
A systematic genome-wide account of binding sites for the model transcription factor Gcn4
Abstract
Sequence-specific DNA-binding transcription factors are central to gene regulation. They are often associated with consensus binding sites that predict far more genomic sites than are bound in vivo. One explanation is that most sites are blocked by nucleosomes, such that only sites in nucleosome-depleted regulatory regions are bound. We compared the binding of the yeast transcription factor Gcn4 in vivo using published ChIP-seq data (546 sites) and in vitro, using a modified SELEX method (“G-SELEX”), which utilizes short genomic DNA fragments to quantify binding at all sites. We confirm that Gcn4 binds strongly to an AP-1-like sequence (TGACTCA) and weakly to half-sites. However, Gcn4 binds only some of the 1078 exact matches to this sequence, even in vitro. We show that there are only 166 copies of the high-affinity RTGACTCAY site (exact match) in the yeast genome, all occupied in vivo, largely independently of whether they are located in nucleosome-depleted or nucleosomal regions. Generally, RTGACTCAR/YTGACTCAY sites are bound much more weakly and YTGACTCAR sites are unbound, with biological implications for determining induction levels. We conclude that, to a first approximation, Gcn4 binding can be predicted using the high-affinity site, without reference to chromatin structure. We propose that transcription factor binding sites should be defined more precisely using quantitative data, allowing more accurate genome-wide prediction of binding sites and greater insight into gene regulation.
Transcription factors are typically associated with consensus DNA binding sites composed of roughly six base pairs, not all of which are fully specified, although there is wide variation (e.g., Harbison et al. 2004; MacIsaac et al. 2006; Badis et al. 2008; Zhu et al. 2009). Such consensus sites occur quite frequently in genomic DNA, not only in regulatory elements, but also in genes and elsewhere. Consensus sites often predict far more transcription factor binding sites than are actually bound in vivo. This observation has led to the proposal that consensus sites in nonregulatory regions are unbound because they are blocked by chromatin (Liu et al. 2006). However, recent measurements of DNA accessibility in yeast and mouse nuclei imply that all consensus sites are likely to be accessible in some cells within a population (Chereji et al. 2019; Oberbeckmann et al. 2019). This general but limited accessibility predicts detectable binding at all consensus sites, albeit reduced relative to sites in nucleosome-free DNA. An alternative explanation is that consensus site sequences derived from ChIP-seq data may be too degenerate in some cases, such that only a subset of the predicted sites are true sites. We have investigated this question using the well-studied yeast Gcn4 transcription factor as a model.
Gcn4 is a critical transcriptional regulator in Saccharomyces cerevisiae, conserved throughout yeast species, and required for amino acid biosynthesis in response to amino acid starvation (Natarajan et al. 2001; Hinnebusch 2005). Gcn4 was one of the first eukaryotic transcription factors to be studied in detail. Gcn4 is a dimer in solution (Hope and Struhl 1987). Structural studies using peptides corresponding to its C-terminal basic leucine zipper (bZIP) DNA-binding domain indicate that it is partially disordered until it binds to its cognate site (Weiss et al. 1990; Bracken et al. 1999; Gill et al. 2016). Gcn4 was originally thought to recognize a 6-bp consensus sequence, TGACTC (Arndt and Fink 1986), or a 7-bp sequence (TGACTCT), based on studies of the HIS3 gene promoter (Hill et al. 1986). However, mutational and structural studies of Gcn4 complexed with DNA indicate that Gcn4 prefers to bind to the closely related Activator Protein-1 (AP-1) consensus site, RTGA(G/C)TCAY (Oliphant et al. 1989; Ellenberger et al. 1992). Subsequent studies using global approaches in vivo (ChIP-chip [Harbison et al. 2004] and ChIP-seq [Rawal et al. 2018]) and in vitro (protein binding to DNA microarrays [Zhu et al. 2009]) provide support for a similar consensus site.
Recently, we used ChIP-seq to detect Gcn4 binding in vivo after treating cells with sulfometuron (SM) to induce amino acid starvation. We observed Gcn4 binding at 546 sites in the yeast genome (Rawal et al. 2018). Although most of the ChIP-seq peaks overlap with AP-1 consensus sites, about two-thirds of the 1754 AP-1 consensus sites are unoccupied in vivo. These unoccupied motifs could be blocked by nucleosomes, which impede Gcn4 binding in vivo (Devlin et al. 1991; Yu and Morse 1999). On the other hand, ChIP-seq data reveal that Gcn4 binds to some sites within open reading frames (ORFs), which are not located in pre-existing nucleosome-depleted regions (Rawal et al. 2018). A supervised machine learning approach indicates that a strong match to the consensus motif is the most important determinant of Gcn4 binding in vivo and that the second most important feature is proximity to a nucleosome dyad. However, this does not account for the fact that most consensus sites are not bound by Gcn4 in vivo.
To explore why Gcn4 binds in vivo to fewer than one-third of its consensus sites, we designed an in vitro method to determine preferred binding sites for a DNA-binding protein, called G-SELEX (Fig. 1A). The approach is based on DIP-chip (Liu et al. 2005) and SELEX methods (selective evolution of ligands by exponential enrichment [Darmostuk et al. 2015; Bayat et al. 2018]). Recombinant Gcn4 attached to magnetic beads is used to select short genomic DNA fragments containing a binding site. After three rounds of selection and amplification, the selected DNA fragments are subjected to Illumina paired-end sequencing. G-SELEX identifies all genomic binding sites and provides quantitative data for binding at each site in the absence of other proteins. We compared Gcn4 binding in vitro with our published data for Gcn4 binding in vivo (Rawal et al. 2018) to determine the strict, exact, high-affinity binding site for Gcn4, which accounts for high occupancy binding in vivo, with only a relatively minor contribution from chromatin.
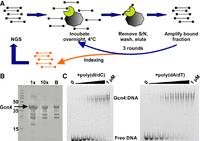
The G-SELEX method, purification and DNA binding of Gcn4. (A) Overall schematic of G-SELEX method: Purified genomic DNA is sonicated, ligated to the Illumina paired-end adaptor (black lines capped with black squares), and amplified by PCR. An excess of DNA is incubated with Gcn4 (green semicircle) bound to Ni-NTA beads (gray octagon) at 4°C overnight. Gcn4-bound DNA is eluted from the beads, purified, amplified, and used for a second round of Gcn4 binding. After three rounds, the eluted DNA is indexed and sequenced. (B) Purification of recombinant Gcn4. Purified Gcn4 (0.3 μL and 3 μL) analyzed by SDS-PAGE. The major band accounts for ∼50% of the protein (by densitometry). Gcn4 containing a 6xHis tag binds tightly to the Ni-NTA magnetic beads used for the experiments (lane “B”: loaded beads). (C) Gcn4 binds to a fluorescently labeled, double-stranded 24-mer containing the AP-1 site located in the ARG1 promoter (EMSA). DNA (10 nM) was incubated for 30 min at room temperature with 0, 2.5, 5, 10, 20, 40, 80, 160, 320, 1000 nM Gcn4 and 500 ng of unlabeled poly(dI/dC) (left) or poly(dA/dT) (right) before loading in a polyacrylamide gel.
