
Limited allele-specific gene expression in highly polyploid sugarcane
Abstract
Polyploidy is widespread in plants, allowing the different copies of genes to be expressed differently in a tissue-specific or developmentally specific way. This allele-specific expression (ASE) has been widely reported, but the proportion and nature of genes showing this characteristic have not been well defined. We now report an analysis of the frequency and patterns of ASE at the whole-genome level in the highly polyploid sugarcane genome. Very high depth whole-genome sequencing and RNA sequencing revealed strong correlations between allelic proportions in the genome and in expressed sequences. This level of sequencing allowed discrimination of each of the possible allele doses in this 12-ploid genome. Most genes were expressed in direct proportion to the frequency of the allele in the genome with examples of polymorphisms being found with every possible discrete level of dose from 1:11 for single-copy alleles to 12:0 for monomorphic sites. The rarer cases of ASE were more frequent in the expression of defense-response genes, as well as in some processes related to the biosynthesis of cell walls. ASE was more common in genes with variants that resulted in significant disruption of function. The low level of ASE may reflect the recent origin of polyploid hybrid sugarcane. Much of the ASE present can be attributed to strong selection for resistance to diseases in both nature and domestication.
Sugarcane has remarkable potential as a food and bioenergy crop being a C4 grass with high photosynthetic efficiency and biomass yield (Henry 2010), grown mainly for the dual purpose of producing sugar and ethanol. Currently, sugarcane comprises 86% of the sugar crops cultivated worldwide (OECD/FAO 2019). Biofuel obtained from sugarcane may have a key role in addressing climate change concerns (Goldemberg 2007; Souza et al. 2017). Continuing sugarcane breeding efforts are necessary to meet the increasing demands for sugar and ethanol, and a better understanding of molecular processes relevant to carbon partitioning will in turn guide molecular breeding of this crop (Wang et al. 2013).
Genomic analyses in sugarcane (Saccharum spp.) are limited by the unique combination of several layers of genomic complexity. All known Saccharum accessions are highly polyploid (D'Hont et al. 1998), with evidence of both allo- and autopolyploidy in the evolutionary history of the genus (Kim et al. 2014). Cultivated sugarcane clones are interspecific hybrids between Saccharum officinarum and Saccharum spontaneum, with varying contributions from each genome, multiple aneuploidy events, and recombination between the parental genomes (D'Hont et al. 1996). Hybrids show a variable number of chromosome copies per homology group (Grivet and Arruda 2002), with different total numbers of chromosomes in different genotypes (Piperidis et al. 2010), and large genomes (D'Hont and Glaszmann 2001). However, there is still a prevalence of 2n = 12x among hybrids (Le Cunff et al. 2008; Piperidis and D'Hont 2020). Pompidor et al. (2021) recently proposed the existence of three founding genomes in the genus Saccharum, two of them unevenly found in the sugar-rich S. officinarum, and the third being observed in the wild S. spontaneum.
Genomic resources are being developed for sugarcane (Kandel et al. 2018; Diniz et al. 2019). However, because of the amalgam of obstacles described above, research lags behind that for other major crops (Thirugnanasambandam et al. 2018). One challenge is the assembly of a genomic sequence that fully represents a complete hybrid genome. Despite the recent publication of multiple (partial) sugarcane genome sequences (Garsmeur et al. 2018; Zhang et al. 2018; Souza et al. 2019), sorghum is still a valuable genomic resource and has been extensively used as a genome reference for sugarcane (Grivet and Arruda 2002; Dillon et al. 2007). These two genomes are highly conserved, particularly in the transcribed regions (Wang et al. 2010). Using diploid sorghum as a reference sidesteps issues owing to the multiple alignment of reads to several chromosome copies.
Polyploidy causes a multitude of changes in cell structure and function, both in allo- (Comai 2000; Renny-Byfield and Wendel 2014) and autopolyploids (Yant and Bomblies 2015). It is usually accompanied by a so-called genomic shock and an increase in allelic diversity, which can consequently cause changes to gene expression profiles (Chen and Ni 2006; Feldman and Levy 2009; Baduel et al. 2018). Such alterations include allele-specific expression (ASE), a phenomenon whereby the different alleles of a given gene are unevenly expressed (Gaur et al. 2013). This excessive abundance of one allele stems from differential expression and/or degradation of mRNA molecules originated from different chromosome copies. Allelic imbalance can occur owing to variation in cis-acting regulatory regions, nonsense-mediated decay, and epigenetic imprinting, among other factors (Castel et al. 2015; Chen et al. 2018).
ASE has been evaluated in diploid and polyploid plants, such as Arabidopsis (Zhang and Borevitz 2009), maize (Springer and Stupar 2007), autotetraploid potato (Pham et al. 2017), and allohexaploid wheat (Powell et al. 2017). It has been associated with heterosis in hybrid rice, as a likely cause of dominance and overdominance effects (Shao et al. 2019) with evidence that genes with ASE were under selective pressure through breeding. Hybridization in maize is a cause of ASE, and although not frequent, it can arise from nonsyntenic genes and contribute to hybrid vigour (Baldauf et al. 2020). There is also evidence of conserved ASE between maize and rice orthologs, with accompanying evidence of positive selection (Waters et al. 2013). The joint availability of high-throughput RNA sequencing (RNA-seq) assays and methods for quantitative genotyping of single-nucleotide variants (SNVs) has allowed ASE to be studied in sugarcane (Vilela et al. 2017; Sforça et al. 2019; Cai et al. 2020; Correr et al. 2021). However, these previous investigations focused on a small number of genes, rely on limited genomic sampling, or suffer from potential sources of bias in the genotype calls. There are as yet no genome-wide analyses of ASE in sugarcane that make use of high-depth whole-genome sequencing (WGS) data. Hybridization events in sugarcane include the likely occurrence of allopolyploidy, interspecific hybridization during early breeding endeavors, and crosses between elite genotypes to obtain full-sib progenies, which are all possible causes of ASE. Artificial selection after interspecific hybridization, mainly for high sugar yield and disease resistance, has potentially affected the expression patterns of individual alleles in varying ways. Also, because many genes are involved in the accumulation of sucrose from top to bottom internodes in sugarcane (Whittaker and Botha 1997; Botha and Black 2000), it is conceivable that ASE may play a role in this carbon partitioning process.
