
Genomic architecture constrained placental mammal X Chromosome evolution
Abstract
Susumu Ohno proposed that the gene content of the mammalian X Chromosome should remain highly conserved due to dosage compensation. X Chromosome linkage (gene order) conservation is widespread in placental mammals but does not fall within the scope of Ohno's prediction and may be an indirect result of selection on gene content or selection against rearrangements that might disrupt X-Chromosome inactivation (XCI). Previous comparisons between the human and mouse X Chromosome sequences have suggested that although single-copy X Chromosome genes are conserved between species, most ampliconic genes were independently acquired. To better understand the evolutionary and functional constraints on X-linked gene content and linkage conservation in placental mammals, we aligned a new, high-quality, long-read X Chromosome reference assembly from the domestic cat (incorporating 19.3 Mb of targeted BAC clone sequence) to the pig, human, and mouse assemblies. A comprehensive analysis of annotated X-linked orthologs in public databases demonstrated that the majority of ampliconic gene families were present on the ancestral placental X Chromosome. We generated a domestic cat Hi-C contact map from an F1 domestic cat/Asian leopard cat hybrid and demonstrated the formation of the bipartite structure found in primate and rodent inactivated X Chromosomes. Conservation of gene order and recombination patterns is attributable to strong selective constraints on three-dimensional genomic architecture necessary for superloop formation. Species with rearranged X Chromosomes retain the ancestral order and relative spacing of loci critical for superloop formation during XCI, with compensatory inversions evolving to maintain these long-range physical interactions.
The X Chromosome is the most well-studied chromosome in mammals (Morgan and Bridges 1916; Lyon 1961; Ohno 1967; Penny et al. 1996; Ross et al. 2005; Engreitz et al. 2013; Tukiainen et al. 2017; Miga et al. 2020). One of the hallmark characteristics of the X Chromosome is the remarkable degree of conservation in gene content across placental mammals, a property hypothesized by Susumu Ohno to have evolved to maintain dosage relationships between X-linked genes and their autosomal counterparts during the early stages of sex chromosome differentiation (Ohno 1967; Lyon 1992). This pattern of conservation is unparalleled by any autosome (Murphy et al. 2005; Kim et al. 2017). Another notable feature of the mammalian X Chromosome is the extent of linkage (i.e., gene order) conservation (Nadeau 1989) displayed between phylogenetically distant lineages (Murphy et al. 1999; Quilter et al. 2002; Raudsepp et al. 2004; Rodríguez Delgado et al. 2009). Linkage conservation does not fall within the scope of Ohno's original prediction (Ohno 1967) as intrachromosomal rearrangements presumably would not disrupt dosage levels to the same degree as would interchromosomal translocations. Linkage conservation is also not as pervasive as the conservation of gene content, as some placental mammals exhibit X Chromosome rearrangements relative to the placental mammal ancestor, although these are generally more rare and phylogenetically restricted (e.g., mouse, rat) (Amar et al. 1988; Piumi et al. 1998; Robinson et al. 1998; Sandstedt and Tucker 2004; Park et al. 2013; Proskuryakova et al. 2017).
X Chromosome linkage conservation may be an indirect result of selective pressures to maintain gene content, or conversely, intrachromosomal rearrangements could be directly selected against because they disrupt some critical biological process like X-Chromosome inactivation (Rodríguez Delgado et al. 2009). X-Chromosome inactivation (XCI) in placental mammals involves the spread of the XIST RNA in a proximity-dependent manner governed by three-dimensional chromatin architecture (Wang et al. 2018). The resulting inactive X Chromosome (Xi) also forms a unique bipartite structure divided by the macrosatellite DXZ4 (Deng et al. 2015). DXZ4 escapes inactivation and achieves this large-scale structural change through formation of long-range interactions between other XCI escapee loci: XIST, ICEE, and FIRRE (Chadwick 2008; Darrow et al. 2016). Interaction between these loci is attributed to sharing of two distinct features. The first is that each locus is enriched for CTCF binding motifs, which act to anchor loops resulting from chromatin extrusion to these regions in a polarity-dependent manner (Rao et al. 2014). Second, each of these loci are colocalized to the peri-nucleolar envelope through interaction with FIRRE lncRNA, resulting in the well-described association between the nucleolus and Barr body (Yang et al. 2015; Jégu et al. 2017). Because CTCF motif directionality heavily influences how superloops form and are associated, maintenance of the Xi bipartite structure is sensitive to any structural perturbation that would alter the conventional orientation of these loci (Bonora et al. 2018). Therefore, it follows that selection against such large-scale structural changes may have led to the extensive collinearity observed across the majority of placental mammal X Chromosomes. The spatial requirements for the formation of the bipartite structure during XCI may also potentially select for compensatory rearrangements to maintain the order and spacing of these interacting loci. However, the extent of conservation of the bipartite structure outside of rodent and primate lineages remains unexplored.
