
Rapid and accurate alignment of nucleotide conversion sequencing reads with HISAT-3N
Abstract
Sequencing technologies using nucleotide conversion techniques such as cytosine to thymine in bisulfite-seq and thymine to cytosine in SLAM seq are powerful tools to explore the chemical intricacies of cellular processes. To date, no one has developed a unified methodology for aligning converted sequences and consolidating alignment of these technologies in one package. In this paper, we describe hierarchical indexing for spliced alignment of transcripts–3 nucleotides (HISAT-3N), which can rapidly and accurately align sequences consisting of any nucleotide conversion by leveraging the powerful hierarchical index and repeat index algorithms originally developed for the HISAT software. Tests on real and simulated data sets show that HISAT-3N is faster than other modern systems, with greater alignment accuracy, higher scalability, and smaller memory requirements. HISAT-3N therefore becomes an ideal aligner when used with converted sequence technologies.
Nucleotide conversion (NC) techniques coupled with sequencing technologies are powerful tools that probe the chemical intricacies of nucleic acids by making detectable chemical modifications to nucleic acids, thereby gaining insight into highly dynamic cellular processes (Lister et al. 2009; Nishikura 2016; Muhar et al. 2018). Bisulfite sequencing (BS-seq) (Frommer et al. 1992) is one of the most well-known NC sequencing technologies. It uses bisulfite treatment to convert unmethylated cytosine to thymine in DNA molecules while leaving the methylated or hydroxymethylated cytosine (5mC or 5hmC) untouched. BS-seq data have been used to qualitatively and quantitatively interrogate DNA methylation locations in genomic DNA for many years (Frommer et al. 1992). More recently, TET-assisted pyridine borane sequencing (TAPS) (Liu et al. 2019) technology has overcome the problem of producing low-complexity sequences, which is produced in the destructive bisulfite treatment of BS-seq. Another NC technique that has gained traction is known as thiol (SH)-linked alkylation for metabolic sequencing of RNA (SLAM seq), which introduces 4-thiouridine (s4U) into living cells to replace uracil occasionally during the process of transcription in nascent RNA transcripts (Herzog et al. 2017). During the reverse transcription stage of the sample preparation process, the s4U is paired with guanine instead of adenine, thus introducing guanine into the reverse strand. Sequencing reads are then generated with cytosine in place of thymine in the original sequence during PCR. Standard RNA-seq (Mortazavi et al. 2008) can be used to detect adenosine-to-inosine RNA editing events by converting the modified adenosine (inosine) to guanosine during the reverse transcription. As sequencing technologies rapidly advance, NC technologies are likely to be combined with single-cell sequencing technology such as scBS-seq (Smallwood et al. 2014) and scSLAM-seq (Erhard et al. 2019), conveniently enabling researchers to understand various cellular processes at single-cell resolution. These NC sequencing technologies necessitate alignment strategies different from those in commonly used aligners such as Bowtie 2 (Langmead et al. 2009; Langmead and Salzberg 2012), BWA (Li and Durbin 2009), STAR (Dobin et al. 2013), and HISAT2 (Kim et al. 2015, 2019), in which converted nucleotides are treated as mismatches, often leading to the placement of sequences at incorrect genomic locations or failing to align them altogether. The inability to handle NC sequences leads to substantial biases, including the omission of underexpressed genes before downstream analysis.
To handle this erroneous alignment issue, several alignment programs were developed for aligning one specific type of NC sequencing data such as BS-Seq or SLAM seq reads. Bismark (Krueger and Andrews 2011), a widely adopted aligner primarily developed for BS-seq data, uses a three-nucleotide (nt) alignment strategy; that is, all cytosines in both the reference genome and sequencing reads are converted to thymines before alignment, so that there are only three nucleotide types (A, G, T) used in the alignment process. Bismark then uses Bowtie 2 or HISAT2 as an underlying sequence aligner. These sequence aligners accurately handle reads that can be uniquely mapped; however, they produce a randomly selected alignment when a read is mapped to multiple locations, with the mapping accuracy generally inversely proportional to the number of mappable locations. Because Bismark internally runs at least four instances of a sequence aligner (e.g., Bowtie 2), it requires a substantial amount of memory (e.g., >20 GB for human DNA sequences) and requires additional steps to handle and combine alignment outputs, which consume >80% of total runtime. SLAM-DUNK (Neumann et al. 2019), an aligner primarily developed for SLAM seq reads, uses NextGenMap (Sedlazeck et al. 2013) as an underlying aligner. NextGenMap's k-mer table-based alignment strategy enables fast mapping of reads, but NextGenMap is not designed to handle RNA-seq reads, especially those spanning multiple exons. This shortcoming of SLAM-DUNK could lead to incorrect alignment for such multiexon spanning reads.