We aimed to gauge the genome-wide extent of ASE in culms of elite sugarcane hybrids at different developmental stages and how structural and functional properties of genes affect the rate of ASE in this complex polyploid.
Results
Genome-wide study of polymorphisms in sugarcane hybrids by high sequencing depth
A set of seven sugarcane hybrids were selected from a larger set of 24 hybrids with extensive phenotypic variation (Supplemental Fig. S1) and used to study ASE. According to the phenotypic characterization detailed by Perlo et al. (2020), KQ228 was chosen to represent a high-yield (as measured in tons of cane per hectare), high early-season sugar genotype. In contrast, although SRA5 was also high yielding, it showed low sugar and high fiber content. Q155 and KQB09-20432 (KQB09) represented high-sugar and high-fiber genotypes, respectively. SRA1 showed both low sugar and low fiber content. Finally, MQ239 and Q186 had intermediate behavior with regard to both traits. The seven hybrids are representative of current elite breeding germplasm and are thus all related with multiple recurring common ancestors (Supplemental Fig. S2). We noted a peculiarity in the genetic background of KQB09. The paternal grandfather of KQB09, Hainan92-9, is a S. spontaneum. Although KQB09 is a direct descendant of KQ228, this unique background makes it the most genetically dissimilar among the studied hybrids.
The entire genomes of the seven hybrids were sequenced to identify polymorphic sites. KQ228 and SRA5 were sequenced at a depth of coverage of 100× per chromosome copy, or 1200× of the monoploid genome, considering a polyploid genome of 10 Gbp with 12 copies of each chromosome (D'Hont and Glaszmann 2001; Le Cunff et al. 2008). For the other genotypes, 240× of the monoploid genome was obtained (Supplemental Table S1). The high-quality reads were aligned against the sorghum genome reference with between 37.1% and 38.5% of the reads aligning. The specificity of alignment against genic regions was high, with little effect of filtering out low-quality alignments, in agreement with the fact that the genes are low-copy, conserved regions. The effective depth of coverage after alignment was close to the expected values of 1200× and 240×, depending on the genotype (Supplemental Fig. S3A). This approach was especially successful in unambiguously assigning reads to conserved single-copy grass genes (Supplemental Fig. S3B).
A total of 9,321,181 polymorphic sites were identified in the genic regions of the seven genotypes, of which 6,533,916 were biallelic SNVs and 192,157 were multiallelic SNVs. The majority of the remaining sites were insertions and deletions, indicating that the redundancy brought about by the multiple chromosome copies in sugarcane possibly allows for a substantial number of indels to be present. After filtering out low-quality polymorphisms 5,748,251 biallelic SNVs remained. Classifying these sites based on their predicted impact on gene structures resulted in the following distribution of predicted effects: 13,619,337 (79.82%) modifier effects, 1,799,543 (10.55%) SNVs with low impact, 1,608,014 (9.42%) with moderate effects, and 36,392 (0.21%) with high predicted impact. The number of effects is larger than the number of SNVs because each site can be annotated multiple times when in close proximity to multiple genes. For SNVs present within coding regions, 1,567,565 (48.96%) were annotated as silent, or synonymous substitutions; 1,611,587 (50.33%) caused amino acid changes (missense substitutions); and 22,716 (0.71%) were nonsense mutations. A total of 2,140,328 of the 5.75 million high-quality SNVs were polymorphic only in comparison to the sorghum reference but were fixed in these sugarcane genotypes. Because we can only assess ASE for heterozygous sites, these loci were not used for downstream analyses.
Discrete clusters of allele doses revealed by WGS and RNA-seq
The expression data set used corresponded to an RNA-seq assay of sugarcane stalks at five different developmental stages. For each of the genotypes, triplicate libraries were sequenced for internodes of different ages (collection time points C1 and C2) and varying maturity (internodes 5, 8, and 22; the latter also denoted by Ex-5; for details, see Methods). Sequencing generated from 56.0 million to 85.1 million raw reads per library, of which 50.6% to 84.7% remained after preprocessing to remove low-quality reads and contaminant ribosomal RNA, with a median yield of 81.8% (Supplemental Table S2). Hierarchical clustering was used to assess the variation between biological replicates and thus to diagnose potential issues during library prep and sequencing (Supplemental Fig. S4). Samples of lower quality were finally discarded: one biological replicate of KQ228 C2 In5, SRA1 C1 In5, Q186 C2 In5, and Q186 C2 InEx-5.
Alignment rates of the RNA-seq libraries against sorghum were higher than for the WGS data, ranging from 72.4% to 86.0% (Supplemental Table S2). After quantifying allele-specific abundances for each biallelic SNV, poorly covered genomic regions and lowly expressed genes were filtered out, and from 512,827 to 851,295 heterozygous sites per treatment level were analyzed (Table 1).