Another chromosomal feature parallels the conserved collinearity of most mammalian X Chromosomes: the recombination landscape. Specifically, cat, dog, pig, and, to a somewhat lesser extent, human, all share a massive recombination cold spot that spans roughly the central one-third of the X Chromosome and extends tens of megabases distally from the centromere (Wong et al. 2010; Li et al. 2019). Li et al. demonstrated that the broader landscape of recombination cold spots and hot spots was conserved in species with X Chromosome collinearity, with orthologous boundaries demarcating marked rate shifts at several points along the chromosome. The hot spots flanking the cold spots possess some of the highest recombination rates in the cat genome (Li et al. 2016). The largest and most centrally located cold spot is associated with recurrent bouts of strong selective sweeps and high levels of genetic differentiation, across different mammal orders (Montague et al. 2014; Ai et al. 2015; Dutheil et al. 2015; Nam et al. 2015; Figueiró et al. 2017; Lucotte et al. 2018; Li et al. 2019). Nam et al. (2015) showed that the recurrent bouts of selective sweeps observed in multiple hominid primate species were targeted toward X-linked ampliconic genes in the ancestor of great apes. These authors also suggested that the reduced recombination rates observed flanking ampliconic loci could explain the reduction in diversity and the efficacy of selection in these same regions. Like linkage conservation, it is unclear what physical constraints or functional loci may drive the large reduction in recombination rate over such a large portion of the X Chromosome. Whether the conserved recombination landscape represents an additional consequence of constraints on dosage compensation has not been previously addressed in the context of physical chromosomal features.
Multispecies comparative approaches have the power to identify selective pressures that shape these unique aspects of X Chromosome evolution. Mueller et al. (2013) conducted the first fine-scale comparative X Chromosome comparison using the two highest-quality mammalian X Chromosome assemblies, from human and mouse, to systematically test Ohno's predictions. They found that, whereas most X-linked protein coding genes had orthologs in the other species (82% and 77% in human and mouse, respectively), there appeared to be rapid turnover of a subset of loci: independently acquired multicopy and ampliconic genes (presumably from autosomal progenitor genes) with testis-specific expression. Ampliconic genes are distinguished from multicopy genes in that they reside in segmentally duplicated sequences that share >99% identity. If ampliconic gene content accounts for, or is the result of, the striking patterns of reduced variation with large recombination cold spots on the X Chromosome in different mammalian lineages, then an accurate depiction of the history of gene conservation and divergence is necessary to understand the level of constraint acting on these regions.
The excessive lineage-specific acquisition of novel ampliconic and multicopy gene family members hypothesized by Mueller et al. could have been biased by the limited phylogenomic sampling available at the time. The other (i.e., dog and horse) placental mammal X Chromosome sequences that were available for comparison were early draft assemblies and contained hundreds to thousands of sequence gaps. This fragmentation is a consequence of the highly repetitive, and often large, ampliconic regions that cannot be accurately assembled using standard second-generation sequencing methods, particularly in diploid genome assemblies (Eichler et al. 2004; Alkan et al. 2011; Huddleston et al. 2014; Chaisson et al. 2015; Khost et al. 2017). The relatively poor continuity of the dog and horse genomes that were used to infer patterns of evolutionary conservation with the human and mouse could have led to inferences of gene absence that were misinterpreted as lineage-specific acquisition.
Here, we sought to address several outstanding questions about mammalian X Chromosome architecture and ampliconic gene family evolution by expanding the phylogenetic sampling of high-quality genomes in a comparative analysis. First, we wanted to determine if any of the ampliconic or multicopy gene families previously interpreted as being independently acquired in human or mouse were in fact ancestral eutherian genes or members of ancestral gene families that went undetected in early draft assemblies from other placental mammals. Specifically, we tested the hypothesis that the independent specialization of ampliconic gene repertoires proposed by Mueller et al. (2013) was influenced by (1) the high rates of gene family birth and death in the mouse lineage, and (2) the limited additional taxon sampling from fragmented draft quality genome assemblies. Second, we examined whether physical and/or functional properties might explain the unparalleled linkage conservation seen on the mammalian X Chromosome. Third, we asked whether the remarkable number of intrachromosomal rearrangements that occurred in the mouse lineage facilitated the substantial shift in the ampliconic gene catalog of this species. Finally, we searched for structural correlates that might explain why evolutionarily divergent mammals share large, physically orthologous recombination cold spots (Li et al. 2019) and if these reflect some constraint imposed by X-Chromosome inactivation.
Results
Domestic cat BAC clone sequencing, assembly, and annotation
We sequenced and assembled 83 domestic cat X-linked BAC clones using Pacific Biosciences (PacBio) long-read sequencing platform. These clones were selected from regions containing the largest gaps in the felCat8.0 X Chromosome assembly (which was built from a combination of short-read types: Sanger + Roche454 + Illumina), and from regions where, based on genome alignments, one would predict the presence of orthologs of human and/or mouse ampliconic genes. During the course of this project, the felCat9.0 assembly was released (Table 1). felCat9.0 utilized long-read data, and the number of X Chromosome gaps was reduced by two orders of magnitude when compared to felCat8.0 (Table 1). The assembled clones were mapped to the recently completed felCat9.0 assembly (Buckley et al. 2020). As such, many of the clones that were targeted to fill gaps in felCat8.0 were mapped to contiguous regions in felCat9.0. This provided a means to confirm the accuracy of our clone-based assembly approach (Supplemental Table S1).