To overcome these limitations and improve speed and accuracy for NC read alignments, we have developed HISAT-3N, an extension of HISAT2 that implements the 3-nt alignment algorithm. Compared with other 3N alignment programs, which are tied to a specific NC sequencing technology, HISAT-3N is the first general program that can handle any NC reads including BS-seq and SLAM seq, RNA, or DNA. Users can study the dynamic cellular processes accurately and efficiently with HISAT-3N. Thus, HISAT-3N is the ideal option for handling NC alignment.
Results
We evaluated the performance of HISAT-3N and compared it to other commonly used NC aligners: Bismark, BS-Seeker2 (Guo et al. 2013), and BSMAP (Xi and Li 2009) for BS-seq. Note that we include BS-Seeker2 in our evaluation instead of its successor, BS-Seeker3 (Huang et al. 2018), as BS-Seeker2 has better alignment capabilities with BS-seq reads of various C-to-T conversion rates. Also, unlike other evaluated programs, BS-Seeker3 aligns reads in only one direction. The ability to align in both directions, namely, forward and reverse complement, is an important feature to be supported in aligners as nondirectional BS-seq libraries are often used. We used two data sets to evaluate the BS-seq alignment performance of the programs: (1) 10 million simulated 100-bp paired-end DNA reads with a 50% C-to-T conversion rate and a 0.2% per-base sequencing error rate to evaluate the performance for nondirectional whole-genome bisulfite-seq read alignment, and (2) 78 million real paired-end whole-genome BS-seq reads (NCBI Sequence Read Archive [SRA; https://www.ncbi.nlm.nih.gov/sra] accession SRR3469520) (The ENCODE Project Consortium 2012). The real reads are trimmed to remove adaptor before alignment. We used the simulated data sets to estimate the alignment rate and accuracy of each aligner. We define alignment rate here as the number of aligned read pairs (or reads) divided by the total number of read pairs (or reads). Alignment accuracy refers to the number of correctly aligned read pairs (or reads) divided by the total number of read pairs (or reads). Multialigned read pairs (or reads) are classified as correctly aligned if any one of multiple alignments is correct.
HISAT-3N exhibits higher alignment accuracy in simulated BS-seq data compared with Bismark, BS-Seeker2, and BSMAP (Table 1). Using HISAT-3N with only the 3N index, 98.52% of paired-end reads are mapped to the correct locations. Using HISAT-3N with both the 3N and repeat indexes improves alignment accuracy to 99.36% and requires only 10% more processing time. BSMAP was the fastest (11 min), closely followed by HISAT-3N (13 min). HISAT-3N is 7 and 23 times faster than Bismark and BS-Seeker2, respectively. We also tested the alignment speed and rate on experimentally derived real data. When the input data size is large (for instance, 78 million paired-end reads), the speed advantage of HISAT-3N becomes clear (Table 2). HISAT-3N only took about 1.5 h to map the data, whereas Bismark and BS-Seeker2 required ∼17 h and 50 h, respectively. BSMAP implements an algorithm of creating every possible combination of C-to-T conversions in reads, which apparently makes the program much slower in aligning longer reads, as found in our analysis of BSMAP with 125-bp-long BS-seq read data (Table 2), in which BSMAP takes 50%–70% more time than HISAT-3N.
Performance comparison for HISAT-3N, Bismark, BS-Seeker2, and BSMAP on 10 million simulated 100-bp paired-end BS-seq reads
Performance comparison for HISAT-3N, Bismark, BS-Seeker2, and BSMAP on 78 million real whole-genome paired-end BS-seq reads
Then we evaluated the performance of HISAT-3N and SLAM-DUNK for SLAM seq. Our test data sets for SLAM seq alignment include (1) 10 million simulated 100-bp single-end reads generated from 3′ regions of transcripts using realistic settings, for example, with a 2% T-to-C conversion rate and a 0.2% per-base sequencing error rate, and (2) 45 million experimentally derived single-end SLAM seq reads (SRA acccesion SRR5806774) (Muhar et al. 2018). HISAT-3N also showed high alignment accuracy for the simulated SLAM seq data (Table 3). SLAM-DUNK runs faster than HISAT-3N for alignment. However, the alignment rate and accuracy for HISAT-3N are substantially higher than those for SLAM-DUNK. Although SLAM-DUNK has a similar alignment rate to HISAT-3N, 26.58% of the reads are partially mapped (not aligned end to end). Furthermore, because SLAM-DUNK cannot handle spliced alignment, there is a higher chance of aligning reads to incorrect positions. The alignment accuracy for HISAT-3N with repeat index reaches 99.81% versus 95.59% for SLAM-DUNK with 1000 alignment searching.