Results
Recombinant Gcn4 binds tightly to the AP-1 motif in the ARG1 promoter
The results of recombinant Gcn4-6xHis-FLAG purification are shown in Figure 1B. Gcn4 is prone to degradation, as indicated in prior studies (Gartenberg et al. 1990), and such degradation is apparent in our findings. However, because the 6xHis tag was inserted in the C terminus, where the DNA-binding and leucine zipper dimerization domains also reside, the degraded forms of Gcn4-6xHis-FLAG should not interfere with its ability to bind the AP-1 consensus sequence (TGACTCA). Indeed, as shown in Figure 1C, electrophoretic mobility shift assays (EMSAs) indicate that Gcn4 binds tightly to a fluorescently labeled DNA probe corresponding to the AP-1 site in the ARG1 promoter in the presence of unlabeled poly(dI/dC) or poly(dA/dT) as competitor, with an apparent Kd of ∼80 nM (Supplemental Fig. S1).
G-SELEX shows significant binding of Gcn4 to known and predicted Gcn4 sites
We developed a modified SELEX method to identify biologically relevant Gcn4 binding sites in the yeast genome by using genomic DNA fragments instead of the usual oligonucleotide libraries (“G-SELEX”). We prepared an input library of short genomic DNA fragments by sonication and subsequent gel purification to obtain a narrow insert size range (∼50 to ∼150 bp with a mean of ∼85 bp) and added Illumina adaptors. A small amount of recombinant Gcn4 was immobilized by attaching it to magnetic Ni-NTA beads via its C-terminal 6xHis tag (Fig. 1A). The Gcn4-beads were incubated with library DNA overnight to reach equilibrium and then washed briefly before elution of the bound DNA. Eluted DNA was amplified and subjected to two more rounds of selection before Illumina sequencing. Gcn4-bound sites were identified using MACS2 (Zhang et al. 2008), resulting in a total of 2359 peaks that are common to all three biological replicates (the pairwise Pearson's correlations for the 2359 peak maxima in the three replicates are 0.86, 0.92, and 0.98). The combined data were normalized to the genomic average (set at 1). The background was generally very low.
Previous ChIP-seq studies of Gcn4 binding in vivo demonstrated that, upon induction, Gcn4 binds at promoters as well as unconventional (UC) sites within open reading frames of its target genes to activate transcription in vivo (Rawal et al. 2018). We found that G-SELEX captures many of the same sites identified by ChIP-seq, including UC sites. Data for ARG1, a well-established Gcn4 target gene involved in amino acid biosynthesis (MacIsaac et al. 2006; Uluisik et al. 2011), are shown in Figure 2. All three G-SELEX replicate experiments show a strong peak in the ARG1 promoter in the same location as the Gcn4 ChIP-seq peak in induced cells reported previously (Rawal et al. 2018). Many examples of promoter and UC sites are presented in Supplemental Figure S2. In particular, we note that the UC peaks observed in MCH1 and STP2 are located within their ORFs, rather than their promoters, as expected (Rawal et al. 2018). Thus, G-SELEX correctly identifies Gcn4 binding sites that occur within promoters and ORFs. We present a systematic analysis of the G-SELEX peaks below.
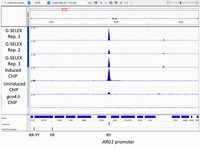
Gcn4 binding profiles in the vicinity of the ARG1 gene (G-SELEX and ChIP-seq data). All data were normalized against a genomic average of one, and tracks were plotted in Integrative Genomics Viewer. Gcn4 binds to the ARG1 promoter site in vitro (G-SELEX: biological replicate experiments indicated by the top three tracks), confirming findings from ChIP-seq data (bottom three tracks: induced cells, uninduced cells, and cells lacking Gcn4 [Rawal et al. 2018]). Vertical black bars indicate AP-1 motif locations with motif type (see below). More examples are provided in Supplemental Figure S1.
G-SELEX detects many more Gcn4 binding sites than ChIP-seq
Prior computational analysis of the S. cerevisiae genome suggests that it contains 1754 potential Gcn4 binding sites, which were curated for comparison with ChIP-seq data (Rawal et al. 2018). However, ChIP-seq uncovered occupancy peaks in only 546 locations, 471 of which contained a consensus motif very similar to the AP-1 site (Fig. 3A; Rawal et al. 2018)—that is, Gcn4 binds only 471 of 1754 consensus sites in vivo. The discrepancy is still more pronounced for the G-SELEX data, given that 2359 G-SELEX peaks were identified. Clearly, G-SELEX detects far more Gcn4 binding sites in vitro than those occupied in vivo.
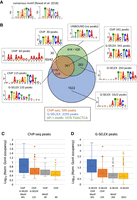
Qualitative and quantitative comparison of Gcn4 binding in vivo (ChIP-seq data) and in vitro (G-SELEX data). ChIP-seq data for Gcn4 are from Rawal et al. (2018). (A) Consensus motif (both strands) for Gcn4 binding from Rawal et al. (2018). (B) Qualitative comparison of ChIP-seq and G-SELEX peak overlaps and with the genomic locations of AP-1 motifs (TGACTCA). Venn diagram: ChIP-seq (orange), G-SELEX (blue), and AP-1 motifs (green). Some ChIP peaks overlap with two G-SELEX peaks and/or two AP-1 motifs, as indicated by two numbers. Motifs were derived using MEME. Quantitative comparison of different classes of (C) ChIP-seq and (D) G-SELEX peaks defined by the Venn diagram are listed below each plot. Gcn4 occupancy data were normalized to the genomic average (set at 1).
