasted to database InterProscan, Swiss-Prot, and RefSeq for identifying the protein domain and assigning gene symbol and name. At last, noncoding RNAs (ncRNAs) were annotated using the method adapted from Ensembl (http://useast.ensembl.org/info/genome/genebuild/ncrna.html).
Details of the procedures are provided in Supplemental Methods (Genome annotation).
Orthology assignment
Orthology of genes was assigned by a clustering of sequence similarity followed by gene-tree construction in each gene cluster. Sequence similarity was indexed by BLAST (Camacho et al. 2009) score between protein sequences of each two genes. Clustering was implemented by hc_cluster (Supplemental Fig. S15; Ruan et al. 2007). Then the gene tree of each cluster was constructed using TreeBeST (http://treesoft.sourceforge.net/treebest.shtml).
Orthology between chromosomes from two species was depicted using a Circos diagram (Krzywinski et al. 2009) based on orthologous genes with conserved synteny.
Details of the procedures are provided in Supplemental Methods (Orthology assignment).
Phylogenetic analysis
Phylogenetic tree was reconstructed first using the maximum-likelihood method implemented in RAxML (Stamatakis 2014), then we confirmed the topology using Bayesian inference implemented in MrBayes (Ronquist et al. 2012). The input data were protein sequences of 1425 one-to-one orthologs with conserved synteny. We also inferred the divergence time of species using MCMCTree under a relaxed-clock model (Yang 2007; Inoue et al. 2011). Two time calibrations were set: O. latipes–T. nigroviridis (∼96.9–150.9 Ma) and Clupeiformes-Cypriniformes (∼185–225 Ma) (Near et al. 2012; Lin et al. 2016; Hughes et al. 2018).
Details of the procedures are provided in Supplemental Methods (Phylogenetic analysis).
Dynamics of gene family size
Gene groups clustered by Hcluster_sg were taken as gene families. Together with the phylogenetic tree built by RAxML and MCMCTree, they were transferred to CAFE5 for gene family size analysis (Zenodo https://doi:10.5281/zenodo.3625141, as developed on GitHub [https://github.com/hahnlab/CAFE5]). To estimate the birth-death parameter λ, we only selected gene families that were present in more than seven species and in each species had less than 100 gene family members. Then with the previously estimated λ, gene family contraction and expansion dynamics were accessed for the remaining families. Before λ estimation, an error model was estimated to account for genome assembly and annotation error. λ was estimated using Gamma modeling with two categories.
RAD-tag sequencing and analysis of sex-specific markers
RAD-tag libraries were built from genomic DNA of 29 females and 27 males and sequenced on the HiSeq 2500 platform. The reads were demultiplexed and then determined present or not in each female and male individual in RADSex (R Core Team 2013; Feron et al. 2021). A tile plot describing the number of reads in the number of female/male individuals was then generated and used to reveal the sex-determination system of the species. Reads that present only in male were then aligned to the genome to locate the sex-determination region.
Details of the procedures are provided in Supplemental Methods (RAD-tag sequencing and analysis of sex-specific markers).
RNA extraction and transcriptome sequencing
Total RNA was isolated using TRIzol Reagent (Thermo Fisher Scientific) according to the supplier's recommendation. Custom sequencing (BGI) of TruSeq libraries generated 30–35 million 150-bp paired-end clean reads for each sample on the BGISEQ platform.
Differential gene expression analysis
Clean reads were aligned to genome assembly using STAR (–runMode alignReads –quantMode GeneCounts) (Dobin et al. 2013) and counted and normalized for each gene using DESeq2 (Love et al. 2014). Genes with more than 10 normalized reads count in a tissue were defined as “expressed divergence time estimate of splitfined.” Expression with up-regulation of log2 fold change >2 in one tissue than in the other was termed as “enhanced expression” in the former tissue. We pooled the data of early- and late-stage embryos and trophotaeniae considering they are tightly grouped together in both hierarchical clustering and correspondence analysis (Supplemental Fig. S16). For cross-comparison of detected genes, data set of human, Poeciliopsis, and seahorse were retrieved from previous studies (for details on how the data sets were produced, see Supplemental Table S21).
Details of the procedures are provided in Supplemental Methods (Differential gene expression analysis).
Positive selection
Protein and CDS sequences of genes were downloaded from NCBI (Supplemental Table S22). Sequences were aligned using MUSCLE (Edgar 2004), and the format was transformed using an in-house script (Supplemental Code). Positive selection on a gene of the species was detected using “Environment for Tree Exploration” (ETE3) toolkit (Huerta-Cepas et al. 2016), which automates CodeML and Slr analyses by using preconfigured evolutionary models, the species tree, and alignment of the orthologous coding sequences. A gene was identified as a positively selected gene when it contains sites (probability > 0.95) of either class 2a (positive selection in marked branch and conserved in rest) or class 2b (positive selection in marked branch and relaxed in rest).
Details of the procedures are provided in Supplemental Methods (Positive selection).
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession numbers PRJNA768329 and PRJNA745519. The genome assembly generated in this study has been submitted to the NCBI Assembly database (https://www.ncbi.nlm.nih.gov/assembly) under accession number PRJNA768329. The genome annotation has been submitted to figshare (https://figshare.com/articles/dataset/Girardinichthys_multiradiatus_201028_tar_gz/15173181).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank the Long Read Team of the DRESDEN-concept Genome Center, Deutsche Forschungsgemeinschaft (DFG), Next Generation Sequencing (NGS) Competence Center, part of the Center for Molecular and Cellular Bioengineering (CMCB), Technische Universität Dresden, and The Max Planck Institute of Molecular Cell Biology and Genetics (MPI-CBG), for their contribution. M.S. was supported by the DFG (SCHA 408/10-1). C.M.G. received funding from Programa de Apoyo a Proyectos de Investigación e Inovación Tecnológica (PAPIIT) (PAPIIT IN210718), the science funding agency of the Universidad Nacional Autonoma de Mexico. M.P. was funded by the Federal Ministry of Education and Research (grant 01IS18026C).
Author contributions: M.S., C.M.G., and G.M. conceived and designed the project. E.G.A.L. and C.M.G. provided the materials. B.W. and S.W. extracted DNA and RNA samples. S.W. conducted the sequencing, M.P. did the assembly, and K.D. performed the annotation. K.D., S.K., I.dC., and M.S. analyzed the sequencing data. Y.G. and R.F. performed the RADSex analyses. K.D., R.F., S.K., S.W., and M.P. prepared figures and tables. C.M.G., M.S., and K.D. wrote the manuscript draft, and Y.G. revised it. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275826.121.
- Received May 28, 2021.
- Accepted January 10, 2022.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