Comparison of felCat8.0 and felCat9.0 X Chromosome assemblies and the improved v9.1 X Chromosome assembly
We were able to span six of the 54 remaining X Chromosome gaps in felCat9.0 assembly, including two large ampliconic regions incorporating ∼467 kb of new BAC-clone sequence data. We also substantially improved the sequence content around 12 additional gaps, including eight within ampliconic regions, incorporating ∼980 kb of new BAC-clone sequence data (Supplemental Tables S1, S2). Twenty-five of the remaining assembly gaps are within or adjacent to (<100 kb away) newly identified ampliconic sequence that was resolved in the felCat9.0 assembly, and five are within regions that align to human X Chromosome ampliconic sequence. Although the remaining gaps will lead to an underestimate of the amount of ampliconic sequence on the domestic cat X Chromosome, these gap sizes are estimated to be <600 kb based on BAC-end sequence alignments, suggesting that the final amount of domestic cat ampliconic sequence is likely to be much closer to that of human (∼6.8 Mb) than the mouse (∼20.6 Mb).
We identified eight novel protein-coding genes (open reading frame >400 nucleotides) within the new BAC clone sequences, five of which displayed testis-biased expression (Supplemental Table S3). Seven genes occur within ampliconic sequence, and one is immediately adjacent to orthologous sequence that is ampliconic in human (∼135 Mb), although the cat sequence is not ampliconic (Supplemental Table S4). One of these novel genes encodes a putative olfactory receptor (98% similarity to an annotated cheetah gene), and a second shares 91% identity (49% query coverage) to a mountain lion cancer/testis antigen 1-like paralog. A third gene shares 70% nucleotide identity with a domestic dog unnamed protein-coding gene. The remaining five novel genes originated from a single BAC clone (FCAB-331E2) that was mapped near the telomeric end of the long arm (∼129 Mb) adjacent to a region that is ampliconic in human but not in cat (Supplemental Table S4). These last five genes had no significant matches to the NCBI nonredundant protein sequence database.
Comparative gene content and chromosome architecture
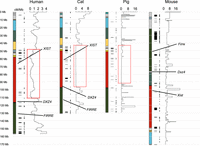
We also analyzed the X Chromosome assembly and gene content for the pig, a representative of a fourth divergent placental mammal order, Cetartiodactyla. This long sequence read assembly has comparable metrics of contiguity to the cat genome and also included a substantial BAC-finishing component (Ward et al. 2020). Pairwise nucleotide alignments between the X Chromosomes of four species confirmed conservation of gene order between the human, cat, and pig across nearly the entire length of the chromosome (Fig. 1; Supplemental Table S4). The same gene order is also shared by other placental mammals representing divergent ordinal lineages, including horse, dog, and elephant (Raudsepp et al. 2004; Murphy et al. 2005; Rodríguez Delgado et al. 2009). This supports the conclusion that this gene order represents the configuration found in the ancestor of placental mammals. In contrast, no fewer than seven inversions distinguish the mouse X Chromosome from human and most other placental mammals (Supplemental Table S5; Pevzner and Tesler 2003a,b).

Pairwise X Chromosome alignments between human and cat, human and pig, and human and mouse.
The number of annotated X-linked protein-coding genes ranged from 774 genes in the pig to 943 genes in the mouse. The mouse X Chromosome was an outlier in all features, having the largest number of genes, the largest amount of ampliconic sequence content, and the largest number of lineage-specific gene gains and lineage-specific gene losses (Fig. 2; Table 2). The values for human, cat, and pig were more similar: they showed fewer gene gains, fewer instances of lineage-specific gene loss, and less total ampliconic sequence content. Mueller et al. (2013) found that lineage-specific gene gain events were enriched for ampliconic loci, and we observed the same pattern in all four species, although ampliconic gains were far less frequent than previously estimated (Supplemental Tables S6–S9). Multicopy loci were the most common category of lineage-specific gains. Because the majority of ampliconic genes are members of multicopy gene families, the previous categorization of repetitive genes into two classes becomes somewhat arbitrary from the perspective of gene gain and loss, because the boundaries of ampliconic sequence vary across species when the gene content may not (Fig. 3).
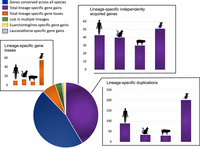
X-linked genes annotated in four mammal species (n = 1330) and their histories across lineages. Lineage-specific gains are broken down by those acquired via lineage-specific duplication and those that were independently acquired.

Annotated comparative alignments of two orthologous X Chromosome regions. (A) Alignment corresponding to ∼51–53 Mb of the human X Chromosome. The mouse X Chromosome has been rearranged within this region, and the two distant regions are shown. (B) Alignment corresponding to 154–155.6 Mb of the human X Chromosome. Genes located within the distal portion of the alignment are found at noncontiguous regions within the mouse X Chromosome. Ampliconic regions are delineated by black bars within the ideograms, and lineage specific loci are shown in red.