We determined the overlaps between the ChIP-seq data (binding in vivo), the G-SELEX data (binding in vitro), and the locations of the exact AP-1 motif (TGACTCA, rather than the consensus AP-1 site used previously [Rawal et al. 2018]). The result is presented as a Venn diagram (Fig. 3B). Of the 546 ChIP-seq peaks, 456 are also detected by G-SELEX. Of these, 341 peaks common to ChIP-seq and G-SELEX contain an exact match to the AP-1 motif; the other common peaks do not contain an AP-1 motif (115 ChIP-seq peaks overlap with 133 G-SELEX peaks; the ChIP-seq peaks are generally wider than the G-SELEX peaks and so may include more than one G-SELEX peak). Of the 90 ChIP-seq peaks not detected by G-SELEX, 30 contain an AP-1 motif. The vast majority of G-SELEX peaks (1622; 69%) are not detected in vivo and lack an AP-1 motif. In addition, 263 G-SELEX peaks with AP-1 motifs are not detected in vivo. Finally, 426 AP-1 motifs are not bound by Gcn4 in vitro or in vivo (all are present in the input library, except for one that is deleted in our strain). Clearly, there are large discrepancies between site occupancies in vivo and in vitro and between predicted site occupancies based on the AP-1 motif.
To begin to address these discrepancies, we analyzed the motifs present in the various classes of site represented in the Venn diagram using MEME (Fig. 3B; Bailey et al. 2009). The 341 peaks common to ChIP-seq and G-SELEX containing the AP-1 motif return the AP-1 motif, as expected, but a strong preference for a 5′-A flanking the AP-1 motif is also indicated for both sets of data (ATGA[C/G]TCA). The 115 ChIP-seq peaks that overlap with G-SELEX peaks but lack an exact AP-1 motif yield a degenerate AP-1 motif, similar to that reported previously (Rawal et al. 2018), as do the corresponding G-SELEX peaks (note that the motif for the G-SELEX peaks is on the opposite strand). The 60 ChIP-seq peaks that are not detected by G-SELEX give a poly(A) motif, also reported previously (Rawal et al. 2018). There are 30 ChIP-seq peaks not detected by G-SELEX, which yield an AP-1 site flanked on both sides by a pyrimidine base: (C/T)TGA(G/C)TCA(C/T). The 1622 G-SELEX sites lacking the AP-1 motif and bound only in vitro return an AP-1 half-site motif with an additional 5′-A (ATGAC). The 263 G-SELEX peaks containing a motif, but not observed by ChIP-seq, yield an AP-1 motif with a preference for a 3′-purine base (TGA[G/C]TCA[A/G]). Finally, the MEME motif for the unbound sites differs from the AP-1 site in that it specifies an extra 5′-T, suggesting that a 5′-T interferes with Gcn4 binding (opposite to the preference for 5′-A at bound sites). In summary, only 341 AP-1 motifs out of a total of 1078 exact matches (32%) are bound in vivo and in vitro. Of the remaining 737 AP-1 motifs, 30 are bound in vivo but not in vitro, 263 are bound in vitro but not in vivo, and 426 are not bound in vitro or in vivo. Clearly, the AP-1 motif is not always bound by Gcn4, even in vitro.
To gain further insight, we calculated the occupancies for each set of peaks within the Venn diagram (Fig. 3B), presented as box plots (Fig. 3C,D). In the case of the ChIP-seq data (Fig. 3C), the 341 peaks also detected by G-SELEX and containing an exact AP-1 motif have a much higher average site occupancy than the peaks in the other categories. The analysis of common peaks with a degenerate AP-1 motif indicates that these half-sites are generally bound much more weakly, as would be expected, although there are some exceptions. The ChIP-seq-only peaks with a motif indicate a somewhat weaker average occupancy, whereas the ChIP-seq peaks giving the poly(A) motif are not much above background (the genomic average is set at 1). In the case of the G-SELEX data (Fig. 3D), data for the common peaks also indicate much higher Gcn4 occupancies in vitro than the peaks in the other categories, including the large number of AP-1 half-sites, again with a few exceptions. Thus, the large majority (69%) of G-SELEX peaks represent relatively weakly bound half-sites. We also note that 18 ChIP and 12 G-SELEX peaks contain two motifs; they have higher occupancies than the large majority of peaks with only one motif (Supplemental Fig. S3). In summary, these data suggest that Gcn4 binds a subset of AP-1 motifs with much higher affinity in vivo and in vitro.
Half-site binding is generally weak
We performed a detailed analysis of half-sites (Supplemental Table S1). There are 25,838 half-site motifs in the genome (exact matches), as defined by the MEME motif in Figure 3B (ATGAC). Excluding the 291 half-sites belonging to full sites reduces the total to 25,547. After excluding half-sites present in peaks containing full sites and those in the rDNA locus, we determined that ∼12% are bound in vitro (G-SELEX) and ∼3% in vivo (ChIP-seq). As expected, bound half-sites have higher occupancy than unbound sites both in vitro and in vivo because peaks were identified using a cut-off value (Supplemental Fig. S4A). MEME analysis for unbound and bound half-sites in vitro and in vivo returned the original half-site motif in all four cases, with no difference in flanking base pairs (Supplemental Fig. S4A). Because this observation does not explain why some half-sites are bound and others are not, we tested whether the number of half-sites in each peak is important, given that multiple half-sites could each contribute some weak binding (Supplemental Fig. S4B). G-SELEX peaks with more than three half-sites have higher occupancy than those with fewer half-sites, but this effect is not observed in the ChIP-seq data (although only seven ChIP peaks have more than three half-sites). The number of half-sites is therefore a contributory factor. It is important to note that even the bound half-site peaks are generally very weak compared with full site peaks (cf. Fig. 3C,D).
The high-affinity Gcn4 binding site is RTGACTCAY
To explain these data, we reasoned that a high-affinity Gcn4 binding site must contain more base pairs than just the AP-1 motif. Accordingly, we analyzed the contribution of the 5′ and 3′ flanking base pairs to Gcn4 binding. We calculated the normalized peak coverage for each AP-1 motif in the G-SELEX data. The AP-1 motifs were then subdivided into 16 groups according to their 5′ and 3′ flanking nucleotides (5′-mTGACTCAn-3′). Box plots showing the distribution of Gcn4 occupancies for each class of motif indicate that motifs with m = A or G and n = T or C are generally much more strongly bound than the others (Fig. 4A; note log scale). We refer to these motifs as “AC”, “AT”, “GC”, and “GT”, named for their flanking bases. Therefore, the Gcn4 binding site appears to be RTGACTCAY, where R is a purine base and Y is a pyrimidine base. This conclusion was confirmed by regrouping the motifs into the three possible types, termed RY, RR/YY (which are equivalent when both strands are considered), and YR (Fig. 4B). RY motifs are much more strongly bound than RR/YY motifs, which are more strongly bound than YR motifs, with some overlap (Fig. 4B). Analysis of the unbound AP-1 motifs (Fig. 3B) reveals that 67% are YR motifs; the rest are RR/YY. Unbound motifs include 78% of all YR motifs, including 93% of TA, 91% of TG, 79% of CA, and 43% of CG motifs, consistent with the MEME motif (Fig. 3B), indicating that a 5′-T inhibits Gcn4 binding.