Results of the allele-specific expression (ASE) tests
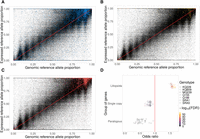
An initial exploratory data analysis to investigate the relationship between genomic and expressed proportions of the reference allele at each site revealed several features of ASE in sugarcane. The genomic allele proportions showed 11 well-defined clusters, with each cluster centered directly over a fraction of 12 (Fig. 1). This agrees with there being 11 heterozygous classes, with one to 11 doses of the reference allele in a dodecaploid genome. The majority of SNVs showed a single dose of the alternative allele; that is, the major allele was present in 11 copies for most variant sites. Some loci did not cluster tightly with any of the groups, including several with an apparent dose greater than 11 or less than one. In addition to simple random noise, this may indicate events of aneuploidy and/or copy number variants.
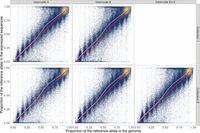
Relationship between expressed and genomic proportions of the reference allele for genotype KQ228. Different internodes are shown in separate panels. The red line indicates the null hypothesis of perfect identity between the expressed proportion of the reference allele (Υ; in the y-axis) and the corresponding genomic proportion (denoted by Γ; x-axis). Each point represents one single-nucleotide variant (SNV), and lighter colors indicate a higher density of points. The smoothed trend of observed points is shown in pink. Notice the majority of sites with high proportion of the reference allele in the genome. To highlight the discrete nature of clusters, we only show high-depth sites, with 1000 or more genomic reads and 80 or more RNA-seq reads.
Frequency of ASE in sugarcane
A strong overall agreement between genomic and expressed allele ratios was observed. The fraction of reads carrying the reference
allele in the RNA-seq data set was directly proportional to the corresponding WGS fraction (Fig. 1). There was very limited bias toward the reference allele, which is a common concern in ASE studies (Castel et al. 2015). Another feature of many of the SNVs was that they showed exclusive expression of one allele, as seen by the masses of points
forming the horizontal lines at 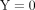 and
and  . These loci are the likely cause of the departure of the smoothed trend (in pink) from the null expectation (Fig. 1, red diagonal line). These observations were consistent for all genotypes and internodes (Supplemental Fig. S5).
. These loci are the likely cause of the departure of the smoothed trend (in pink) from the null expectation (Fig. 1, red diagonal line). These observations were consistent for all genotypes and internodes (Supplemental Fig. S5).
A Bayesian hierarchical beta-binomial model (Correr et al. 2021) confirmed that most SNVs showed no evidence of ASE, with 19,154 to 56,956 heterozygous sites yielding significant tests (Table 1; Supplemental Fig. S6). Similar fractions of SNVs with ASE were found for all genotypes and internodes, except for the four cases in which a lower-quality sample was discarded. Uniformity was also seen when the heterozygous loci were gathered at the gene level. Between 2311 and 5902 genes had at least two SNVs with significant test results and were classified as genes showing allele-specific expression. They correspond to 13.6% to 31.7% of the effectively sampled genes, with an average of 24.1% (Table 1). The full set of genes with ASE, for the 35 combinations of genotypes and internodes, is provided in Supplemental Table S3.
Factors contributing to ASE
When comparing genes showing ASE for the various treatments, a higher similarity was found among samples from the same genotype than from the same internode (Fig. 2). The overlap of genes with ASE for different internodes of the same genotype was 61.6% on average, ranging from 57.8% (SRA1) to 63.9% (KQ228). In some cases, the overlap was as high as 74.6%, as seen for the pair KQ228 C2 In8 and KQ228 C2 InEx-5. On the other hand, the overlap among samples of the same internode ranged from 52.3% (C1 In5) to 57.5% (C2 In8), with an average of 54.8%. Combinations involving different genotypes and different internodes still showed noticeable overlap, ranging from 31.8% to 61.7% (average of 49.5%). Some genes consistently showed ASE for different treatments, whereas there was an added effect of internode and an even greater contribution of the genotype factor.
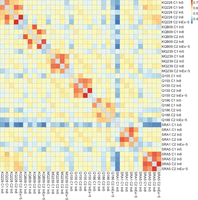
Percentage of genes with allele-specific expression (ASE) detected in common for all genotype × internode combinations. We found higher similarity among different internodes of the same genotype (hot colors in the diagonal blocks), and there was also similarity between the same internode of different genotypes (the light orange cells off the main diagonal).
Clustering the treatments based on the outcomes of ASE tests, that is, the patterns of individual genes with or without ASE, confirmed the closer proximity of samples from the same genotype (Supplemental Fig. S7). The exception was a cluster composed of the more immature internodes from five of the seven genotypes, which were placed to the left of the dendrogram. This indicates that early in stalk development, when all genotypes divert carbon to synthesize cell walls, similar sets of genes may show ASE regardless of the genetic background. Although KQB09 has a unique genomic composition, with recent contribution from S. spontaneum, samples from its more mature culms clustered closer to its parent KQ228.
A more detailed comparison of heterozygous SNVs in common for KQ228 and SRA5 showed a strong positive correlation between the genomic allele proportions observed in each genotype (ρ = 0.92, P-value < 10−15) (Fig. 3A). Rarely was the difference between their most likely doses greater than three or four chromosome copies. A similar trend for the relationship was found between the expressed allele proportions, with ρ = 0.88, P-value < 10−15 (Fig. 3B). Some loci did show substantial differences between the proportions of the reference allele in the two transcriptomes.
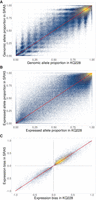
Comparison between KQ228 and SRA5, the two genotypes sequenced at higher depth. (A) Relationship between genomic allele ratios (doses) for shared variants, showing overall agreement between the two genotypes. (B) Relationship between allele proportions in their expressed sequences. (C) Comparison of expression bias for shared SNVs with significant ASE, showing prevalence of higher expression of the reference allele in both genotypes.