Content summary of X Chromosome assemblies
Mueller et al. (2013) reported 3.15 Mb and 19.4 Mb of ampliconic sequence for human and mouse, respectively. Our reanalysis based on the same criteria (see Methods), but with newer genome assemblies, increased the amount of ampliconic sequence to ∼6.8 Mb and ∼20.6 Mb, respectively (Supplemental Tables S10, S11). The cat X Chromosome contains ∼5.1 Mb of ampliconic sequence, with the majority of amplicons located within the middle one-third of the chromosome (Fig. 4; Supplemental Table S12). The pig X Chromosome contains only ∼1.9 Mb of ampliconic sequence, which is also concentrated within the middle third of the chromosome (Fig. 4; Supplemental Table S13).
Interspecific comparison of four mammalian X Chromosomes. Ideograms of the human, cat, pig, and mouse X Chromosomes with areas of conserved synteny shown in colored bands. For each X Chromosome, the ampliconic regions are shown to the right as black bars, and the locations of lineage-specific gene gains are to the right of these bars depicted as black dots. Regional rates of recombination are plotted along the length of each X Chromosome, and the shared recombination cold spot is outlined in the dashed red box for the human, domestic cat, and pig.
The chromosomal positions of ampliconic regions were frequently conserved across ordinal lineages. For example, the human ampliconic region located at 51.67–52.92 Mbp shares orthologous genes with both the domestic cat and the pig, although each lineage does appear to have acquired novel genes within the syntenic region (Fig. 3A). Furthermore, ampliconic human genes shared by other species fall within ampliconic sequence in some species but fall outside of ampliconic boundaries in others (e.g., NUDT10, EZHIP [previously known as CXorf67], NUDT11) (Fig. 3A). An ampliconic region near the distal end of the human X Chromosome (∼154 Mb), contains 30 genes conserved across all four species, but only 15 of these genes fall within ampliconic sequence in the domestic cat, and none are found in ampliconic sequence within the pig and mouse chromosomes (Fig. 3B; Supplemental Table S4).
Differences in genome annotation quality between human and the pig and cat may contribute to an underestimation of both ancestral and lineage-specific ampliconic genes within the latter two species. Given this variability in annotation quality, we classified ancestral ampliconic gene regions based on two criteria: (1) conserved gene content, or presence of similar gene family members; and (2) physical proximity to ampliconic sequence found within at least one other species (i.e., within 1 Mb of aligned conserved genes between species). Using these criteria, we identified 17 of 23 human ampliconic regions (82%) that were ancestral, 14 of 17 cat ampliconic regions (94%) as ancestral, and 15 of 20 pig ampliconic regions (75%) as ancestral. In contrast, only 12 of 21 (54%) mouse ampliconic regions were identified as ancestral (Supplemental Table S14).
On the human X Chromosome, 68% of the ampliconic genes encode proteins from the cancer-testis antigen (CTA) family (Simpson et al. 2005; Chen et al. 2011; Fratta et al. 2011). CTA proteins are named as such because they are expressed in a variety of human tumors, but their normal expression is generally restricted to the male germ line and have poorly understood functions. X-linked CTA genes are predominantly expressed in spermatogonia, the mitotically proliferating germ cells in the testis (Fratta et al. 2011). We searched for previously undetected multicopy or ampliconic CTA genes in the new long-read X Chromosome assemblies of cat and pig and evidence of orthologs in other placental mammals using the Ensembl gene family database. Fourteen of 16 human multicopy or ampliconic CTA gene families were present in one or more placental mammal orders (Table 3; Supplemental Figs. S1–S15), indicating that the majority of ampliconic gene families arose early in placental mammal evolution, and that most human ampliconic gene families were not recently acquired.
Evolutionary conservation of human CTA ampliconic gene families
There are two apparent exceptions: SPANX and CSAG families do not have any clear orthologs outside of primates, and these two proteins are postmeiotically expressed (Chen et al. 2011). However, Hansen et al. (2008) provided evidence that the VCX, SPANX, and CSAG families evolved rapidly but shared amino acid and promoter sequence homology to one another. These authors further suggested that these three primate gene families share a common X-linked ancestor with the murine ampliconic Spanx and Cypt orthologous gene families.
In contrast, the mouse X Chromosome possesses only 50% of the ancestral ampliconic CTA gene families (Table 3), having lost orthologs of the CSAG, CT45, GAGE, MAGEC, PAGE, SAGE, SPANX, and XAGE gene families. Most of the independently acquired mouse ampliconic genes are shared by other members of the family Muridae (rats and mice) or Muridae + Cricetidae with an origin ∼30 million years ago (Supplemental Tables S4, S8). The majority of the evolutionarily recent and expanded gene families are postmeiotically expressed. However, three of the largest ampliconic gene families, Rhox, Xlr, and Slx, previously described as independently acquired in the mouse lineage, actually represent X-linked mouse lineage duplications of an ancient X-linked SYCP3L gene family that is shared across mammals from several superordinal clades (Table 3; Supplemental Table S4).
