
Ultrafast functional profiling of RNA-seq data for nonmodel organisms
- Peng Liu1,
- Jessica Ewald1,
- Jose Hector Galvez2,3,
- Jessica Head1,
- Doug Crump4,
- Guillaume Bourque2,3,
- Niladri Basu1 and
- Jianguo Xia1,2
- 1Faculty of Agricultural and Environmental Sciences, McGill University, Montreal, Quebec H9X 3V9, Canada;
- 2Department of Human Genetics, McGill University, Montreal, Quebec H3A 0C7, Canada;
- 3Canadian Center for Computational Genomics, McGill University, Montreal, Quebec H3A 0G1, Canada;
- 4Environment and Climate Change Canada, National Wildlife Research Centre, Ottawa, Ontario K1A 0H3, Canada
Abstract
Computational time and cost remain a major bottleneck for RNA-seq data analysis of nonmodel organisms without reference genomes. To address this challenge, we have developed Seq2Fun, a novel, all-in-one, ultrafast tool to directly perform functional quantification of RNA-seq reads without transcriptome de novo assembly. The pipeline starts with raw read quality control: sequencing error correction, removing poly(A) tails, and joining overlapped paired-end reads. It then conducts a DNA-to-protein search by translating each read into all possible amino acid fragments and subsequently identifies possible homologous sequences in a well-curated protein database. Finally, the pipeline generates several informative outputs including gene abundance tables, pathway and species hit tables, an HTML report to visualize the results, and an output of clean reads annotated with mapped genes ready for downstream analysis. Seq2Fun does not have any intermediate steps of file writing and loading, making I/O very efficient. Seq2Fun is written in C++ and can run on a personal computer with a limited number of CPUs and memory. It can process >2,000,000 reads/min and is >120 times faster than conventional workflows based on de novo assembly, while maintaining high accuracy in our various test data sets.
Genomics data, including RNA-seq, have become a core component of life science research. Although the majority of RNA-seq data have been derived from studies on model organisms, such data are increasingly being realized from studies on nonmodel organisms (da Fonseca et al. 2016; Matz 2018). Compared to well-established RNA-seq pipelines and web-based platforms developed for model organisms (Lohse et al. 2012; Zhou et al. 2019), there are several unique challenges when dealing with RNA-seq data from nonmodel organisms, including the lack of reference genomes and high-quality annotations, as well as the difficulties in obtaining large sample sizes especially from experimental settings. In general, RNA-seq studies in nonmodel organisms tend to have a relatively simple experimental design in which the main objective is to identify differentially expressed genes (DEGs) and perturbed pathways between study groups (da Fonseca et al. 2016).
Gene- or pathway-level analysis for species that do not have reference genomes relies heavily on the construction and annotation of their transcripts (Martin and Wang 2011; Eldem et al. 2017; Voshall and Moriyama 2018). The conventional RNA-seq workflow involves the use of multiple software tools to conduct raw reads quality checks, read error correction, transcriptome de novo assembly, transcriptome quality assessment, transcriptome annotation, and downstream analysis, including identification of DEGs and pathway enrichment analysis (Martin and Wang 2011; Eldem et al. 2017; Voshall and Moriyama 2018). Although the downstream statistical analysis is relatively straightforward, raw data processing remains a key obstacle. In particular, transcriptome de novo assembly is a complex, time-consuming task and requires extensive computational resources (Martin and Wang 2011; Eldem et al. 2017; Voshall and Moriyama 2018). Several transcriptome de novo assemblers have been developed, such as the established tools Trinity (Haas et al. 2013; https://github.com/trinityrnaseq/trinityrnaseq/wiki) and SOAPdenovo-Trans (Xie et al. 2014), and more recently developed tools such as Bridger (Chang et al. 2015), BinPacker (Liu et al. 2016), and TransLiG (Liu et al. 2019). However, analysis with these tools can take several days or even weeks to complete on a high-performance computer. Additionally, the assembled transcriptomes of nonmodel organisms often suffer from many false positives and false negatives, and no single assembler can deliver the best results for all scenarios (Hölzer and Marz 2019; Liu et al. 2019). Another key step in the conventional RNA-seq workflow is transcriptome annotation. The established procedure is to perform DNA-to-protein BLASTX via translated search, as it can overcome the large evolutionary divergence among homologous sequences especially when compared to the DNA-to-DNA BLASTN approach (Conesa et al. 2016; Ye et al. 2019). This method has been implemented in several programs such as Blast2GO (Conesa and Götz 2008) and Trinotate (https://github.com/Trinotate/Trinotate.github.io/wiki). However, these implementations are also time-consuming and computationally intensive. The computational skills and computing resources involved in executing de novo assembly and annotation represent huge barriers to entry for using RNA-seq in research on nonmodel organisms.