When there was a significant excess of a given allele in both genotypes, the preferential allele expression was often in the same direction (Fig. 3C). In most cases, the bias was toward the reference allele, as seen by the heavier mass of points in the first quadrant. These observations are likely reflections of their similar genetic background. This may be in part because of the shared domestication history of Saccharum genotypes, as well as to strong selective pressures in the recent breeding of sugarcane.
ASE frequency in defense response, cell wall, and stress-response genes
The frequency of occurrence of ASE in genes involved in particular cellular functions was investigated. Gene Set Enrichment Analysis (GSEA) (Mootha et al. 2003; Subramanian et al. 2005) provided evidence of enriched Gene Ontology (GO) terms (The Gene Ontology Consortium et al. 2000). Two functional terms appeared as consistently enriched for most treatments, namely, ADP binding (GO:0043531) and defense response (GO:0006952) (Supplemental Table S4; Supplemental Fig. S8). These terms are in fact closely related, as many resistance proteins have nucleotide-binding domains, and many genes were simultaneously annotated with both terms. Enrichment of UDP-glycosyltransferase activity (GO:0008194) was found for most genotypes and internodes. UDP-glycosyltransferases are a large protein family with multiple roles, including the biosynthesis of many cell wall compounds. The terms sulfotransferase activity (GO:0008146) and sulfation (GO:0051923) were also enriched for many treatments. Additional GO terms were significantly enriched in particular treatments, but no discernible pattern was observable, except for an apparent excess of terms for KQ228 and SRA5, the genotypes sequenced at higher depth. The terms O-methyltransferase (GO:0008171) and aromatic compound biosynthetic process (GO:0019438) were enriched in the fiber-rich genotype SRA5, particularly in its upper internodes. Manual inspection revealed that genes annotated with these terms and showing ASE are potentially involved in the synthesis of cell wall components including a gene similar to ZRP4 (Zea Root Preferential), which is involved in the synthesis of suberin and possibly of lignin (Held et al. 1993; Bosch et al. 2011). Strong ASE was observed for a gene similar to herbicide safener binding protein, an O-methyltransferase predicted to be involved in the synthesis of lignin precursors (Scott-Craig et al. 1998).
A direct comparison of the contrasting KQ228 and SRA5 genotypes also showed how ASE may contribute to phenotypic variation. A total of 389 genes were found that consistently showed ASE in at least four of the five sampled internodes in KQ228 but in no more than one internode in SRA5. Conversely, 399 genes displayed consistent SRA5-specific ASE. Mapping both gene lists to the KEGG pathways (Kanehisa and Goto 2000) showed that only one of the KQ228-specific genes was assigned to the phenylpropanoid pathway, namely, a β-glucosidase that catalyzes the synthesis of coumarinate (BGLU, EC 3.2.1.21) (Fig. 4). In contrast, the SRA5-specific genes included five additional genes in this pathway, responsible for multiple steps in the biosynthesis and polymerization of monolignols, the precursor molecules of lignin (Vogt 2010). These included trans-cinnamate 4-monooxygenase (C4H, EC 1.14.14.91), 4-coumarate-CoA ligase (4CL, EC 6.2.1.12), shikimate O-hydroxycinnamoyltransferase (HCT, EC 2.3.1.133), cinnamoyl-CoA reductase (CCR, EC 1.2.1.44), and a peroxidase (PER, EC 1.11.1.7). It is conceivable that consistent ASE in these genes is important for the fiber-rich phenotype of the SRA5 cultivar. Binning genes into MapMan functional groups (Thimm et al. 2004) showed additional SRA5-specific genes related to cell wall organization, such as Cyt-P450 hydroxylase scaffold protein (membrane steroid binding protein [MSBP]), cellulose synthase, endo-1,4-beta-glucanase, and leucine-rich repeat extensin. Eleven genes with KQ228-specific ASE were also associated with cell wall organization but involved in hemicellulose and pectin metabolism. These include extensin beta-1,2-arabinosyltransferase, glucuronosyltransferase, callose synthase, endo-beta-1,4-xylanase, beta-1,3-galactosyltransferase, mannan synthase, beta-galactosidase, and xylosyltransferase. Genes associated with post-Golgi vesicle trafficking, such as SNARE proteins and Rab GTPases, which are important for transporting cell wall components, were found in higher numbers in SRA5 (18 vs. nine in KQ228).
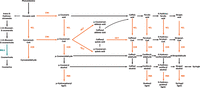
Genes in the phenylpropanoid pathway with consistent ASE for SRA5 and KQ228. Enzymes in orange correspond to genes with consistent ASE in the internodes of SRA5 but not in KQ228. The metabolic step in blue indicates different genes with consistent ASE in either genotype. (BGLU) β-Glucosidase; (C4H) trans-cinnamate 4-monooxygenase; (4CL) 4-coumarate-CoA ligase; (HCT) shikimate O-hydroxycinnamoyltransferase; (CCR) cinnamoyl-CoA reductase; (PER) peroxidase.
Sugar and starch metabolism is tightly linked to the phenotype of sucrose accumulation. No gene with SRA5-specific ASE was annotated with this term, but we observed six such genes in KQ228: triose phosphate/phosphate translocator, glucose transporter, UDP-D-glucose 4-epimerase, starch branching enzyme, starch synthase, and cytosolic glucanotransferase DPE2. Possibly related solute transporters, including ABCB transporters, hexose transporter, and organic phosphate/glycerol-3-phosphate permease, were found in excess in KQ228 (21 vs. 10). This shows that ASE also affects sucrose accumulation in sugarcane. The genes with consistent ASE in both genotypes also included multiple genes involved in RNA and protein biosynthesis, including large and small ribosomal subunit components, mRNA quality control, and various transcription factors.