Performance comparison between HISAT-3N and SLAM-DUNK on 10 million simulated 100-bp single-end SLAM seq reads
When testing aligners with real SLAM seq data (Table 4), we found that HISAT-3N maintained a high alignment rate (97.25%), although it ran slightly slower than SLAM-DUNK. SLAM-DUNK aligned 99.17% of the reads when we set the maximum number of alignments to 1000. Based on the results of our simulation reads, the alignment accuracy for SLAM-DUNK is only 95.59%; thus, we infer that SLAM-DUNK results for the real reads could include many incorrect alignments.
Performance comparison between HISAT-3N and SLAM-DUNK on 45 million real single-end SLAM seq reads
Discussion
We have used and expanded the HISAT2 index and alignment algorithms to develop HISAT-3N, which is specifically designed for NC sequencing reads. HISAT-3N has five key advantages over other NC aligners such as Bismark and SLAM-DUNK, making HISAT-3N versatile and easily used: (1) HISAT-3N's 3-nt alignment algorithm supports virtually any type of NC sample preparation protocol rather than being specific to one technology; (2) HISAT-3N uses the same index to map different types of NC reads (e.g., BS-seq, TAPS, and SLAM seq); (3) HISAT-3N seamlessly handles 3-nt DNA or RNA data; (4) HISAT-3N runs much faster owing to bypassing the time-intensive steps of writing and reading the intermediate alignment results from disk; and (5) HISAT-3N provides an option to report all multimapped reads regardless of the number of mapped positions (e.g., 1000 alignment positions), which considerably increases the alignment accuracy.
HISAT-3N uses the same conversion index for handling BS-seq, SLAM seq, and any other conversion approaches as outlined in Table 5, thus making HISAT-3N protocol independent and more versatile and convenient for use. To the best of our knowledge, all known NC sequencing technologies, including SLAM seq, BS-seq, TAPS, TAB-seq (Yu et al. 2012), oxBS-seq (Booth et al. 2012), and standard RNA-seq, convert cytosine to thymine (or vice-versa) or convert adenine to guanine (or vice-versa); thus, the REF-3N and REF-RC-3N indexes (Methods) are sufficient for unifying alignment of all known NC sequencing data types. HISAT-3N also allows for incorporation of splice sites, single-nucleotide polymorphisms, and small insertions/deletions using a graph index structure available in the HISAT2 program. Like most other sequence aligners, the HISAT-3N index only needs to be built once and is used for aligning samples of different kinds without the need to build other indexes.
Summary of several nucleotide conversion sequencing methods
The information reduction of the genome and reads that is characteristic of the 3-nt algorithm obfuscates the true placement of the reads. Indeed, reads are mapped to more locations of the 3N reference compared with when the original reads are mapped to the original reference. We show this effect by aligning 10 million simulated 100-bp single-end DNA reads. The 4-nt alignment with HISAT2 shows that only 5.65% of reads can be mapped to more than one location. The 3-nt alignment with HISAT-3N shows 6.78% of reads are mapped to more than one location. Most aligners randomly report one or a subset of locations when reads are mapped to multiple locations. If users require an aligner to report all alignments, then the aligner may use 10 times more space for saving alignment outputs with a substantially increased runtime. We apply HISAT2's repeat index to resolve this multimapping problem (Fig. 1). This function first identifies one sequence to serve as a representative of all its identical sequences from the genome, which we designate as a repeat sequence. Reads are then directly aligned to that repeat sequence, producing one alignment per read. HISAT2 provides application programming interfaces (APIs) that enable the rapid retrieval of the genomic locations corresponding to the repeat alignment. The whole-genome index searching strategy (Fig. 1A, left) needs to search each mapping position one by one, which can be very time consuming. The repeat index searching strategy (Fig. 1A, right) can result in unique mapping and then expand the repeat mapping information to retrieve all the mapping positions. For multiple mapping, searching the repeat index can be substantially faster than the whole-genome index. This repeat mapping process only adds 10% more runtime compared with when exclusively 3N indexes are used. More specifically, on a subset of simulated 10 million single-end BS-seq reads, using the repeat index is three times faster than aligning reads using the two REF-3N indexes if the first 1000 mapping locations are searched and outputted (Fig. 1B). The repeat index enables the analysis for repetitive genomic regions such as methylation patterns; HISAT-3N provides an option to report multiple alignments, with the maximum number of alignments specified by the user. We plan to develop new statistical methods that incorporate multimapped reads for analyzing methylation patterns for such genomic regions.