Given the diverse challenges outlined above, there is an unmet need to develop a straightforward and computationally efficient tool for handling RNA-seq data from nonmodel organisms. For transcriptomics studies in nonmodel organisms that focus on mRNAs or protein-coding genes, we propose a new processing and analysis strategy of directly translating RNA-seq reads into all possible short amino acid (aa) sequences and then comparing these with protein references to identify their possible functional homologs. Several superfast DNA-to-protein aligners, including DIAMOND (Buchfink et al. 2015), MMseqs2 (Steinegger and Söding 2017), and Kaiju (Menzel et al. 2016), have been developed or used to map DNA reads directly to microbial protein databases, thus skipping genome assembly and directly quantifying the functional capabilities of the sample's microbiome. Bacterial genomes are densely packed with protein-coding genes and free of introns (Bentley and Parkhill 2004), characteristics that are largely shared by eukaryotic RNA-seq reads. Thus, theoretically it should be possible to directly quantify the expression of protein-coding genes from eukaryotic RNA-seq reads using similar approaches and algorithms, although we are not aware of any existing tools that do this.
Here, we present Seq2Fun, an ultrafast, assembly-free, all-in-one tool for functional quantification of RNA-seq reads for nonmodel organisms, to address the problems identified above. In addition to describing the underlying algorithm, we use both simulated and real data sets from a variety of species to show that Seq2Fun outperforms the conventional RNA-seq analysis workflow based on transcriptome de novo assembly in both accuracy and computational efficiency. We also demonstrate with a case study how Seq2Fun can be used to analyze RNA-seq data from an organism without a reference genome.
Results
Workflow of Seq2Fun
Seq2Fun uses a novel strategy of directly translating RNA-seq reads into all possible amino acid sequences and searches for homologous protein sequences in a well-curated database. Seq2Fun is written in the high-performance language C++. To further achieve high computing efficiency in terms of both speed and memory footprint, our implementation employs an FM-index data structure (Ferragina and Manzini 2000) and only retains the information necessary for quantifying the expression of protein-coding genes involved in the KEGG pathways. There are three main steps underlying Seq2Fun: (1) quality control of raw reads; (2) translation of cleaned reads into all possible amino acid sequences and alignment to a multispecies protein database; and (3) generation of KEGG ortholog (KO) expression abundance tables and summary figures for downstream analysis (Fig. 1; Supplemental Figs. S1, S2).
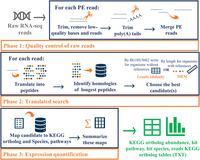
Overview of the Seq2Fun workflow. Seq2Fun accepts raw RNA-seq reads and generates various expression count tables. There are three main phases: quality control; translated search; and expression quantification. Seq2Fun starts by loading read pack (n = 10,000 raw RNA-seq reads), followed by trimming, adaptor and poly(A) tail removal, overlapped paired-end reads merging, and sequence error correction; cleaned reads are translated into all possible amino acid sequences, and the longest fragments are subjected to search in a protein database based on FM-index to identify the most likely functional homologs either by maximum exact match (MEM) or Greedy mode. Each matched read is assigned with protein ID(s), followed by mapping each protein ID with the KEGG ortholog ID, and finally summing each KEGG ortholog to produce a KEGG ortholog abundance table, pathway hit table, species hit table, and KEGG ortholog reads table. An HTML report is also generated to summarize and visualize read qualities and results tables. Cleaned reads labeled with mapped KEGG orthologs are also retrieved.
Seq2Fun can run in two modes: maximum exact match (MEM) or Greedy mode. The MEM mode only allows exact matches between query and reference sequences and therefore is appropriate for organisms that have very closely related species in the database. The Greedy mode allows mismatches between query and reference sequences to help overcome evolutionary divergence among homologous sequences and is more suitable for organisms that do not have a closely related reference genome in the database. More descriptions are available in the Supplemental Materials (Supplemental Methods 3.1). To achieve a balance between speed and accuracy of reads quantification, various KEGG ortholog protein databases have been built for different groups including eukaryotes, animals, plants, and fungi, as well as sub-groups such as mammals, birds, reptiles, amphibians, fishes, and arthropods.