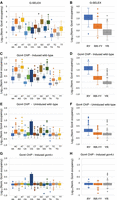
The high-affinity Gcn4 binding site is RTGACTCAY: Comparison of AP-1 site binding as a function of flanking base pairs. All 1078 AP-1 sites (TGACTCA) were subdivided and named according to their flanking base pairs (e.g., “AT” indicates the motif ATGACTCAT). Data were normalized to the genomic average (=1) and decompressed using a log scale. (A) G-SELEX data for each of the 16 possible types of AP-1 site. (B) G-SELEX data for the three possible motif types with R = A or G and Y = C or T. (C,D) Gcn4 ChIP-seq data for induced wild-type cells. (E,F) Gcn4 ChIP-seq data for uninduced wild-type cells. (G,H) Gcn4 ChIP-seq data for induced gcn4Δ cells.
The same analysis was performed on the ChIP-seq peaks containing an AP-1 motif, with similar results (Fig. 4C,D). In induced cells, Gcn4 occupancy is much higher at RY sites than at other sites, except for “CG” sites (the YR motif CTGACTCAG), which apparently have intermediate occupancy. In uninduced cells, the same qualitative binding pattern is observed as in induced cells, but the overall binding is much lower (Fig. 4E,F; note the same y-axis scale as induced cells in Fig. 4C), except for the CG sites, which remain prominent. The signal for CG sites is most likely an artifact, because these sites give a relatively strong ChIP-seq signal at CG motifs even in cells lacking Gcn4 (gcn4Δ) (Fig. 4G,H). They correspond to 31 CG motifs located in Ty1 transposable element repeats which are distributed around the genome. Regrouping the motifs as RY, RR/YY, and YR shows that RY sites are much more strongly bound in induced cells than are the others, with some exceptions (Fig. 4D). In addition, RR/YY sites are more strongly bound than YR sites (Fig. 4D). Clearly, the high-affinity Gcn4 binding site is RTGACTCAY in vitro and in vivo.
The distribution of RY, RR/YY, and YR motifs encoded in the yeast genome is biased against RY motifs and in favor of YR motifs: 166 RY (15%), 546 RR/YY (51%), and 366 YR (34%); the expected distribution is 25%, 50%, and 25%, respectively. Removal of the 31 Ty1 motifs from the calculation does not account for this bias. Because RY sites are bound much more tightly than YR sites, the bias against RY motifs is likely to be biologically important. In fact, all but one of the 166 RY motifs in the yeast genome (99%) are present in the 341 peaks common to both ChIP-seq and G-SELEX, and all (100%) are bound in vivo. The exception (a site on Chromosome 12 at nt 676941) appears to have mutated from ATGACTCAC in the reference genome to AcGACTCAC in the strain used for G-SELEX, according to our preliminary sequencing data. The common peaks also contain 183 RR/YY motifs (34%) and 11 YR motifs (3%). We note that 18 of the 341 common ChIP-seq peaks contain two AP-1 motifs; two peaks contain a second RY motif, 10 peaks contain an additional RR/YY motif, and six peaks contain both a YR motif and an RY or an RR/YY motif, suggesting that the YR motif may not be bound in these cases, potentially reducing the number of YR motifs bound in common peaks to just five. We conclude that the high-affinity Gcn4 site is RTGACTCAY and that all 166 genomic copies of this site are bound in vivo. The remaining sites bound in vivo are lower occupancy sites corresponding almost entirely to RR/YY motifs. Peak by peak summaries of the ChIP-seq data and the G-SELEX data, as well as the data for the 1078 AP-1 motifs, are provided in Supplemental Tables S2–S4, respectively.
It is worth noting that, of the 13 “UC” genes in which Gcn4 binds inside the ORF and for which the Gcn4 site was previously confirmed to be functional in vivo by mutation (Rawal et al. 2018), eight contain RY motifs (POS5, SOL1, SPO21, HIS2, ROT2, VPS41, HMG2, and YFR045W) and two have RR/YY motifs (TYR1 and HOS4). The exceptions are GYP8 (which has a weak RR/YY peak in the downstream CAF16 promoter), BIO4 (which has a YR motif associated with a weak Gcn4 peak [Rawal et al. 2018]), and COG1. The latter gene has an ORF peak observed in vitro and in vivo associated with an RY motif containing one mismatch (ATGACTAAT), mutation of which affects induction of the downstream SDT1 gene (Rawal et al. 2018); SDT1 contains an RR/YY motif associated with a second peak, also observed in vitro and in vivo (Supplemental Fig. S2). It is conceivable that these two sites, ∼400 bp apart, are both involved in induction of SDT1.
Conversion of native RY sites to YR sites substantially weakens Gcn4 binding in vitro
Gcn4 peak height is not necessarily an accurate measure of relative affinity. It is a measure of relative occupancy, with the caveats that PCR artifacts may distort the data to some extent, and that, in the case of G-SELEX, differences may be amplified by each round of selection. In addition, in vivo, Gcn4 binding may be blocked by other DNA-binding proteins, such as histones. That these caveats are not trivial is suggested by the fairly wide range of occupancy observed for each set of identical AP-1 motifs (Fig. 4A,C). On the other hand, the difference in occupancy levels between the RY motifs and all others is obvious (Fig. 4B,D).
As a direct test to determine whether a 5′ purine and 3′ pyrimidine make substantial contributions to Gcn4 binding affinity, we performed EMSA experiments using probes generated from the ARO1 and STP2 binding sites (Supplemental Fig. S2). Gcn4 binds to the RY sites in the ARO1 promoter and the STP2 ORF with similar affinities, with Kd ∼ 80 nM in both cases (Fig. 5; Supplemental Fig. S1), similar to that for the RY motif in the ARG1 promoter (Fig. 1C). Mutation of the RY motif to a YR motif increases Kd to ∼320 nM in both cases, corresponding to a fourfold decrease in binding affinity (Fig. 5; Supplemental Fig. S1). It should be noted that the measured value of Kd can depend on experimental conditions, such as ionic strength, competitor concentration, probe length, and electrophoresis conditions. We conclude that Gcn4 binds substantially more tightly to RY motifs than to YR motifs.