Mouse lineage-specific evolutionary breakpoint regions (EBRs) were frequently associated with ancestral ampliconic sequence that flanked or spanned the EBR (Figs. 3, 4; Supplemental Table S4). For example, two mouse ampliconic regions located at ∼3–5 Mb and ∼149.3 Mb coincide with an ancestral placental ampliconic region located between 51 and 53 Mb in human (Figs. 3, 4; Supplemental Tables S4, S14). However, the ampliconic gene content is entirely different in the two species, with loss of an ancestral MAGED cluster and the emergence of two testis-specific ampliconic gene families in mouse: Btbd35 and Ott-like. Thus, one ancestral amplicon gave rise to two unlinked mouse amplicons that each became populated by novel protein-coding genes. The other end of the mouse lineage-specific inversion corresponds with a different ampliconic region shared between the human and pig X Chromosomes (Supplemental Tables S4, S14). We also generated recombination rate profiles along the length of each X Chromosome to examine the effect of local recombination rate on ampliconic gene retention (Fig. 4; Kong et al. 2002; Ma et al. 2010; Li et al. 2016; Simecek et al. 2017). Ampliconic regions were concentrated within the large recombination cold spots conserved in human (∼39%), cat (80%), and pig (∼70%) (Li et al. 2019).
Evolution of genomic elements involved in X-Chromosome inactivation
To determine if the bipartite structure formed during primate and rodent female XCI was conserved in laurasiatherian mammals, we generated a domestic cat Hi-C contact map using Hi-C data phased from an F1 Bengal hybrid (Bredemeyer et al. 2021). The resulting domestic cat haplotype map confirmed formation of a bipartite structure with DXZ4 retaining its role as the hinge region, indicating this unique structural conformation of the inactive X Chromosome was an ancestral feature in the common ancestor of boreoeutherian mammals and likely all placental mammals (Fig. 5A). In contrast, the alternative Asian leopard cat haplotype was not organized into super domains and instead exhibited robust A/B compartmentalization and TAD organization, characteristic of an active X Chromosome (Supplemental Fig. S16). This discrepancy between haplotypes suggests possible skewing of XCI in favor of a domestic cat Xi in the F1 Bengal hybrid, a phenomenon previously described in interspecific rodent crosses (Deng et al. 2015; Darrow et al. 2016).
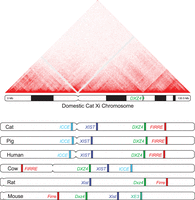
Spatial organization of loci previously associated with X Chromosome structural organization. (A) Hi-C contact map of the domestic cat inactive X Chromosome reveals conservation of the unique bipartite structural conformation and role of DXZ4 as a hinge region between superdomains (resolution = 500 kb, balanced normalization). (B) Interspecific comparison of long-range interacting loci reveals that relative position and linear spacing along the X Chromosome is conserved across highly divergent mammalian clades.
Next, we tested the hypothesis that mammals with X Chromosome rearrangements relative to the ancestral order would, through compensatory inversions, retain the same order and spacing of the four Xi escapee loci involved in superloop formation due to interaction constraints during XCI—here, termed the “inversion compensation hypothesis.” The four loci retain the same order and relative spacing in the mouse genome (Fig. 5B), despite approximately eight X Chromosome inversions that are estimated to have occurred on the ancestral branch leading to mouse (Pevzner and Tesler 2003b). One of these loci, the macrosatellite ICCE, was lost in the murid rodent ancestor of the mouse and rat. ICCE is known to interact with DXZ4 during XCI (Westervelt and Chadwick 2018). A lineage-specific mouse-specific amplicon, XE3, occurs ∼ 47.4-Mb from Dxz4 (Fig. 5B), nearly the same proportional distance between DXZ4 and ICEE in human, cat, and pig. It is noteworthy that XE3 also shares similar active epigenetic features (i.e., H3K4me2) with other loci that escape XCI, suggesting it may have acquired a convergent MSR function in the mouse lineage due to the loss of ICCE (Darrow et al. 2014).
As a phylogenetically independent test of the inversion compensation hypothesis, we examined the order and spacing of these same four loci in two other high-quality assemblies. DXZ4 is a complex macrosatellite that is not fully assembled in most genome assemblies but is consistently located adjacent to the PLS3 gene in genomes where it is resolved. Therefore, we used PLS3 as a proxy for the location of DXZ4 in the cattle genome. The cow X Chromosome is distinguished from other cetartiodactyls by multiple rearrangements that are known to have occurred in the ancestor of Bovini (Proskuryakova et al. 2017). Despite this, the four interacting loci that escape inactivation also remained in the same order observed in the other placental mammals, although the spacing between FIRRE and DXZ4 is much larger than in other species (Table 4; Fig. 5B). A similar pattern was also apparent in rat for the three loci that are conserved (Dxz4, Firre, and Xist), with the proportional distances between the loci being very similar to human, cat, and pig (Table 4).