The MEM and Greedy modes of Seq2Fun were evaluated using both simulated and real RNA-seq data sets from mouse (Mus musculus), chicken (Gallus gallus), zebrafish (Danio rerio), and roundworm (Caenorhabditis elegans) (Supplemental Table S3). For these benchmark tests, the Seq2Fun MEM mode aligns the translated reads to a database containing only that species’ protein sequences, whereas the Greedy mode aligns translated reads to a custom multispecies protein sequence database that was modified to exclude sequences from that species (Supplemental Tables S1, S2). All Seq2Fun analyses were conducted with default parameters (e.g., number of mismatches, minimum matching length, minimum score) that were chosen based on a parameter sensitivity analysis, the results of which are described in the Supplemental Materials.
Computational efficiency
Seq2Fun (Greedy mode) consumed as little as 0.4 GB of RAM (mouse data set), whereas the conventional workflow (Trinity) typically requires 50 GB of RAM (1 GB RAM per million reads) (Grabherr et al. 2011). The Seq2Fun Greedy mode was 113 (0.4 GB RAM), 125 (2.27 GB RAM), 86 (0.6 GB RAM), and 50 (0.7 GB RAM) times faster than the conventional workflow for the mouse, chicken, zebrafish, and roundworm data sets, respectively (Table 1). Transcriptome de novo assembly accounted for 49%–78% of the computational time using Trinity (Haas et al. 2013), whereas annotation to the KEGG ortholog database accounted for 4%–19% using KofamScan (Aramaki et al. 2020). These results indicate that Seq2Fun is computationally efficient and can run on a personal computer that has a fair amount of resources (e.g., eight threads and 16 GB RAM).
Performance assessments based on simulated and real data sets
Annotation and quantification
We first evaluated the performance of Seq2Fun on simulated data sets. The recall of Seq2Fun using both genes and reads from the data sets of the four organisms was highest for MEM mode, followed by Greedy mode, and both were higher than the conventional workflow results (Table 1; Supplemental Table S4). There was little difference among precision across all evaluated methods for both the reads and gene-level results for the simulated data (Table 1; Supplemental Table S4). The R2 values were generally higher for Seq2Fun (0.85–1.00) in both MEM and Greedy modes for the simulated data sets, compared to the values of the conventional workflow (0.58–0.97). Further investigations indicated that some outliers contributed to these differences (Supplemental Results).
We next evaluated Seq2Fun on real data sets. The recall of Seq2Fun using both genes and reads from real data sets from all four organisms was highest for MEM mode, followed by Greedy mode, and both were higher than the results from the conventional workflow (Table 1; Supplemental Table S4). The read-level results had lower precision compared to the gene-level results for all three tools, and this difference was particularly noticeable for the conventional workflow (Supplemental Table S4). Although there was little difference in gene-level precision among the three tools, the read-level precision was lower for the conventional workflow compared to the Seq2Fun results (Table 1; Supplemental Table S4).
Compared to the simulated data, there were more variations in the real data sets for the explained variations measured by R2 values (Table 1). There were no consistent differences between the MEM and Greedy Seq2Fun modes, which had high R2 values ranging from 0.78 to 0.96. In contrast, the R2 values from the conventional workflow were substantially lower for all four data sets, ranging from 0.37 to 0.55 (Table 1). The lower R2 values of the conventional workflow compared to Seq2Fun can be explained by inconsistent coverage of KEGG orthologs in the assembled transcriptome. For example, we found that 54 KEGG orthologs (counts > 100) were identified in the reference results but not in the conventional results, whereas 17 KEGG orthologs were present in the conventional results and not in the reference results of the zebrafish data set.
Gene- and pathway-level analysis
Only the chicken, zebrafish, and roundworm's real RNA-seq data sets were used to conduct differential gene expression analysis and pathway enrichment analysis because the experimental design of the mouse data set lacked biological replicates and thus was not amenable for proper statistical analysis. Both modes of Seq2Fun had higher R2 values than the conventional workflow in all cases, except for the gene-level fold change in the chicken data set, where the conventional workflow had a higher value than Seq2Fun MEM (Table 2). In general, the Seq2Fun MEM and Greedy modes identified more reference genes and pathways than the conventional workflow for all three organisms (Table 2).