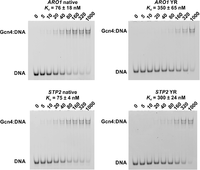
Electrophoretic mobility shift assays for Gcn4 binding site probes show weakened binding when the native RY site is converted to a YR site. Probes (30 bp with 3′-6-FAM labels) containing the RY site in the ARO1 promoter or the RY site in the STP2 ORF (left panels). The right panels show ARO1 and STP2 probes in which the native RY motif is converted to a YR motif (see Supplemental Table S5 for the sequences). DNA at a final concentration of 10 nM was incubated with 500 ng of unlabeled poly(dA/dT) as competitor and increasing Gcn4 concentrations (shown in nM above each lane).
Gcn4 binding in vitro correlates quantitatively with binding in vivo
We have shown that the majority (84%) of ChIP-seq peaks are also detected by G-SELEX (456 of 546 peaks) and that those common peaks containing an AP-1 motif (341 peaks = 62%) are much more prominent than the others (Fig. 3B,C). This qualitative correlation in peak overlap suggests that there might be a quantitative correlation between Gcn4 binding in vivo and Gcn4 binding in vitro. In fact, a good correlation is revealed by a scatterplot of the normalized G-SELEX peak height against the normalized induced Gcn4 ChIP-seq peak height for each of the 1078 AP-1 motifs (R = 0.43) (Fig. 6A). Although this correlation is far from perfect, it indicates that motif binding affinity is a major determinant of Gcn4 occupancy in vivo. The residual variance may be accounted for by differential amplification (see above) and by chromatin (see below). The distributions of RY, RR/YY, and YR motifs in the scatterplot are broadly as expected from their relative affinities for Gcn4 (Fig. 6A).
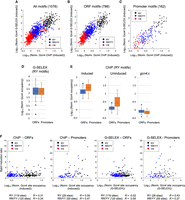
Gcn4 motif binding in vivo correlates with binding in vitro and with gene induction levels. (A) Correlation between Gcn4 binding in vivo and in vitro for all AP-1 motifs (black circles, RY motifs; blue circles, RR/YY motifs; red circles, YR motifs). A log–log plot was used to decompress the data (values of R are for linear data). (B) AP-1 motifs located in ORFs. (C) AP-1 motifs located in promoters (defined as located within 600 bp upstream of the transcription start site [TSS] unless present in an ORF). (D) RY motifs in ORFs and promoters are occupied at similar levels in vitro (G-SELEX data). (E) Gcn4 binds to RY motifs in promoters at higher levels than to RY motifs in ORFs (ChIP-seq data for induced and uninduced wild-type cells and for induced gcn4Δ cells). (F) Gcn4 motif binding in vivo (induced wild-type cells) correlates with gene induction (defined by the ratio of bound Pol II [Rpb3 subunit] in induced and uninduced cells measured by ChIP-seq), whereas Gcn4 binding at promoter sites in vitro correlates with induction ratio, but Gcn4 binding at ORF sites does not.
Gcn4 occupancy is higher at promoter RY sites in vivo but not in vitro
We showed previously that, in vivo, Gcn4 consensus sites in ORFs average lower Gcn4 occupancy than consensus sites in promoters, consistent with reduced accessibility in ORFs due to nucleosomes (Rawal et al. 2018). It seemed likely that this would also be true of AP-1 motifs. We divided up the 1078 motifs according to their genomic locations: 788 (73%) are found within ORFs (including introns), 162 (15%) are in promoters, 37 (3%) in Ty1 or Ty4 transposable elements, and one in each of the two rDNA repeats in the reference genome. The remaining 89 sites (8%) are located in long intergenic regions (>600 bp upstream of a start codon and not in another defined element) or between the stop codons of convergent genes. The close proximity of yeast genes means that they account for a large fraction of the genome (73%), indicating that motifs are not enriched in, or depleted from, ORFs.
We examined motif distributions with respect to ORFs and promoters. Of the 788 motifs located inside ORFs, there are 121 RY (15%), 405 RR/YY (51%), and 262 YR (33%)—very similar to the genomic fractions of these motifs (15%, 51%, and 34%, respectively). Of the 162 motifs inside promoters, 29 are RY (18%), 95 are RR/YY (59%), and 38 are YR (23%), indicating that the high-affinity RY motifs are not enriched in promoters. On the other hand, RR/YY motifs are overrepresented in promoters at the expense of YR motifs. Separate scatterplots for motifs in ORFs (Fig. 6B) and promoters (Fig. 6C) show that the correlations between Gcn4 occupancy in vivo and in vitro (R = 0.54 and 0.59, respectively) are better than for all motifs (R = 0.43) (Fig. 6A). In all three cases, RY motifs are more strongly bound than RR/YY motifs, which are more strongly bound than YR motifs.
The promoters containing an RY motif include almost all of the genes encoding the enzymes for arginine biosynthesis, including ARG1, ARG3, ARG4, ARG7, ARG8, ORT1, and CPA2, as well as the CAN1 arginine permease; the exceptions are CPA1 and ARG5,6, which have RR/YY motifs, and ARG2, which has a YR motif in its ORF (Supplemental Fig. S5). Promoters containing an RY motif also include other very strongly induced genes such as HIS1, HIS4, ARO1, GGC1, HOM3, and ILV6 (Natarajan et al. 2001).
We compared Gcn4 binding to RY motifs in ORFs and promoters in vitro and in vivo quantitatively using box plots. In vitro, similar ranges of Gcn4 binding to ORF motifs and promoter motifs are observed (Fig. 6D), whereas, in vivo, Gcn4 binds more strongly to promoter sites than to ORF sites, on average (Fig. 6E). Quantitatively, the difference between the means or medians is approximately twofold, lower than expected given that the average nucleosome occupancy of ORFs is ∼75% (Chereji et al. 2019; Oberbeckmann et al. 2019) which formally predicts an approximately fourfold reduction in accessibility, although there is some variation due to nucleosome phasing and there is a tendency for Gcn4 to bind in linkers (Rawal et al. 2018). This observation is consistent with the proposal that Gcn4 sites in ORFs are partly blocked by nucleosomes in vivo. On the other hand, ORF sites in genes whose full-length transcripts are induced actually have somewhat higher Gcn4 occupancies than induced genes with Gcn4 sites in promoters (Rawal et al. 2018). Most importantly, it is clear that ORF sites are not completely blocked by chromatin.