Chromosome locations of interacting loci during X-Chromosome inactivation
Discussion
In this study, we conducted a fine-scale multispecies comparison of placental mammal X Chromosome gene content to reevaluate the hypothesis that a majority of X-linked ampliconic genes have been independently acquired in different ordinal lineages. Our study increased the number of species that were included in previous studies (Mueller et al. 2013), taking advantage of long-read-based assemblies that were not previously available. We also increased the scope of the comparison, including the domestic cat and pig from the superordinal clade Laurasiatheria, the sister clade of Euarchontoglires (which includes rodents and primates), which provides a more comprehensive sampling of placental mammals from which to draw conclusions regarding ancestral gene content.
Our results demonstrate that the majority of ampliconic genes and sequence in cat, pig, and human occur in positional orthology across the X Chromosome, and each species possesses one or more members of the same CTA gene family. Ancestral ampliconic genes were also found to be enriched for CTA genes that are expressed in early spermatogenesis and are restricted to the X-conserved region, the portion of the X Chromosome conserved between placental mammals and marsupials (Spencer et al. 1991). In contrast, the more recently acquired human and mouse X-linked ampliconic genes are all expressed in later stages of spermatogenesis (Mueller et al. 2008). Many of the mouse lineage-specific genes arose within ancestral ampliconic sequence. Ampliconic regions are unique in that they are typically large hypomethylated domains which may evolve as a mechanism to regulate this unique class of germline-specific genes (Ikeda et al. 2013). We speculate that these epigenetic features are conducive to the maintenance of germline-specific gene expression and may have provided the necessary environment for their recurrent emergence.
At least one subset of ampliconic CTA genes, Mageb1-3, is hypothesized to be involved in XCI (Li et al. 2019) and suggests that CTA gene families evolved early during placental mammal evolution coincident with the evolution of sex chromosome silencing mechanisms. Therefore, it appears unlikely that lineage-specific acquisition of ampliconic genes contributed to the KPg radiation of placental orders (Mueller et al. 2013), which other studies have instead linked to other environmental factors (Meredith et al. 2011; Springer et al. 2019). Rather, ancestral placental X-linked ampliconic gene families were characterized by random gene loss, retention, and expansion in different ordinal lineages following their diversification in the Paleogene.
The mouse ampliconic gene repertoire is exceptional in having both lost and expanded a comparatively large number of preexisting X-linked gene families (Fig. 2). Most of this activity occurred during the last 30 million years when the murid X Chromosome was rearranged through a series of inversions. Mammals from each ordinal lineage later acquired a smaller repertoire of novel ampliconic gene families, some which have evolved rapidly (e.g., SPANX, VCX, CSAG, Cypt) and have unclear sequence orthology. However, some authors have concluded that these four gene families have X-linked origins (Hansen et al. 2008), and therefore many apparent independently acquired genes (Fig. 2) may have undetectable X-linked orthologs due to rapid sequence divergence. We conclude that the majority of multicopy or ampliconic genes on extant placental mammal X Chromosomes are derived from ancient X-linked gene families and were thus not independently acquired.
The extent of conserved linkage (the conservation of gene order) among mammalian X Chromosomes is far greater than any ancestral autosomal synteny blocks (Murphy et al. 2005, 2007; Kim et al. 2017). Rodríguez Delgado et al. (2009) speculated that the conserved X Chromosome collinearity observed in most placental mammals may have been influenced by selective constraints on XCI. Li et al. (2019) showed that the landscape of X Chromosome recombination rate was conserved across several placental mammal orders and paralleled many of the genic and structural features of XCI. Here, we extend and integrate these two observations by providing evidence that the conservation of X linkage was driven by constraints that maintained the order and spacing of macrosatellite loci involved in superloop formation and the bipartite structure during XCI (Darrow et al. 2016; Bonora et al. 2018).
This hypothesis is bolstered by three compelling observations. First, all species with rearranged X Chromosomes possess the same order and, in nearly all cases, spacing of macrosatellite repeats found in the ancestral placental X Chromosome configuration. Manipulation of the order and orientation of these loci would reverse the directionality of the CTCF binding motifs embedded within macrosatellite repeat units, preventing formation of the long-range superloops required for formation of the Xi chromatin conformation (Bonora et al. 2018). Second, X-linked satellite arrays are epigenetically distinct in that they reside in euchromatic bands amid the heterochromatic background of the inactive X Chromosome, reflecting the chromosome-wide architecture that is under evolutionary constraint for three-dimensional folding (Chadwick and Willard 2004; Chadwick 2008; Horakova et al. 2012). Third, the largest conserved linkage block (∼45–50 Mbp) includes the X-Chromosome inactivation center (XIC) and is characterized by very low rates of recombination across >90% of its length in pig and cat. This block also possesses the highest density of ampliconic regions (>74%) and is flanked distally by the smallest conserved linkage block that includes DXZ4, which is critical to formation of the bipartite structure during XCI. The breakpoints flanking Dxz4 were reused during the recent evolution of murid rodents, which maintained the position and orientation of Dxz4 relative to other macrosatellite loci with which it forms superloops during XCI.