Benchmark on gene expression and pathway analysis
Case study
To demonstrate a typical use case in the environmental life sciences for an organism that does not have a reference genome, Seq2Fun (Greedy mode) was used to analyze RNA-seq data from double-crested cormorant (DCCO) embryos. DCCO embryos were exposed to ethinylestradiol (EE2), a synthetic estrogen that is the active substance in some forms of birth control, via egg injection at a high (n = 4) and low dose (n = 5) (Farhat et al. 2020). Controls (n = 5) were exposed to DMSO solvent. Liver tissue was collected at mid-incubation for whole transcriptome RNA-seq (detailed experimental descriptions are in Methods). For comparison purposes, we also ran a conventional workflow on the DCCO data set. The results showed that Seq2Fun was about 50 times faster than the conventional workflow. It finished the whole data set analysis (14 samples and 75 million reads) within 24 min using about 1.9 GB RAM and eight threads, compared to 20 h and 50 GB RAM with the conventional workflow. Results obtained from Seq2Fun showed that there were 881 differentially expressed genes across both doses, whereas the conventional workflow generated 794 DEGs. ORA resulted in 11 significant pathways, whereas GSEA produced 24 (Fig. 2). In comparison to results generated using a conventional de novo assembly workflow, the fold changes computed by both software had an R2 of 0.83. Comparing the top 300 differentially expressed genes resulted in an overlap of 213 (71%) and 229 (76.33%) when sorting genes by P-value and fold change, respectively. Comparing the top 30 pathways resulted in an overlap of 22 (73.33%) and 20 (66.67%) when sorting pathways by P-value for ORA and GSEA, respectively. The conventional workflow is not the “gold standard” in the same way as the reference genome-based workflow, and thus these overlap percentages should not be interpreted as measures of accuracy. However, these results do show that Seq2Fun is detecting largely the same perturbed genes and pathways as the conventional workflow, which may be comforting to prospective users.
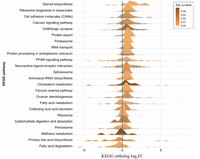
Significant pathways identified from RNA-seq data of double-crested cormorant (DCCO). The pathways are visualized by ridgeline plots. The distribution for each pathway is colored according to the pathway's adjusted P-value. The vertical gray lines indicate the log2FC values of all genes in the enriched pathways.
The significantly enriched pathways in Figure 2 are consistent with an estrogenic mode of action. For example, exposure to estrogenic substances has been consistently shown to cause a decrease in cholesterol levels in plasma (Bravo et al. 1999; Parini et al. 2000). Cholesterol is synthesized from fatty acids within peroxisomes, mitochondria, and the endoplasmic reticulum and is an immediate precursor to several important biomolecules, including steroid hormones and bile acids. Many of the pathways perturbed are closely related to cholesterol synthesis and metabolism, especially including “Steroid biosynthesis,” “Peroxisome proliferator-activated receptor (PPAR) signaling pathway,” “Neuroactive ligand-receptor interaction,” “Cholesterol metabolism,” “Ovarian steroidogenesis,” “Fatty acid metabolism,” “Peroxisome,” “Primary bile acid biosynthesis,” and “Fatty acid degradation.” Additionally, some of the pathways that are most consistently up- or down-regulated match the directionality observed in previous studies. For example, it has been observed that genes involved in cholesterol synthesis are up-regulated to compensate for the decreased cholesterol levels (Fink et al. 2005). These lower levels also induce a decrease in bile acid excretion, causing it to accumulate in the liver and down-regulate genes involved in bile acid synthesis (Stieger et al. 2000). In Figure 2, it is clear that the “Steroid biosynthesis” pathway, which directly produces cholesterol, is up-regulated and the “Primary bile acid biosynthesis” pathway is down-regulated.