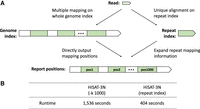
Repeat index enables faster 3-nt read alignment. (A) HISAT-3N aligns reads using two different strategies: (1) HISAT-3N can directly align reads to the whole genome using the genome index and output their mapped locations (A, left), and (2) HISAT-3N can use a repeat index to uniquely align reads to the repeat sequences regardless of how many locations to which they align on the genome (A, right). (B) Runtime comparison between direct mapping and repeat mapping strategy. The test data are 10 million simulated single-end BS-seq reads (0.2% per-base sequencing error rate).
HISAT-3N achieves >98% alignment accuracy for both simulated BS-seq and SLAM seq reads, which is substantially higher than the other aligners that we tested. Furthermore, we compared HISAT-3N with HISAT2 on the same simulated reads without NC (Supplemental Tables S1, S2). As we expected, HISAT-3N and HISAT2 have nearly identical alignment rate and alignment accuracy, showing that the 3N alignment algorithm in HISAT-3N can handle regular (no conversion) sequencing reads without decreasing accuracy. In this study, HISAT-3N's performance on aligning RNA-seq reads in identifying RNA-editing events is not evaluated, given the low frequency of RNA-editing events and the absence of real, high-quality benchmark data. In principle, HISAT-3N is certainly capable of aligning RNA-seq reads with editing events, especially reads that include edits near splice sites. Inherited from HISAT2, HISAT-3N also supports the graph genome with SNPs, including deletions of any length and insertions of ≤30 bp (e.g., a human reference genome with over 10 million SNPs). This could further increase the alignment accuracy of HISAT-3N.
Not only is HISAT-3N between 7 and 23 times faster than other NC sequence aligners like Bismark and BS-Seeker2, HISAT-3N scales better than these aforementioned programs. When HISAT-3N multithreading is increased from 16 to 24 threads, HISAT-3N is 11- and 36-fold faster compared with Bismark and BS-Seeker2, respectively (Table 6). Because Bismark and BS-Seeker2 have to open four Bowtie 2 simultaneously for the alignment process and only one thread for the alignment result filtration, the alignment speed is partially independent on the number of CPUs used. To use more CPUs for the filtering, Bismark and BS-Seeker2 need to open four additional Bowtie 2 and consume ∼16 GB more memory, making them impractical for a common personal computer. Although the other programs run 1%–20% faster with 24 threads than with 16 threads, HISAT-3N runs 50% faster with 24 threads than with 16 threads, showing the most scalability.
Scalability comparison between HISAT-3N, Bismark, BS-Seeker2, and BSMAP on 10 million simulated 100-bp paired-end BS-seq reads (0.2% per-base sequencing error rate)
Here we present HISAT-3N, a rapid, versatile sequence aligner that processes reads generated by all known NC sequencing technologies, including BS-seq, SLAM seq, TAPS, oxBS-seq, TAB-seq, scBS-seq, and scSLAM-seq. HISAT-3N combines a 3-nt alignment strategy with the major alignment improvements present in HISAT2 to rapidly perform all nucleotide conversions in memory without storing and reading intermediate alignment results as is performed by other aligners. This implementation substantially improves the processing speed and makes HISAT-3N an ideal choice for analyzing NC sequencing technology data in the modern era.
Methods
Design principle
HISAT-3N's 3-nt alignment method consists of four major steps: prealignment index building, read sequence conversion, 3-nt alignment, and result filtration. Here we use BS-seq data as an illustrative example without loss of generality, as other NC sequencing data types are similarly handled (Fig. 2). HISAT-3N first builds two HISAT2 indexes from the reference human genome (GRCh38), denoted here as REF. The first index is built on REF with cytosine changed to thymine, denoted as REF-3N. The second HISAT2 index is built on the reverse complement of REF with cytosine changed to thymine, denoted as REF-RC-3N. Although we have chosen cytosine to thymine to be our convention, the opposite conversion, namely, thymine to cytosine, works equally well for handling BS-seq data.
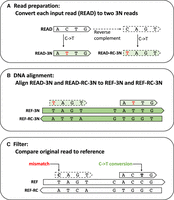
HISAT-3N alignment steps for BS-seq reads. (A) HISAT-3N converts each input read (READ) to two 3N reads: READ-3N and READ-RC-3N. READ-3N is READ with all thymine replaced by cytosine. READ-RC-3N is the reverse complement of READ, plus the replacement of cytosine with thymine. (B) HISAT-3N maps the two 3N reads to both REF-3N and REF-RC-3N references using prebuilt indexes. (C) After the 3-nt alignment, HISAT-3N compares the original read sequence (READ) to the original 4-nt references (REF and REF-RC) to identify unmethylated cytosine positions and recalculate an alignment score accordingly.