Another ancestral DXZ4-interacting partner, the ICCE macrosatellite, was lost from an intron of the mouse and rat Nbdy gene (Westervelt and Chadwick 2018). We speculate that this loss may have occurred because its new, derived chromosome location was too distal to effectively form superloops with Dxz4. However, a novel tandem repeat locus, XE3, arose on the mouse X Chromosome at nearly the same physical and proportional distance from Dxz4 that separates DXZ4 and ICCE in other placental mammals. Although there is no evidence that XE3 forms superloops with Dxz4, Firre, and Xist, it is striking that XE3 also coincides with a band of euchromatic histone modifications and also appears differentially packaged on the active and inactive X Chromosomes (Westervelt and Chadwick 2018). Given the convergent chromosome position and epigenetic features of murid XE3 and ICCE found in other placental mammals, we speculate that XE3 may have evolved to compensate for a function lost with the ICCE tandem repeat. Additional studies of the XE3 locus would be informative in this regard.
The long-term reduction in recombination rate across the largest X Chromosome linkage block is remarkable. One hypothesis to explain its persistence is that the conserved flanking hot spots have acted as recombination “sinks” that substantially suppressed recombination within the intervening sequence while maintaining linkage of critical loci that extend outward to initiate the spreading of XIST signal. Evolutionary constraints to maintain the spacing and distribution of cis-interacting loci may also have limited the expansion of ampliconic sequence that was acquired within the largest recombination cold spot. A release from the ancestral physical constraints on ampliconic sequence acquisition in the form of chromosome rearrangements may have permitted the expansion of ampliconic gene families specialized for spermiogenesis, as the murid rodent X Chromosome acquired a markedly new chromosome architecture during its ∼30–40-million-yr radiation. Because ampliconic sequences are often located within large hypomethylated domains found at mouse X Chromosome evolutionary breakpoints, we hypothesize that this epigenetic environment may have favored evolutionary breakpoints as engines of genetic novelty during murid rodent evolution. The future generation of high-quality, gapless assemblies from additional mammalian species with rearranged X Chromosomes will provide opportunities to test these findings.
In summary, we demonstrated that the majority of multicopy or ampliconic X-linked genes in the finished human and mouse genome assemblies are derived from ancient X-linked gene families present in the ancestral placental mammal genome. This conclusion highlights the importance of both broad taxonomic sampling and inclusion of high-quality genome assemblies and annotations when attempting to infer ancestral versus lineage-specific patterns of gene gain and loss in the early stages of mammalian evolution. Ancestral ampliconic CTA gene families have been marked by extensive gene gain and loss in different ordinal lineages. They are enriched for CTAs that are expressed in early spermatogenesis, whereas the recently acquired human and mouse X-linked ampliconic genes are all expressed in later stages of spermatogenesis (Mueller et al. 2008). The conservation of gene order observed across the majority of placental mammals is likely attributable to strong selective constraints on the three-dimensional genomic architecture necessary for X-Chromosome inactivation. Species with rearranged X Chromosomes have retained the ancestral order and relative spacing of loci critical for superloop formation during X-Chromosome inactivation, suggesting that selection for compensatory inversions evolved to maintain these long-range physical interactions.
Methods
BAC clone sequencing, assembly, and annotation
We conducted sequencing and assembly of BAC clones from the Felis catus female BAC library FSCC from Amplicon Express. Clones were chosen based on the mapping location of the BAC end sequences (BES) aligned to the domestic cat felCat8.0 genome assembly. Briefly, clones with both BES mapping to either side of a gap within the assembly, or a single end uniquely mapped adjacent to a gap, were selected for sequencing. Selected clones were cultured and DNA was extracted using standard protocols. Clone DNA was pooled into three separate groups to minimize potential overlap of orthologous BAC regions, and sequenced using the PacBio Sequel system.
Given the previously described disparities in the abilities of different assembly pipelines and parameters to accurately reconstruct complex regions of the genome (Khost et al. 2017), we assembled each pool separately using both the Celera 8.3rc2 (Myers et al. 2000; Koren et al. 2012, 2013; Berlin et al. 2015) and Canu (Koren et al. 2017) pipelines with a variety of parameters. Raw PacBio reads were mapped to each assembly with BLASR using default settings (Altschul et al. 1990), and the resulting alignment was used to refine each assembly using Arrow.
In order to remove any sequences not originating from the domestic cat genome, each assembly was aligned to the Escherichia coli genome using BLAST (Altschul et al. 1990) in Geneious (Kearse et al. 2012), and the resulting alignments were examined by eye to confirm and remove any contaminants. The vector sequence used in the BAC library was then mapped to the remaining sequences using LASTZ (version 1.02.00) (Harris 2007), in order to identify and remove any vector present in the assembled sequences.
Next, we downloaded all available BAC-end sequences from NCBI and mapped these to each assembly, allowing us to identify ends of separate BAC clones that had been assembled together due their overlap within the genome. These assemblies were then aligned using MAFFT (Katoh and Standley 2013) and were visually inspected to identify any major disparities. If none were found, the consensus sequence from the alignment was used as the representative sequence for the clone. In those cases where the different assembly pipelines produced assemblies with major disparities, the consensus sequence for the longer sequences was used, as misassembly of ampliconic regions usually results in the collapse of adjacent segmental duplications.