Discussion
The biggest challenges of the conventional RNA-seq workflows are the key steps of transcriptome de novo assembly and gene annotation. These steps require a high-performance server with extensive computational resources, typically with more than 100 GB of RAM. These resources are not easily accessible to the majority of labs working on nonmodel organisms (Eldem et al. 2017) and thus represent a major barrier for the community to scale-up wider use of RNA-seq data. Here, we developed, tested, and validated Seq2Fun, which adopted a novel strategy for RNA-seq analysis to help overcome these challenges. In doing so, we demonstrated that Seq2Fun consumes as little as 2.27 GB RAM and is up to 125 times faster than a conventional workflow based on Trinity (Haas et al. 2013), one of the most widely used software for de novo transcriptome assembly. To minimize RAM requirements, Seq2Fun uses a relatively small but comprehensive database and does not load all reads into memory. It breaks input data into packs of 10,000 reads, and each thread is employed to process one read pack at a time, such that the overall RAM usage is well controlled and is not increased with larger input data. Moreover, unlike de novo transcriptome assembly, each read (or pair of reads for PE data) can be processed independently in Seq2Fun. It is worth noting that Seq2Fun has achieved a faster, more efficient, and more accurate way to conduct functional analysis of RNA-seq data for nonmodel organisms. It is used to complement and not to replace the conventional workflows.
The development of Seq2Fun for functional quantification of RNA-seq reads for eukaryotic organisms was inspired by the core algorithm of Kaiju (Menzel et al. 2016), which is designed for taxonomic classification using shotgun metagenomic sequences. While developing Seq2Fun, we made several important improvements to the core algorithm that resulted in significant performance gains. For instance, Seq2Fun has a very efficient I/O and can process 2.46 million PE reads/min, whereas Kaiju processes >1 million PE reads/min without raw reads quality checks. This is because Seq2Fun can seamlessly generate abundance tables from raw reads without any intermediate steps, and the protein database is always stored in memory without repeatedly reloading when samples are processed in batches. In addition, Seq2Fun automatically joins overlapped PE reads into a single longer read, which may yield longer amino acid fragments. This could partially contribute to the higher recall and precision values. Finally, Seq2Fun directly generates five levels of output files, including a gene abundance table, a species hit table, a pathway hit table, a read mapping table, and an HTML report containing summary figures and tables (Supplemental Fig. S2) under single sample profiling mode. This report is ready for primary interpretation without any further analysis efforts.
For most nonmodel organisms, biological understanding of study outcomes is limited to protein-coding genes with functional annotations held within databases including KEGG pathways (Kanehisa and Goto 2000), Gene Ontology (The Gene Ontology Consortium et al. 2000; The Gene Ontology Consortium 2019) or PANTHER classification system (Mi et al. 2019). Therefore, developing Seq2Fun databases to focus on functionally annotated genes such as KEGG orthologs largely meets the preferred needs of most scientists studying nonmodel organisms (Supplemental Table S1). In addition to supporting pathway-level analysis, the single sample profiling mode in Seq2Fun generates an HTML report that summarizes the most abundant KEGG orthologs, KEGG pathways, and hit species, which may facilitate quick interpretation of study results.
Overall, the outcomes of our various tests indicate that Seq2Fun is highly accurate and stable for gene quantification. There were relatively small variations of R2 values for the Seq2Fun MEM and Greedy modes compared to the conventional workflow across all evaluated data sets. The variation in the conventional workflow results is mostly likely caused by some moderately to highly expressed genes (Supplemental Materials), indicating that these genes are difficult to assembly and/or quantify by the conventional workflow. The sensitivity analysis showed that Seq2Fun is robust to small variations in the parameters (e.g., minimum scores, number of mismatches, minimum fragment length) (Supplemental Figs. S3–S10).
Limitations and next steps
Transcriptome de novo assemblers typically have the ability to discover novel genes including protein-coding and long noncoding genes, as well as novel isoforms of previously known genes (Martin and Wang 2011). This is not possible for Seq2Fun as it is only designed for identification and quantification of known protein-coding genes from a database at a high speed and low computational cost. There are also several other steps of Seq2Fun in which information is not fully utilized in order to optimize the speed. In its current version, Seq2Fun only implements a DNA-to-protein search strategy and not DNA-to-DNA searching. It is possible to implement both search strategies via a tiered search approach. This would not only improve the computational efficiency of the currently implemented DNA-to-protein search but also would enable mapping to any genes, including those from noncoding genes (Franzosa et al. 2018; Ye et al. 2019). We are currently developing the tiered search which will be available in a future version of Seq2Fun.
Obtaining assembled gene sequences for specific genes could be useful in some research contexts. To partially compensate for the lack of assembled transcripts in the Seq2Fun results, we have implemented a feature to support targeted gene assembly. In this feature, users can specify a KEGG ortholog of interest to retrieve all reads mapped to this gene. These reads can then be used for de novo assembly with Trinity, which takes only several minutes per gene.