HISAT-3N uses FASTA- or FASTQ-formatted sequencing reads as inputs that can be compressed or uncompressed, single end or paired end. For each read, HISAT-3N generates two 3N reads (Fig. 2A): (1) READ-3N, read with cytosine changed to thymine; (2) READ-RC-3N, read that is first converted to its reverse complement, followed by changing cytosine to thymine. Note that in general READ-3N and READ-RC-3N are not reverse complements of each other.
Then HISAT-3N uses HISAT2 to align two versions of a read, READ-3N and READ-RC-3N, to both REF-3N and REF-RC-3N, involving a total of four alignment processes (READ-3N to REF-3N, READ-3N to REF-RC-3N, READ-RC-3N to REF-3N, and READ-RC-3N to REF-RC-3N) (Fig. 2B). Depending on sequencing data types, HISAT-3N performs nonspliced alignment for DNA-seq reads (e.g., BS-seq) and spliced alignment for RNA-seq reads (e.g., SLAM seq) (Fig. 2B; Supplemental Fig. S1, respectively).
After HISAT-3N aligns 3N reads to the 3N references, HISAT-3N compares the original read, READ, to the original genome reference, REF, for each alignment of 3N reads in order to identify converted nucleotide locations and mismatches (Fig. 2C). HISAT-3N then sorts the alignment results by alignment score (a function of the number of mismatches and indels) and reports the alignments with the best alignment score in the SAM format (Li et al. 2009). During the output process, HISAT-3N adds extra SAM tags (Supplemental Methods) to indicate the number of conversions and the aligned strand (REF or REF-RC). HISAT-3N directly performs this alignment readjustment of nucleotide conversions and mismatches directly in computer memory without storing and reading intermediate alignment results. Thus, this postprocessing step of HISAT-3N is relatively fast, taking <10% of its total runtime. HISAT-3N also provides a script to create a 3N-conversion table for methylated and unmethylated cytosines drawing from the sorted SAM alignment file, as described in the Supplemental Methods.
Repeat index and alignment
To build a repeat index, a set of identical sequences that are ≥40 bp and found in at least five locations of REF-3N and REF-RC-3N are combined into one repeat sequence. During the alignment process, if a read or its partial segments are mapped to five or more locations using 3N indexes (Fig. 1), the read will be directly mapped to the repeat sequences using the repeat index, resulting in one repeat alignment per read. Then, HISAT-3N expands the repeat alignment result and outputs the alignment information in standard SAM format. The repeat index and HISAT-3N's alignment algorithm enable reporting of all alignments for reads originating from repetitive genomic regions.
Testing environment
We tested the alignment programs using a computer system with 24 CPU cores (two Intel Xeon E5-2680 v3) and 256 GB of memory. Each aligner is configured to use 16 threads so that each thread is assigned one CPU core on this system. Bismark and BS-Seeker2 run four instances of Bowtie 2 for the alignment process, and each instance of Bowtie 2 uses four threads, resulting in a total of 16 threads.
Read adapter trimming
We trimmed the adapter sequence on real reads using Trim Galore! (Martin 2011; https://github.com/FelixKrueger/TrimGalore) and filtered out reads <40 bp to generate our real BS-seq and SLAM seq data. We also trimmed poly(A) sequences of the real SLAM seq reads using Trim Galore! (Supplemental Methods).
Software availability
HISAT-3N is open-source software. The source code for HISAT-3N can be found at GitHub (https://github.com/DaehwanKimLab/hisat2) and as Supplemental Code. HISAT-3N can be run under either a Linux or Max OS operating system. The project home page is available at https://daehwankimlab.github.io/hisat2/hisat-3n.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported in part by the National Institute of General Medical Sciences (NIH) under grant R01-GM135341 and by the Cancer Prevention Research Institute of Texas (CPRIT) under grant RR170068 to D.K.
Author contributions: Y.Z., C.P., C.B., M.T., and D.K. performed the analysis and discussed the results of HISAT-3N. Y.Z., C.P., and D.K. designed and implemented HISAT-3N. Y.Z. and C.P. performed the evaluations of the various programs. Y.Z., C.P., C.B., M.T., and D.K. wrote the manuscript. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275193.120.
- Received December 29, 2020.
- Accepted June 3, 2021.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