To test whether AP-1 motifs are differentially located relative to nucleosomes, we divided the motifs into RY, RR/YY, and YR types and sorted each set by Gcn4 occupancy in vivo or in vitro (Supplemental Fig. S6A). Heat maps showing nucleosome occupancy in induced cells (Cole et al. 2014) were constructed for each motif type, aligned on the motif, and sorted by Gcn4 occupancy measured by ChIP or by G-SELEX (Supplemental Fig. S6B). In vivo, the most highly occupied RY and RR/YY motifs are located in NDRs; this effect is absent in vitro because there are no nucleosomes, resulting in a different motif sort order. Mean nucleosome occupancy plots for each motif type show that nucleosome occupancy is slightly below average around RY and RR/YY motifs, but not YR motifs (Supplemental Fig. S6C). They also show that there is no obvious tendency for enrichment or depletion of nucleosomes in the immediate vicinity of Gcn4 motifs and that there is no phasing, suggesting that motifs are randomly located relative to nucleosome arrays. We note that superimposition of Gcn4 motif binding on local nucleosome maps implies that Gcn4 may be bound to a site inside a nucleosome; however, the motif could be in a linker if the nucleosome is not in its average position (Chereji et al. 2019).
Gcn4 occupancy at promoters correlates with increases in gene expression
Previously, we calculated a gene induction ratio by dividing the average RNA polymerase II occupancy in induced cells by that in uninduced cells (measured by ChIP-seq for the Rpb3 subunit), and then linking each ChIP-seq peak with the nearest gene (Rawal et al. 2018). To determine if there is a relationship between Gcn4 site occupancy and Pol II induction ratio, we calculated the occupancy of the Gcn4 motif in vivo for each of the 371 ChIP-seq peaks with an exact match to the AP-1 motif and plotted it against the induction ratio, obtaining good correlations for promoter RY and RR/YY sites and weaker correlations for ORF RY and RR/YY sites (Fig. 6F); RY sites did not always give higher induction ratios than RR/YY sites (Fig. 6F), suggesting that Gcn4 occupancy is not the only important factor determining transcriptional output. Similar plots using Gcn4 occupancy in vitro rather than in vivo indicate a good correlation for RY and RR/YY sites in promoters but no correlation for either type of site in ORFs (Fig. 6F). Some ORF sites activate cryptic transcription rather than transcription from the nearest conventional TSS (Rawal et al. 2018), perhaps accounting for the weaker ORF site correlation, because the induction ratio is calculated for the entire gene and includes both sense and antisense transcription, which cannot be distinguished by Pol II ChIP. The lack of correlation between ORF site occupancy in vitro and induction ratio may be partly due to the presence of nucleosomes in vivo, which only affect Gcn4 occupancy in vivo (Fig. 6D,E), whereas promoters are nucleosome-depleted and may behave more like free DNA. We conclude that the Gcn4 occupancy of promoter motifs in vitro and in vivo is a moderately good predictor of the associated increase in gene expression, but that of ORF motifs is not, at least in vitro.
Discussion
The high-affinity Gcn4 binding site is RTGACTCAY
Prior experimental and in silico studies have generally converged upon the AP-1 consensus site as the correct binding site for Gcn4 (Harbison et al. 2004; Zhu et al. 2009; Rawal et al. 2018). However, they failed to address the excess of potential AP-1 sites that did not appear to be bound. For an in vivo experiment, this could be explained away by the limitations of ChIP-seq, or by a lack of sufficient expression to bind every real site, or by nucleosome-limited accessibility. However, in an in vitro method, there are no such limitations. That the G-SELEX experiments confirmed the binding of Gcn4 to the majority of the same sites as a ChIP-seq experiment was welcome confirmation, but the lack of binding to two-thirds or more of predicted AP-1 sites called into question the validity of the AP-1 motif as the sole determinant of Gcn4 binding. By taking a deeper look at the data, we determined that Gcn4 has a strong preference for a 5′ purine and a 3′ pyrimidine flanking the AP-1 site. Essentially the same consensus site was identified by measuring Gcn4 binding to microarrays of synthetic oligonucleotides (Zhu et al. 2009), although other approaches were less accurate (Harbison et al. 2004; MacIsaac et al. 2006). Indeed, this can be seen even in the motif for half-site binding, where the half-site is flanked by a 5′ adenine that scored relatively strongly according to MEME analysis.
The differences between RY sites and all other possible combinations of flanking bases strongly indicate that the high-affinity Gcn4 binding site is RTGACTCAY, as identified previously (Oliphant et al. 1989). We note that separate MEME analysis of RY, RR/YY, and YR motif flanking sequences did not identify any additional bases. An RY site (ATGACTCAT) was used to obtain the first crystal structure of the Gcn4 DNA-binding domain complexed with DNA (Ellenberger et al. 1992). Gcn4 makes specific contacts with the 7 bp comprising the AP-1 motif (TGACTCA), but the authors also noted that a water molecule appears to form a bridge between Gcn4-N235 and the flanking adenine base (i.e., the R in RY), although they considered the assignment tentative because structure resolution was limited (Ellenberger et al. 1992). Gcn4 may also discern differences in DNA conformation among the RY, RR/YY, and YR motifs.
The Gcn4 site in the HIS3 promoter is an interesting case. Pioneering work focused on the HIS3 promoter (Hill et al. 1986), showing that Gcn4 binds to the sequence ATGACTCTT, which differs from the high-affinity Gcn4 binding site identified here by one mismatch within the AP-1 site (underlined). Almost all mutations in this site result in poor growth under amino acid starvation conditions, with little or no HIS3 induction (Hill et al. 1986). However, an exception (the “optimal” binding site), in which the penultimate T is mutated to A (i.e., conversion to the high-affinity site), results in greater resistance to amino acid starvation, increased HIS3 induction relative to wild type, and stronger binding by Gcn4 in vitro (Hill et al. 1986). A weak peak detected at the HIS3 promoter by both G-SELEX and ChIP-seq is consistent with a low-affinity site (Supplemental Fig. S2), but there is good evidence for its biological importance.
Possible explanations for Gcn4 half-site binding
The detection of half-site binding by G-SELEX confirms prior studies that show Gcn4 binds to the half-site sequence, ATGAC (Sellers et al. 1990; Rawal et al. 2018). However, that we detect so many half-sites with our assay is intriguing. One potential explanation is that the partially unstructured DNA-binding domain of one Gcn4 monomer within the dimer folds correctly when it comes into contact with half-site DNA, while the DNA-binding domain of the other monomer does not, resulting in a Gcn4-DNA complex that is stable enough to be detected by G-SELEX. This aspect of Gcn4 binding has been manipulated to create chimeric DNA-binding proteins (Wolfe et al. 2003; Rodríguez et al. 2016). Half-site binding is also detected in vivo (Rawal et al. 2018), though not to the extent seen with G-SELEX. However, the much stronger peaks at full sites in vivo (Fig. 3B) and in vitro (Fig. 3C) clearly indicate that half-site binding is likely to be more transient and that, given sufficient time, Gcn4 will preferentially bind to full sites instead. In fact, this was a part of the rationale for overnight incubation; pilot experiments with shorter incubations (2 h) did not generate the quality of data seen with overnight incubation, suggesting that Gcn4 requires more time to sample the pool of DNA fragments to locate preferred binding sites (i.e., to reach equilibrium).