Because SRA1 showed low sugar and fiber contents, the genes displaying consistent ASE in this genotype were investigated. Compared with the genes with ASE in SRA5, SRA1 showed 163 specific genes enriched for signaling proteins (10 kinases and three phosphatases), multiprocess regulation (two SnRK1-interacting factors, a pyrophosphatase, and a CBL-dependent protein kinase [CIPK]), redox homeostasis (iron superoxide dismutase and glutathione S-transferase), and the metabolism of terpenoids. These functional groups are associated with stress response, in agreement with the poor phenotype observed for SRA1, and show that ASE is associated with response to environmental and developmental stress. Other stress-related genes with SRA1-specific ASE were a glutaredoxin, two multidrug resistance proteins (MATE), two interleukin-1 receptor-associated (IRAK) kinases, peroxin, and a polyamine oxidase.
The type of mutation and gene conservation associated with ASE
Many of the SNVs in genes annotated with enriched GO terms showed exclusive expression of the reference allele (Fig. 5A,B). This maximum form of ASE may imply that transcriptional and post-transcriptional regulatory mechanisms have a potential impact on the expression of different alleles for these genes. A comparable tendency was seen for SNVs predicted to have a large impact on protein structure (Fig. 5C). This classification is assigned to SNVs that affect the stop codon, possibly giving rise to truncated or extended proteins in the case of stop gain and stop loss substitutions, respectively. Such gene products are often not fully functional and can be detrimental to cell activity. It is thus not surprising that we observed a single allele to be exclusively present for many of these loci, providing evidence of absence of transcription or post-transcriptional mRNA decay of the other SNV allele (Rivas et al. 2015). At the genome level, the distribution of estimated allele doses per SNV class showed an increase in the frequency of higher doses of the reference allele for variants of higher predicted impact (Supplemental Fig. S9A,B). In addition to the already higher genomic doses, a higher-than-expected abundance was observed for transcripts carrying the reference allele for high-impact SNVs (Supplemental Fig. S9C).
Enrichment results of genes with allele-specific expression. (A) SNVs in genes annotated with ontology term defense response (GO:0006952). (B) SNVs in genes annotated with ontology term UDP-glycosyltransferase activity (GO:0008194). (C) SNVs predicted to have high functional impact. In all cases, note the excess of SNVs with exclusive expression of the reference
allele (horizontal line at  ). (D) Underrepresentation of single-copy genes among those with ASE (odds ratio < 1); overrepresentation of core genes, conserved
in Liliopsida (odds ratio > 1). Multiple points for the same genotype indicate different internodes.
). (D) Underrepresentation of single-copy genes among those with ASE (odds ratio < 1); overrepresentation of core genes, conserved
in Liliopsida (odds ratio > 1). Multiple points for the same genotype indicate different internodes.
Pham et al. (2017) found that core angiosperm genes responsible for key plant processes were more likely to show ASE in multiple tetraploid potato genotypes. In the present study, genes conserved in a set of 17 monocot species had higher odds (odds ratios > 1.5) of showing ASE than the remaining genes (Fig. 5D). Conversely, we found that genes that are present in single copy in sorghum, rice, and Brachypodium were less likely to show preferential expression of alleles. These observations support the notion that conserved genes controlling key biological processes may be enriched among those with ASE, but that single-copy genes may be under some control mechanism limiting ASE in sugarcane. Lower odds ratios were also seen for sorghum-exclusive paralogs, but the number of genes in these groups of paralogs was small and statistical significance was often not attained.
Discussion
Significant ASE in the culms of elite sugarcane hybrids was limited to, on average, one out of four genes, with most heterozygous loci showing agreement between allele ratios in genomic and expressed sequences. Previous studies have shown evidence for ASE in sugarcane (Vilela et al. 2017; Sforça et al. 2019; Cai et al. 2020), but the current genome-wide high-depth analysis shows that ASE is not widespread in this complex polyploid, probably owing to factors regulating gene expression having comparable effects on the different alleles. Because of the high ploidy levels of sugarcane hybrids, it was essential to make use of high sequencing depths to obtain precise estimates of allele doses (Margarido and Heckerman 2015; Gerard et al. 2018). This is particularly relevant if the possibility of bias and overdispersion in allele read counts is considered. To err on the side of being conservative, four RNA-seq libraries were removed from ASE analyses to avoid potential issues in accurately quantifying the expression of different alleles. Nevertheless, these samples were from distinct treatments, such that the effect on statistical power to detect ASE was limited.
Because read alignment was performed against sorghum, the pipeline included various filtering steps to reduce the occurrence of spurious SNVs. The total RNA-seq alignment rates were on par with those seen when using the current sugarcane genomes (Diniz et al. 2019). Alignment of reads from transcribed regions was relatively straightforward, such that the majority of relevant alignments were unambiguous. Ensuring that a large proportion of the reads were effectively and correctly assigned to their corresponding genomic regions is crucial in ASE studies (Castel et al. 2015). The fact that very little (if any) bias toward the reference allele was observed indicates a good overall performance of the pipeline, underlining the reliability of the results.
A beta-binomial model was adapted to investigate ASE in mixed- and high-ploidy organisms (Correr et al. 2021), and this study further modified this method to model uncertainty in estimating allele doses. This approach allowed nonhomogeneous sequencing depth profiles to be naturally handled by the model, with a corresponding adjustment of the null hypothesis for each polymorphism. This statistical analysis strategy likely had an impact on the fraction of genes identified as showing significant ASE. Although there was evidence of ASE for 13.6% to 31.7% of the genes in sugarcane, other studies have reported widely varying values: 13.5% in Arabidopsis (Zhang and Borevitz 2009), roughly a third to half of the genes in potato (Pham et al. 2017), ∼50% of the genes in maize (Springer and Stupar 2007), and up to 70% among wheat homoeologs (Powell et al. 2017). This variation encompasses differences between diploids, autopolyploids, and allopolyploids, as well as true biological variation in these systems, and includes differences in experimental design and environmental and technical noise. However, a contribution from the distinct data processing and methodologies used in each case cannot be ruled out. Different statistical models and testing strategies, hard thresholds, and other ad hoc criteria can affect the results. In sugarcane, Correr et al. (2021) also observed ASE for ∼40% of the sampled genes. However, this study relied on genotyping-by-sequencing of reduced representation libraries, a strategy that is prone to biases and genotyping errors (Scheben et al. 2017). These issues can then strongly influence conclusions about ASE for a set of the polymorphic sites.