The proteins used in the Seq2Fun database are restricted to protein-coding genes in KEGG pathways, which may not always be the best choice for different studies and applications. To address this limitation, we have added support to allow Seq2Fun to accept customized databases. For instance, users can expand the database to include all protein-coding genes or restrict the database to a smaller number of target genes, such as genes assigned to a specific KEGG pathway or Gene Ontology term (Mi et al. 2019).
Seq2Fun uses a fixed number of mismatches for all proteins and only allows for amino acid substitution and is unable to cope with indels. This could also affect the identification of highly evolutionarily divergent regions within homolog proteins, although it would likely have a limited impact, as their expression levels could be quantified based on other well-behaved fragments. Finally, truly expressed genes should have a relatively uniform distribution of reads across the whole gene. Seq2Fun does not examine the distribution of reads as it processes each read independently. Future versions of Seq2Fun could address these limitations by offering users more options to customize their analysis.
Methods
Seq2Fun consists of three main phases: raw reads quality control; translated search; and abundance table generation (Fig. 1).
Seq2Fun phase I: quality control of raw reads
Seq2Fun cleans the raw reads by adopting the core algorithm from the fastp v0.20.0 software (Chen et al. 2018). First, each read is trimmed at both the 5′ and 3′ end. Next, the poly(G) tail is removed. Poly(G) tails have been reported to be a common issue for Illumina NextSeq and NovaSeq (Chen et al. 2018) and are produced in the late stage as some T and C bases are wrongly assigned to G. Third, low complexity sequences are removed because they can cause artificially high protein hit scores during protein alignment (Edgar 2004). If the uploaded data are paired-end (PE), the overlapping region of each pair is used to correct sequencing errors by assigning mismatched bases to the bases with a higher quality score (>Q30). Next, sequence adaptors and poly(A) tails are identified and removed, and overlapping PE reads are merged into a longer single-end (SE) read. Finally, all reads are converted to FASTA format.
Seq2Fun phase II: translate reads and map to protein database
First, each clean read is translated into amino acid sequences using six reading frames from both directions, which typically results in dozens of peptide fragments. At most, the top six longest fragments are kept for the translated search in MEM mode, although it could generate more fragments with the BLOSUM62 scores (Henikoff and Henikoff 1992) for Greedy mode (Supplemental Fig. S1) but still far fewer than the original algorithm implemented in Kaiju v1.7.3 (Menzel et al. 2016). Although keeping the top longest fragments could remove some true protein fragments, the same methods are applied to all samples and therefore should have a minimal impact on any downstream comparative analysis such as differential expression analysis. In some cases, only keeping the longest fragments could filter out a proportion of fragments that originated from the merged PE reads if start and stop codons are present in the middle of the reads. Therefore, we recap the maximum cutoff length of peptide fragment to be 60 aa (by default though, the user can change this cutoff), which will prevent the filtering out of some true peptide fragments from the merged reads.
Next, the peptide fragments are aligned to the Seq2Fun database, which consists of protein sequences from KEGG pathway genes that were retrieved using the KEGGREST R package v1.12.2 (https://bioconductor.org/packages/KEGGREST/). The size of the protein database was reduced by removing redundant protein sequences that have >99% similarity across species using CD-HIT v4.8.1 (Supplemental Tables S1, S2; Li and Godzik 2006; Fu et al. 2012). Seq2Fun employs the same core reads alignment algorithm as Kaiju v1.7., which has two different modes designed for species with (MEM mode) and without (Greedy mode) a reference genome (Menzel et al. 2016). The MEM mode only allows exact matches between query and subject sequences from the database. It enables a fast search in the database, and the fragment with the longest matching length is retained. It is designed for organisms with reference genomes in the database. Therefore, in this study, MEM mode was used to map RNA-seq reads from mouse, chicken, zebrafish, and roundworm to their own species-specific protein references (e.g., the mouse database for MEM mode consists of 8438 mouse-specific protein sequences) (Supplemental Tables S1, S2), respectively, in order to demonstrate the feasibility of MEM mode. The downside of MEM mode is that it cannot identify homologous protein sequences of the query if there is even a single discrepancy with the sequences in the database. Therefore, the Greedy mode is introduced to allow a small number of amino acid mismatches between the query and subject, which helps Seq2Fun handle evolutionary divergence between species. The peptide fragments are aligned to a database that contains sequences from many different species, and the fragments with the highest BLOSUM62 scores (Henikoff and Henikoff 1992) are retained. A detailed description of the MEM and Greedy modes is available in the Supplemental Materials. In this study, four databases with genome exclusion for Greedy mode were created for mouse (e.g., 64 mammal species excluding mouse), chicken, zebrafish, and roundworm, respectively (Table 1; Supplemental Tables S1, S2), to mimic conditions for analyzing data from an organism without a reference genome. This phase produces a reads-protein ID map.