Utility of the G-SELEX method
Our new method, G-SELEX, combined with published ChIP-seq data, and using MEME to find the motif, allowed us to identify the high-affinity Gcn4 binding site. G-SELEX provides quantitative data in the absence of confounding factors such as the binding of other proteins, including histones. G-SELEX possesses several characteristics that make it useful in determining the binding site of a given protein of interest: (1) The assay requires only a small amount of protein. (2) The DNA used for the experiment is derived from the organism of interest, so any peaks found can be mapped directly to binding sites that should be biologically relevant. (3) The ionic strength of the buffer is in the physiological range, which likely contributes to the low background and high specificity of the assay. The method includes a very short wash step of 90 sec (30 sec after mixing, 60 sec of returning beads to a magnetic rack), which also improves specificity and helps eliminate false positive peaks. The number of reads necessary to obtain useful data is also less than required by other sequencing-based methods; G-SELEX yielded reliable data in our replicates with as few as six million paired-end reads for the S. cerevisiae genome, which should scale accordingly for higher organisms. A potential—though relatively unlikely—drawback to the technique would be an instance where the preferred binding site of a protein of interest is present in the adaptor sequence, which would prevent selection. However, this may be worked around by using a different adaptor and primers.
Nucleosomes do not block high-affinity Gcn4 binding
It may be possible to refine consensus binding sites using existing ChIP-seq data by making full use of the quantitative aspects of the data. Currently, once a threshold is set to identify the peaks, all peaks are treated equally, resulting in an averaged consensus motif that includes many relatively weak peaks (potentially including half-sites) that may or may not be biologically important. In many cases, a weak consensus site occurs so often in the genome that it has little predictive value. Consequently, analysis is often restricted to regulatory regions, which has been justified by arguing that nucleosomes block other potential binding sites. However, our data do not support this assumption. Although RY motifs in ORFs are less occupied in vivo than in vitro, unlike RY motifs in promoters, clearly they are not completely blocked by nucleosomes in vivo. Some have been shown to be biologically important and, as noted above, Gcn4 occupancies at ORF sites in induced genes are even higher than at sites in the promoters of induced genes (Rawal et al. 2018). Thus, re-analysis of ChIP-seq data for other proteins using only the strongest peaks to predict a more accurate motif is likely to be worthwhile.
Biological implications
Recognition by Gcn4 of the additional R and Y base pairs reduces the number of high-affinity binding sites specified in the yeast genome to the point where it would be reasonable to speculate that they might all be biologically important. There are 1078 AP-1 sites in the yeast genome, but only 166 RY sites. All RY sites are occupied by Gcn4 in vivo (and all but one in vitro). Although there are many additional occupied sites in vivo (546 altogether), they are mostly low-occupancy peaks, whereas almost all of the strong peaks correspond to RY motifs.
Our data indicate that there are four classes of Gcn4 binding site, which are, in order of relative affinity: RY, RR/YY, YR, and half-sites. All of these sites have higher affinity for Gcn4 than unrelated DNA, and examples of all classes are observed in vivo as well as in vitro. However, there is a large difference in occupancy between RY sites and the rest. Therefore, we propose that the high-affinity RY sites mediate the primary response to amino acid starvation through Gcn4. We also predict successively weaker responses mediated by RR/YY and perhaps by YR motifs and half-sites, although a functional role for half-sites seems unlikely. The extent to which these binding sites are occupied would be determined by the cellular Gcn4 concentration which, in turn, determines the biological response. Thus, the response of each gene may be determined by the type of Gcn4 motif involved in activation. We propose that the yeast cell tunes its physiological response to amino acid starvation by regulating the Gcn4 concentration, such that increasing Gcn4 levels results first in the occupation of RY sites and then RR/YY sites, progressively increasing the number of genes that are induced, and to different levels.
Methods
Recombinant Gcn4 expression and purification
The GCN4 open reading frame was obtained by PCR using yeast genomic DNA as template and primers 1 and 2 (Supplemental Table S5), digested with NdeI and XhoI, and inserted into the pET21b(+) expression vector cut with the same enzymes. The resulting plasmid (p789) was cut with XhoI and ligated to a synthetic double-stranded oligonucleotide with XhoI sticky ends (oligonucleotides 3 and 4) (Supplemental Table S5) to insert a C-terminal 6xHis tag fused to a FLAG tag prior to the stop codon. The insert sequence of this plasmid (p790) was confirmed. Gcn4-6xHis-FLAG was expressed in Escherichia coli BL21(DE3) cells (Calbiochem) grown in Terrific Broth at 37°C to A600 = 1.0–1.5. Cultures were induced with 0.25 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG) and shifted to 30°C for 4 h. Gcn4-6xHis-FLAG was purified essentially as described previously (Gartenberg et al. 1990), with an initial purification step using Ni-NTA agarose resin (Thermo Fisher Scientific R901-01). Fractions were eluted in 10 mM sodium phosphate buffer, pH 8.0, 300 mM NaCl with 150–250 mM imidazole. Fractions containing Gcn4-6xHis-FLAG were pooled, precipitated with 25% (v/v) ammonium sulfate, incubated overnight with rotation at 4°C, and then spun down for 30 min at 3000g. The pellets were resuspended and exchanged into SP Buffer A (25 mM HEPES pH 7.3, 75 mM NaCl, 0.2 mM EDTA, 1 mM DTT, 1% glycerol) using Amicon Ultra centrifugal filters (EMD Millipore; MWCO = 10,000) until the pellet was fully dissolved in <5 mL buffer. The solution was loaded onto a 5-mL HiTrap SP-HP column (GE Healthcare) and eluted with a 75 mM–1 M NaCl gradient in SP buffer (Gcn4 elutes between 450 and 600 mM NaCl). Fractions containing Gcn4 were exchanged into Mono-Q Buffer A (50 mM Tris HCl pH 8, 75 mM NaCl, 0.2 mM EDTA, 1 mM DTT, 1% glycerol) and concentrated to 2 mL and below 100 mM NaCl using Amicon filters. The solution was loaded onto a 2-mL Mono-Q column (Pharmacia) and eluted with a 75 mM–1 M NaCl gradient in SP buffer (Gcn4 elutes between 250 and 350 mM NaCl). The buffer in Gcn4-containing fractions was exchanged into Mono-Q Buffer A to reduce the NaCl to ∼200 mM NaCl using Amicon filters. The sole peak obtained from Mono-Q was judged to be pure Gcn4-6xHis-FLAG (>95%) by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), although some degradation products were apparent, as observed previously (Gartenberg et al. 1990). The Gcn4 concentration was determined to be 18.5 μM by direct A280 measurement using a calculated extinction coefficient of ε = 12950, and MW = 33,600.