In this context, one advantage of the GSEA method used here is that it does not depend on previous testing for ASE but only on the ranking of genes. Genes with large absolute deviations between genomic and expressed allele ratios had higher ranks, and functional terms with many high-ranking genes were tagged as enriched with ASE. A small number of GO terms showed significant enrichment, with the prominent presence of defense-response genes among those with ASE in many genotype and internode combinations. Many resistance proteins have nucleotide-binding domains, and binding to ADP or ATP induces conformational changes that regulate signaling cascades (DeYoung and Innes 2006; Tameling et al. 2006). The higher frequency of ASE among defense-response genes agrees with observations in allohexaploid wheat, in which many of these genes showed homoeologous expression bias in plants challenged with a pathogenic fungus (Powell et al. 2017). Species in the genus Saccharum differ broadly with regard to their response to pathogen infection, and sugarcane hybridization and breeding have historically focused on selecting for disease resistance (Cheavegatti-Gianotto et al. 2011; Rott et al. 2013). The seven hybrids have been selected for disease resistance and show different patterns of response against a range of pathogens (Supplemental Table S5). Different alleles may respond differently to particular pathogen strains or races, and breeders may have indirectly selected for higher expression of specific alleles. This ASE probably is also linked to the smaller contribution of the S. spontaneum genome to modern hybrids, as a consequence of abnormal chromosome transmission in the initial backcrossing. Selection pressure on disease resistance kept those traits from S. spontaneum in the genome, and for most, they are left in low doses.
No homogeneous enrichment was detected for many functional terms associated with carbon partitioning, such as those involved in sucrose accumulation or cell wall biosynthesis, with the exception of UDP-glycosyltransferase activity. Enrichment was observed for assorted transferase terms (including of hexosyl groups), cellulose synthase (UDP-forming) activity (GO:0016760), plant-type primary cell wall biogenesis (GO:0009833), and mannan synthase activity (GO:0051753) for scattered treatments. This indicates that sizeable ASE may occur at least in some genotypes and at some points during the development and maturation of sugarcane stalks. The identification of high-level GO terms associated with the synthesis of cell wall compounds in SRA5 opens up the possibility that ASE has direct effects on phenotypes of economic interest. A closer inspection of the phenylpropanoid pathway revealed that many genes involved in the biosynthesis of lignin showed significant ASE for the fiber-rich SRA5 genotype but not for the sugar-rich KQ228.
When analyzing high-impact SNVs, which are predicted to have more drastic effects on protein structure, both higher genomic doses and excessive abundance of the reference allele were discovered. The former implies purifying selection against the alternative allele, whereas the latter hints at its silencing or increased mRNA degradation. This effect of the type of mutation on the incidence of ASE has been reported in humans, similarly with a higher frequency of ASE in nonsense variants (Rivas et al. 2015). These observations can be somewhat noisy because SnpEff treats each polymorphism individually, and multiple adjacent variants may have a joint effect on gene products. However, this is mostly relevant to indels, and the fact that only SNVs were used in this study alleviates the problem. Polymorphisms affecting start and stop codons may be the primary cause of ASE for many genes. If this is in fact the case, the genes bearing these SNVs would be expected to show similar expression patterns across tissues and developmental stages. This agrees with the observation that the transcriptomes of different internodes for the same genotype showed more similar patterns of ASE than other combinations of treatments (Fig. 2). Hence, the control of ASE in sugarcane seems to be largely genetic, which has important implications for plant breeding. The expression of favorable alleles can be directly or indirectly leveraged through selection, at least for a subset of the genes, and these effects are expected to be partly passed onto the progeny of crosses between hybrids. Yet, an effect attributable to internodes and collection time points was also observed. Developmental and environmental cues thus appeared to have some contribution to ASE in these experiments. These effects are also partially accessible to selective breeding, to the extent that they may contribute to genotype × internode interaction. They are also relevant from a biological perspective as they enhance understanding of the biology of sugarcane hybrids.
Clusters of genes conserved in the Liliopsida were overrepresented among those with ASE, whereas single-copy grass genes were underrepresented. Fine-tuning of biological processes that are essential for proper functioning of plant metabolism may include mechanisms favoring the expression of some alleles over the others. In contrast, the lower frequency of ASE among single-copy genes suggests that redundancy may be a relevant factor driving ASE in sugarcane. The extended mutational freedom afforded by gene duplication may allow regulatory mechanisms to tweak the expression of individual allelic copies in these high-ploidy genomes.
The high-depth genomic data analyzed supports the presence of 12 chromosome copies for the majority of polymorphic sites in these seven sugarcane hybrids. Despite the extensive variation in ploidy and chromosome number existent in Saccharum, it is compelling to see such consistency among a set of elite hybrids. As is common in sugarcane breeding programs, these hybrids share a narrow genetic base and are often derived from crosses involving repeated parents. Their common genetic background may have increased the likelihood of producing genomes with similar composition, in spite of their substantial phenotypic variation. However, we could neither infer the genomic origin of each allele nor observe any clear differentiation between putative S. officinarum–derived and S. spontaneum–derived alleles based on short-range polymorphisms alone. Although the genus Saccharum may comprise three distinct subgenomes, these founding genomes are apparently very similar to each other, especially for exonic regions, where the average sequence identity is >99% (Pompidor et al. 2021). Given such high similarity among gene copies, it may not be possible to tell the three subgenomes apart when working at the SNV level in transcribed regions.