Seq2Fun phase III: expression quantification
First, cases where reads were mapped to multiple protein IDs are dealt with. Most often, these proteins are homologies from the same or different organisms and share the same KEGG ortholog ID. If this is not the case, the KEGG ortholog with the highest frequency is used. After ensuring that each read is matched to a single KEGG ortholog, the final quantification is a summation of all read-KEGG ortholog matches. The results of Phase III is a three-column KEGG ortholog abundance table for each sample. To better understand the gene composition of each sample, three additional tables are generated containing hit pathways, hit species, and reads KEGG ortholog mapping. Finally, an HTML report is generated summarizing these data in figures and tables, plus a rarefaction curve with sequence depth plotted against the number of mapped KEGG orthologs for each sample, which can be used to determine the minimum informative number of reads for each sample.
Evaluation with simulated data
To obtain data with known abundances for each gene, we simulated four data sets of mouse, chicken, zebrafish, and roundworm using Polyester v1.8.3 (Frazee et al. 2015). Coding sequences assigned to KEGG pathways for these organisms were obtained from the KEGG database (Kanehisa and Goto 2000) using KEGGREST R package v1.12.2 (Supplemental Table S1). Each data set had the following parameters: three biological replicates for each control and treatment group, at least 10 times sequencing coverage for each gene, and fold changes randomly assigned to each gene that ranged from 1 to 10 (Supplemental Table S3). The simulated data sets were formatted as gene abundance tables (generated by Polyester) that represent the “ground truth.”
The raw reads from each of the simulated data sets were analyzed with three different pipelines: Seq2Fun MEM mode, Seq2Fun Greedy mode, and a conventional workflow. The conventional workflow involved raw reads quality control by fastp v0.20.0 (Chen et al. 2018), a de novo assembled transcriptome generated by Trinity v2.10.0 (Haas et al. 2013). TransDecoder v5.5.0 (https://github.com/TransDecoder/TransDecoder/releases) was then used to predict and translate the open reading frames into amino acid fragments of the assembled transcripts, which were annotated by KofamScan v1.3.0 (Aramaki et al. 2020) and quantified by RSEM v1.3.3 (Li and Dewey 2011). MEM mode conducted analysis of mouse, chicken, zebrafish, and roundworm using their own, species-specific protein reference databases. To mimic analyses for a nonmodel organism, Greedy mode analysis of the mouse, chicken, zebrafish, and roundworm data sets used protein databases that excluded those species’ sequences (Supplemental Tables S1, S2).
The results from each of the three pipelines were evaluated based on precision, recall, and abundance fit of the computed KEGG ortholog abundances compared to the “ground truth” results. As Seq2Fun was developed to complement the conventional workflow, evaluation metrics including recall and precision for both genes and reads were used to assess its performance. These evaluation metrics have been widely used in many de novo transcriptome assemblers (Grabherr et al. 2011; Haas et al. 2013; Xie et al. 2014; Chang et al. 2015; Liu et al. 2016, 2019) as well as in Kaiju (Menzel et al. 2016). Recall is defined as the fraction of true positive features (genes or reads) out of the total “ground truth” features (either by Polyester for simulated data or RSEM for real-world data), whereas precision is defined as the fraction of true positive features out of the total number of predicted features (by each tool). All the abundance tables were filtered with the criteria of at least one read per gene and detected in at least 20% of the samples. Coefficient of determination R2 values were calculated from the KEGG ortholog abundance tables.