Electrophoretic mobility shift assays
Oligonucleotides used for EMSAs were labeled with a 3′ 6-FAM on one strand and annealed by heating to 90°C for 5 min followed by slow cooling. A double-stranded 24-mer was used for the ARG1 promoter probe; all other probes were 30-mers (Supplemental Table S5). EMSAs using purified Gcn4-6xHis-FLAG were performed essentially as previously described (Coey et al. 2016), with Gcn4 concentrations ranging from 5 nM to 1 μM. Gcn4 was equilibrated for 30 min with 10 nM probe in 10 mM TrisHCl pH 8.0, 50 mM NaCl, 4 mM MgCl2, 0.2 mg/mL BSA, 1 mM DTT, 5% glycerol before loading 10 μL per lane in a 6% polyacrylamide gel (Invitrogen) run at 50 V for 90 min at 4°C in 0.5× TBE buffer. Gels were imaged on a Typhoon 9400 variable mode imager (GE Healthcare) and analyzed using ImageQuant software (GE Healthcare). Two replicate EMSAs were performed for each probe.
Preparation of genomic DNA for G-SELEX
Genomic DNA from YDC111 (Kim et al. 2006) was sonicated and purified as described (Cole et al. 2014). The DNA was dissolved in 10 mM TrisHCl pH 8.0, 1 mM EDTA, 0.02 mg/ml RNase (Qiagen) and incubated for 2 h at 37°C to digest residual RNA. DNA fragments < 200 bp were purified from a 2% agarose gel using either a Freeze ‘N Squeeze column (Bio-Rad) or a gel purification column (Qiagen). Genomic DNA fragments were ligated to the Illumina paired-end adaptor as described (Cole et al. 2012). Adaptor-ligated DNA was amplified by PCR using Phusion Master Mix (New England Biolabs M0531) with the Illumina PE 1.0 and 2.0 primers (Cole et al. 2012) and purified using a Qiagen PCR column. DNA concentration was determined by measuring A260.
G-SELEX procedure
HisPur Ni-NTA magnetic beads (Thermo Fisher Scientific 88831; 100 μL at 12.5 mg/mL = 125 μg beads) were washed three times in 250 μL of HEPES-buffered saline (HBS: 10 mM HEPES 7.3, 150 mM NaCl, plus cOmplete EDTA-free protease inhibitor cocktail [Roche 11873580001]). The beads were resuspended in 100 μL HBS, combined with purified Gcn4-6xHis-FLAG (40 μL at 18.5 μM), and mixed by rotation for 2 h at 4°C. The beads were washed three times with 1 mL HBS before resuspension in 200 μL 0.5× HBS, 50% glycerol for long-term storage at −20°C. We confirmed that Gcn4-6xHis-FLAG was bound to the beads by SDS-PAGE analysis of the beads and wash supernatants. Approximately 4 μg of amplified genomic DNA were added to a microcentrifuge tube containing a final volume of 200 μL G-SELEX buffer (10 mM HEPES pH 7.3, 100 mM NaCl, 5 mM MgCl2, cOmplete EDTA-free protease inhibitor cocktail. Gcn4-loaded Ni-NTA beads (5 μL; ∼600 ng Gcn4) were added and mixed with the DNA overnight by rotation at 4°C. The Gcn4 concentration was ∼65 nM monomer, the input DNA concentration was ∼15 μM DNA fragments, and there were ∼10 AP-1 site-containing DNA fragments per Gcn4 monomer. After incubation, the tube was gently centrifuged (<2000 rpm) for 1 min. Beads were collected by placing the tube on a magnetic rack for 2 min and the supernatant was removed. The beads were resuspended in 50 μL G-SELEX buffer, incubated for an additional 30 sec, and then returned to the magnetic rack for 1 min before the supernatant was removed. Bound DNA was eluted from the beads by resuspension in 50 μL NEB Buffer 2 containing 1% SDS. DNA was purified from the supernatant, washed, and eluted using Qiagen PCR columns; concentrations were measured by A260. Eluted DNA (50 ng) was amplified using the NEB universal (E7336A) and Illumina PE 2.0 primers with 11 PCR cycles to obtain ∼3.5 μg of DNA for the next G-SELEX round (Fig. 1A). After three G-SELEX rounds, the final eluted DNA was amplified for seven cycles with primers from the NEBNext Multiplex Oligos for Illumina kit (NEB E7335/E7500) and purified using Ampure XP beads (1:1 ratio). These indexed paired-end DNA libraries were sequenced with an Illumina NextSeq 500.
G-SELEX data analysis
Paired reads were mapped to the sacCer3 version of the S. cerevisiae genome using the alignment software Bowtie 2 (Langmead and Salzberg 2012). Occupancy profiles were obtained from the BAM files and normalized as described previously (Rawal et al. 2018). MACS2 (https://pypi.org/project/MACS2/) (Zhang et al. 2008) was utilized to detect Gcn4 peaks in the three replicates. Significant peaks were identified using a Q-value threshold of 10−5 and YDC111 input DNA as a control. A list of the 2359 peaks present in all three data sets was obtained by intersecting the .NarrowPeak BED files generated by MACS2 (using the -wa -u commands in BEDTools [Quinlan and Hall 2010]). G-SELEX Gcn4 occupancy (coverage) data for the three replicate experiments were combined and normalized to the genomic average. Custom MATLAB scripts were used to compare the G-SELEX data with the curated Gcn4 ChIP-seq data (Rawal et al. 2018) and to identify peaks containing AP-1 sites (Supplemental Code). The results are collated in Supplemental Tables S1–S4. Consensus motifs within peak sequences were identified using MEME (https://meme-suite.org/) (Bailey et al. 2009), using a minimum width of 3 bp, a maximum width of 50 bp, and specifying one site per sequence (“oops” setting).
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE180114.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Alan Hinnebusch for helpful comments on the manuscript, Răzvan Chereji for help with the bioinformatics, and Yeonjung Kim for construction of p789. This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health (NIH). This research was supported by the Intramural Research Program of the NIH (NICHD).
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276080.121.
- Received August 5, 2021.
- Accepted December 15, 2021.
This is a work of the US Government.