The combination of high-depth WGS data and RNA-seq has provided a comprehensive view of the incidence of ASE in sugarcane hybrids. It does not appear to be a pervasive feature of gene expression in this complex crop. Using the sorghum genome as a reference focuses the analysis on genes that are conserved between the two species. An interesting future research opportunity is to investigate sugarcane-exclusive genes and assess whether they are less likely to show ASE. This will be possible when a complete (phased) hybrid sugarcane genome becomes available but will also require improvements in long-read RNA-seq to allow the allelic origin of each read to be unambiguously inferred. In addition, this genomic resource will allow polymorphisms in regulatory regions to be explored and, consequently, to identify cis-acting elements that control the occurrence of ASE in sugarcane. This information can then be leveraged into molecular breeding efforts in this important bioenergy crop.
Methods
Biological material and sample collection
Perlo et al. (2020) described a field experiment used to evaluate 24 sugarcane hybrids contrasting in several traits, including fiber content and sugar yield. Later, we performed RNA-seq to assess differential gene expression in internodes collected at five different developmental stages. Briefly, 24 Saccharum hybrids were planted in a 6 × 4 Latin square with three replicates, in August 2017 at the Sugar Research Burdekin Station in Burdekin, Queensland, Australia. Each plot was 4 m long with 1.52 m between rows, and environmental conditions were kept uniform via fertilization and furrow irrigation. The genotypes were phenotypically characterized in March, June, and September of 2018 by measuring the soluble solids content, polarity, and fiber percentage. For RNA-seq, three independent biological replicates of stalks were collected 19 wk (the first collection, or C1) and 37 wk (C2) after planting. Internodes 5 and 8 were sampled in both collections, whereas internode Ex-5 was sampled in C2. The latter corresponds to internode 5 from C1, tagged in intact culms to be collected when more mature. In all cases, samples were collected in the morning hours, sliced into smaller fragments, frozen in liquid nitrogen immediately after cutting, and kept at −80°C until processed. We refer the reader to Perlo et al. (2020) for further details about the trial.
Based on these phenotypic traits, we chose seven genotypes to use in our work. KQ228 and SRA5 showed contrasting behavior in terms of sugar and fiber accumulation and were chosen as the main genotypes to explore herein. We also sampled the pair Q155 and KQB09-20432, which showed similar phenotypes to KQ228 and SRA5, respectively. The remaining genotypes, SRA1, MQ239, and Q186, were selected because of their negative or neutral phenotypic traits.
WGS and quality control
Leaves +4 and +5 of each of the seven genotypes were sampled for WGS, where leaf +1 is the first with a visible dewlap. DNA was extracted using the method described by Furtado (2014), modified by adding 5 mL chloroform instead of the phenol:chloroform:isoamyl alcohol (25:24:1) mixture. Following extraction, DNA integrity was checked with a NanoDrop spectrophotometer assay to measure the 260/280 and 230/260 ratios.
High-quality genomic DNA was used for sequencing libraries following the Illumina protocol. The seven libraries were sequenced on one S4 flowcell of a NovaSeq 6000 platform to obtain 2 × 150-bp paired-end reads. The sequencing run was designed to represent the genotypes at different depths of coverage. Libraries for KQ228 and SRA5 were sequenced at five times the volume of the other genotypes, with expected depths of 100× and 20× per chromosome copy, respectively.
Raw whole-genome reads were trimmed using CLC Genomics Workbench v20.0.4 at a quality limit of 0.01 to remove data with low Phred scores (Supplemental Table S1). We also removed reads with more than two ambiguous bases and discarded reads shorter than 50 bases after trimming.
Read alignment and SNV calling
The Sorghum bicolor genome 454 v3.0.1 (Paterson et al. 2009; McCormick et al. 2018) was used as a reference to identify SNVs. Briefly, alignment of genomic reads to the sorghum genome was performed with Bowtie 2 v2.4.1 (Langmead and Salzberg 2012); duplicate reads were removed; and only primary alignments were kept. The GATK v4.1.8.1 pipeline (DePristo et al. 2011) was used to identify SNVs and call genotypes. We annotated the predicted impact of each SNV based on their relative genomic position with SnpEff v5.0 (Cingolani et al. 2012). Details about SNV calling are in the Supplemental Methods.
RNA-seq, data preprocessing, and alignment
Total RNA was extracted from three replicates of each genotype × internode combination for a total of 105 samples representing 35 treatments (seven genotypes × five internodes), with each sample comprising a pool of four clonal stalks. Culms from each treatment were first individually pulverized; equal amounts of four stools were combined to form each pool; and pools were again kept at −80°C.
Sequencing libraries were constructed with the standard TruSeq RNA protocol, and the cDNA was sequenced (in pools with additional indexed libraries) on the Illumina NovaSeq 6000. Samples were split into four lanes of one S4 flowcell, with a data volume corresponding to 90 samples per lane. With this multiplexing strategy, the expected yield was of more than 50 million paired-end 100-bp reads for each sample (Supplemental Table S2).
The raw reads were processed to remove Illumina adapters, low-quality bases, and contaminating ribosomal RNA reads. For aligning the RNA-seq reads, we used the same sorghum genome reference and the splice-aware aligner HISAT v2.1.0 (Kim et al. 2015). Details about the processing of RNA-seq reads are in the Supplemental Methods.