The assembly for each clone was then incorporated into the X Chromosome scaffold of the PacBio long-read genome assembly (version 9.0) based on the mapping of BAC-end sequences and BLAST alignments. We removed any unincorporated contigs from the genome assembly file that appeared to be covered by our sequenced clones and mapped Illumina whole-genome sequence data from the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) accession number SRR5055389 onto the new genome that included incorporated clone assemblies using BWA-MEM (Li and Durbin 2009). We then used the resulting alignment files and Pilon (Walker et al. 2014) to correct the assembly. The quality of the incorporated BAC clone sequences were then assessed by mapping Illumina whole-genome sequence data from the domestic cat to the updated assembly and subsequently checking mapping statistics using SAMtools (Supplemental Table S3; Li et al. 2009).
After incorporating the assembled BAC clone sequences, we aligned RNA-seq data from two testis (SRR1981105 and SRR3200462), cerebellum (SRR3218718), kidney (SRR3200460), heart (SRR3200471), lung (SRR3200449), and uterus (SRR3200458) tissues from the domestic cat using STAR (Dobin et al. 2013) with default parameters. Transcripts were assembled using Cufflinks (Trapnell et al. 2012). Assembled transcripts originating from the newly incorporated BAC sequences were then assessed for protein-coding potential, requiring a minimum open reading frame of 400 nucleotides and detectable expression in at least two samples. These stringent parameters were chosen to minimize the possibility of falsely inflating the number of novel protein-coding genes within the ampliconic category, as the regions of the felCat9.0 X Chromosome assembly that were improved in this study consisted primarily of ampliconic content. The resulting transcripts were then aligned to the felCat9.0 genome using BLAST (Altschul et al. 1990) to ensure that they were not already present within the felCat9.0 annotation, as well as to assign chromosome coordinates to the loci for incorporation into our multispecies alignments of X Chromosome gene annotations.
Interspecific X Chromosome comparisons
We downloaded the annotation files for the human (GRCh38.p12), domestic cat (Felis_catus_9.0), pig (Sscrofa11.1), and mouse (GRCm38.p6) genomes and manually aligned the X-linked gene annotations (Supplemental Table S4). To confirm the results of our manual alignment of the annotation files, we conducted pairwise alignments of the cat, pig, and mouse X Chromosomes to the human X Chromosome using NUCmer (Delcher et al. 2002).
We identified ampliconic regions by conducting self-alignments for each X Chromosome assembly using NUCmer (Delcher et al. 2002) using the ‐‐maxmatch and ‐‐nosimplify parameters. Alignments were then filtered to remove any self-aligned sequences, or sequences that were <99.0% or <10 kb in length. We then extended and merged ampliconic regions that were within 500 kb of one another, following Mueller et al. (2013) (Supplemental Tables S6–S9). Figures for the annotation alignments, including the locations of ampliconic loci, recombination rates, and novel genes, were constructed with karyoploteR (Gel and Serra 2017).
Expected values for all χ2 analyses were normalized to account for differences in the lengths of ampliconic and nonampliconic regions, or between the length of the recombination desert and the two flanking regions. For example, when testing for the enrichment of novel loci in ampliconic regions, if the X Chromosome was comprised of 5% ampliconic sequence and contained 100 ampliconic genes, the number of novel genes expected to be ampliconic was five and nonampliconic 95. Identification and localization of macrosatellites across the different species was performed manually using Geneious and the NCBI Genome Data Viewer. We began by comparing annotations overlapping macrosatellites in the human reference assembly (GRCh38.p13) to annotated reference genomes for the cat (felCat9), pig (Sscrofa11.1), cow (ARS-UCD1.2), rat (Rnor_6.0), and mouse (GRCm39) using BLAST (Altschul et al. 1990). Following a successful BLAST hit, we manually investigated surrounding regions for enrichment of CTCF binding motifs and tandem repeat structure visualized using self-self dotplots and GC content traces.
We identified additional placental X-linked orthologs for ampliconic/CTA gene families by searching for orthologs in gene trees using the Ensembl database (release 101). We determined X-linked ancestry (ancestral vs. lineage-specific) for each gene by finding chromosome locations/coordinates for chromosome-level genome assemblies.
Hi-C data analysis
F1 Bengal Hi-C data and single haplotype parental assemblies were downloaded from the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) accession numbers PRJNA670214 and PRJNA682572 and phased into parental haplotypes as described in Bredemeyer et al. (2021). Hi-C maps were generated by mapping the domestic and Asian leopard cat Hi-C reads to their respective single haplotype assembly using Juicer v1.5.7 (Durand et al. 2016a) with option -s none selected for compatibility with DNase Hi-C libraries. The resulting maps were visualized using Juicebox v1.11.08 (Durand et al. 2016b).
Data access
The PacBio data and domestic cat X Chromosome assembly generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA717798.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Nicole Foley, Gang Li, Terje Raudsepp, Paul Samollow, Christopher Seabury, Jinhong Wang, and Wesley Warren for helpful discussions, technical support, and/or comments and advice on an earlier draft of this manuscript. This work was supported by Morris Animal Foundation grant D16FE-011 to W.J.M.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275274.121.
- Received January 18, 2021.
- Accepted June 22, 2021.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