Evaluation with real RNA-seq data
We also downloaded real-world RNA-seq data sets for the same four species: mouse, NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJEB4513 (Werber et al. 2014); chicken, NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE86592 (Hwang et al. 2018); zebrafish, European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser/home) under accession number ERP014517 (White et al. 2017); and roundworm, GEO accession number GSE122728 (Viau et al. 2020). Sequencing platforms were Illumina HiSeq 2000, NextSeq 500, or HiSeq 2500 using TrueSeq RNA libraries (Supplemental Table S3). To make sequencing depths equal, all sample reads were first quality checked by fastp v0.20.0 (Chen et al. 2018) before an equal number of reads were randomly sampled by seqtk v1.3 (Supplemental Table S3; https://github.com/lh3/seqtk; http://weizhongli-lab.org/cd-hit/). Finally, we obtained six samples (5 million reads per sample) for mouse and chicken, 40 samples (∼1.28 million reads per sample) for zebrafish (Supplemental Table S3), and 10 samples (5 million reads per sample) for roundworm for downstream analyses. RSEM v1.3.3 (Li and Dewey 2011) was used for reads quantification against reference genes obtained from the KEGG database for these organisms. This generates gene abundance tables that serve as a proxy of the “ground truth” for the real data sets.
The real-world RNA-seq data sets were evaluated using the same methods and metrics as the simulated data (three pipelines; recall, precision, and KEGG ortholog abundance fit). In addition, we conducted differential expression analysis (DEA) and gene set analysis (GSA), which were used to calculate the R2 of DEA and GSA statistics compared to the reference results. DEA was conducted with limma-voom using the R package limma v.3.28.6 (Ritchie et al. 2015). Significant DEGs were defined as KEGG orthologs with an adjusted P-value (FDR method) cutoff of 0.05. Gene set analysis was performed using the R package FGSEA v.1.14.0 with two methods: ORA, that uses a hypergeometric test and the list of DEGs; and GSEA, that uses the entire ranked list of genes based on their log2FC values. For both ORA and GSEA, pathways were considered significant if their adjusted P-values (FDR method) were less than 0.05.
Case study with nonmodel organisms
DCCO embryos were exposed via egg injection (Crump et al. 2020) to EE2, a synthetic estrogen that is the active substance in some forms of birth control, at a high (31.9 µg/g egg) (n = 4) and low dose (2.3 µg/g egg) (n = 5), as well as controls that were exposed to the DMSO solvent (n = 5). Livers were harvested after 14 d exposure and immediately frozen in liquid nitrogen for total RNA extraction. Total RNA was sent to Genome Quebec (Montreal, Quebec, Canada), where sequencing libraries were built with the TruSeq RNA Library Prep Kit (Illumina) and then submitted to Illumina NovaSeq 6000 for 100-bp PE reads sequencing. All raw reads were subsampled to 5 million PE reads/sample before submission to either Seq2Fun (Greedy mode) or the conventional workflow (which was described for chicken) for further analysis. Seq2Fun (Greedy mode with default parameters) used a protein database from the constructed birds database consisting of 87,530 protein sequences sharing 99% similarity, representing 4177 KOs from 24 bird species (Supplemental Tables S1, S2). This database, as well as the mammals and fishes databases, can be downloaded from https://www.seq2fun.ca. DEA and GSA were conducted using the same pipeline as described in the previous section for the chicken, zebrafish, and roundworm data sets.
Computational resources used
All software tools were run on a PC Dell OptiPlex 7050 with eight threads (Intel Core i7-6700 CPU @ 3.40 GHz) with 64 GB RAM (DDR4, size of 16384 MB, speed 2.4 GHz).
Software availability
The source code of Seq2Fun is available in Supplemental Code; all the databases and data sets are available in GitHub (https://github.com/xia-lab/Seq2Fun) and the Seq2Fun website (https://www.seq2fun.ca/).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This research was funded by Genome Canada, Genome Quebec, the Natural Sciences and Engineering Research Council of Canada (NSERC), and the Canada Research Chairs (CRC) Program.
Author contributions: J.X. and P.L. conceived the project; P.L. developed the code and algorithm, collected data, and constructed the databases; P.L. and J.E. analyzed data, generated figures and tables, and constructed the Seq2Fun website; J.H. and D.C. collected RNA-seq data of DCCO and helped to interpret data of the case study; J.H.G. and G.B. helped with de novo transcriptome assembly; J.X., P.L., and J.E. interpreted data and wrote the manuscript. N.B. helped to improve the manuscript. All the authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.269894.120.
-
Freely available online through the Genome Research Open Access option.
- Received August 5, 2020.
- Accepted February 18, 2021.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