Quantification of relative allele proportions
For both the WGS and RNA-seq data sets, we applied the ASEReadCounter tool v4.1.8.1 to estimate the frequency of each allele, in read counts, for each biallelic SNV of each genotype (this tool does not allow for indels or multiallelic sites). At the genomic level, this provided an estimate of the relative allele dose. In this case, we removed PCR/optical duplicates to avoid potential bias in estimating the doses. At the expression level, these counts were used to obtain estimates of the relative expression levels of both alleles. For RNA-seq reads, the duplicate read filter was disabled so as not to bias the estimated allelic proportions for highly expressed genes. In both cases, we disabled the maximum depth limit to ensure that all reads were effectively counted. As a diagnostic metric, we performed hierarchical clustering with R v4.0.5 (R Core Team 2021) to visualize the dissimilarity among samples. To that end, we used SNVs for which the RNA-seq read count was greater than or equal to five for at least half of the samples. Euclidean distances were calculated based on the relative frequency of the reference allele, and the default complete linkage method was used to find clusters.
Assessment of allele-specific expression
Based on the clustering pattern and the amount of rRNA and PCR duplicates, four samples that were of comparatively lower quality than the rest in the RNA-seq data set were excluded. These samples were all from distinct treatments, such that four genotype × internode combinations were represented by two replicates, whereas the remaining 31 treatments had all three replicates. For each of the 35 treatments, we combined the allele counts of the remaining high-quality RNA-seq replicates.
In addition to the raw allele counts, we calculated for each polymorphic site the relative proportion of the reference allele,
both for the genomic and expression data sets. Let rik represent the number of WGS reads carrying the reference allele for SNV i and treatment k, and let gik indicate the total number of genomic reads for the corresponding locus. We then calculated the observed genomic proportion
of the reference allele, denoted by Γ, as 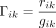 . Similarly for the transcriptome data, we denote by yik the number of RNA-seq reads with the reference allele and by nik the total corresponding read count. The observed expressed proportion of the reference allele is then given by
. Similarly for the transcriptome data, we denote by yik the number of RNA-seq reads with the reference allele and by nik the total corresponding read count. The observed expressed proportion of the reference allele is then given by 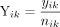 .
.
To test for ASE, the hierarchical beta-binomial model proposed by Correr et al. (2021) was used with modifications. We first modelled yik according to a binomial distribution, 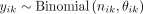 . The a priori distribution of the parameter θik was modelled with a beta distribution,
. The a priori distribution of the parameter θik was modelled with a beta distribution, 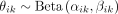 , where αik and βik indicate the genomic dose of the reference and alternative alleles, respectively. Because these doses are unknown, we obtained
estimates by approximating the observed genomic allele proportions, considering a ploidy of 12. In that case, we set αik = [12 × Γik], where the brackets represent the integer closest to 12 × Γik, and βik = 12 − αik.
, where αik and βik indicate the genomic dose of the reference and alternative alleles, respectively. Because these doses are unknown, we obtained
estimates by approximating the observed genomic allele proportions, considering a ploidy of 12. In that case, we set αik = [12 × Γik], where the brackets represent the integer closest to 12 × Γik, and βik = 12 − αik.
According to this model, the posterior distribution of θik is then Beta(yik + αik, nik − yik + βik), but in practice, we used Beta(yik + αik + 0.5, nik − yik + βik + 0.5) to avoid zero counts. For each site, we obtained the highest density interval (HDI) for this posterior distribution with the R package HDInterval v0.2.2 (https://cran.r-project.org/package=HDInterval). To account for the large number of genotype × SNV combinations, the Bonferroni correction (Bonferroni 1936) was applied to the interval mass, with a global significance level of 0.05 and considering an approximation of 100,000 independent tests. This is because neighboring SNVs in a given gene are not independent, such that the number of tests must be between the number of genes and the number of positions tested.
Finally, to call an SNV as showing significant ASE, we checked whether the HDI included the observed genomic proportion Γ of the reference allele. Because this proportion is an estimate and thus subject to random variation, instead of using a point estimate, we used the 95% confidence interval for the binomial distribution calculated with the Wilson method (Wilson 1927). If there was no overlap between this confidence interval and the posterior HDI, the corresponding SNV was deemed to show significant allele-specific expression. Sites with lower genomic depth of coverage have wider confidence intervals, such that this approach effectively models uncertainty and helps to avoid false positives in less-covered regions.
Functional enrichment tests
Genes were first clustered according to three criteria: (1) genes conserved in a set of 17 monocots, (2) sorghum-exclusive paralogs, and (3) single-copy genes in sorghum, rice, and Brachypodium. Poorly covered (fewer than 50 WGS reads) and lowly expressed (fewer than 10 RNA-seq reads) polymorphisms were excluded from the analysis, and a Fisher's exact test was used to test for enrichment of genes with ASE in each cluster. Enrichment of genes annotated with common GO functional terms was performed with GSEAPreranked v4.0.3 (Mootha et al. 2003; Subramanian et al. 2005), with ranking based on the median absolute deviation between expressed and genomic allele ratios. Only genes with 10 or more SNVs and GO terms with five or more genes were considered. See the Supplemental Methods for more details.
Data access
The WGS data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA733812. The RNA-seq data generated in this study have been submitted to the EMBL-EBI European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser/home) under accession number PRJEB44480.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We acknowledge The University of Queensland's Research Computing Centre (RCC) for its support and the Center for Functional Genomics–ESALQ for its computational infrastructure. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior–Brasil (CAPES) grants CAPES-PRINT 88887.466432/2019-00 to G.R.A.M. and CAPES-PRINT 88887.367965/2019-00 to F.H.C. and by grant #2015/22993-7, São Paulo Research Foundation (FAPESP). Australian Research Council and Sugar Research Australia provided funding to R.J.H. and F.C.B.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275904.121.
- Received June 17, 2021.
- Accepted December 19, 2021.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








