
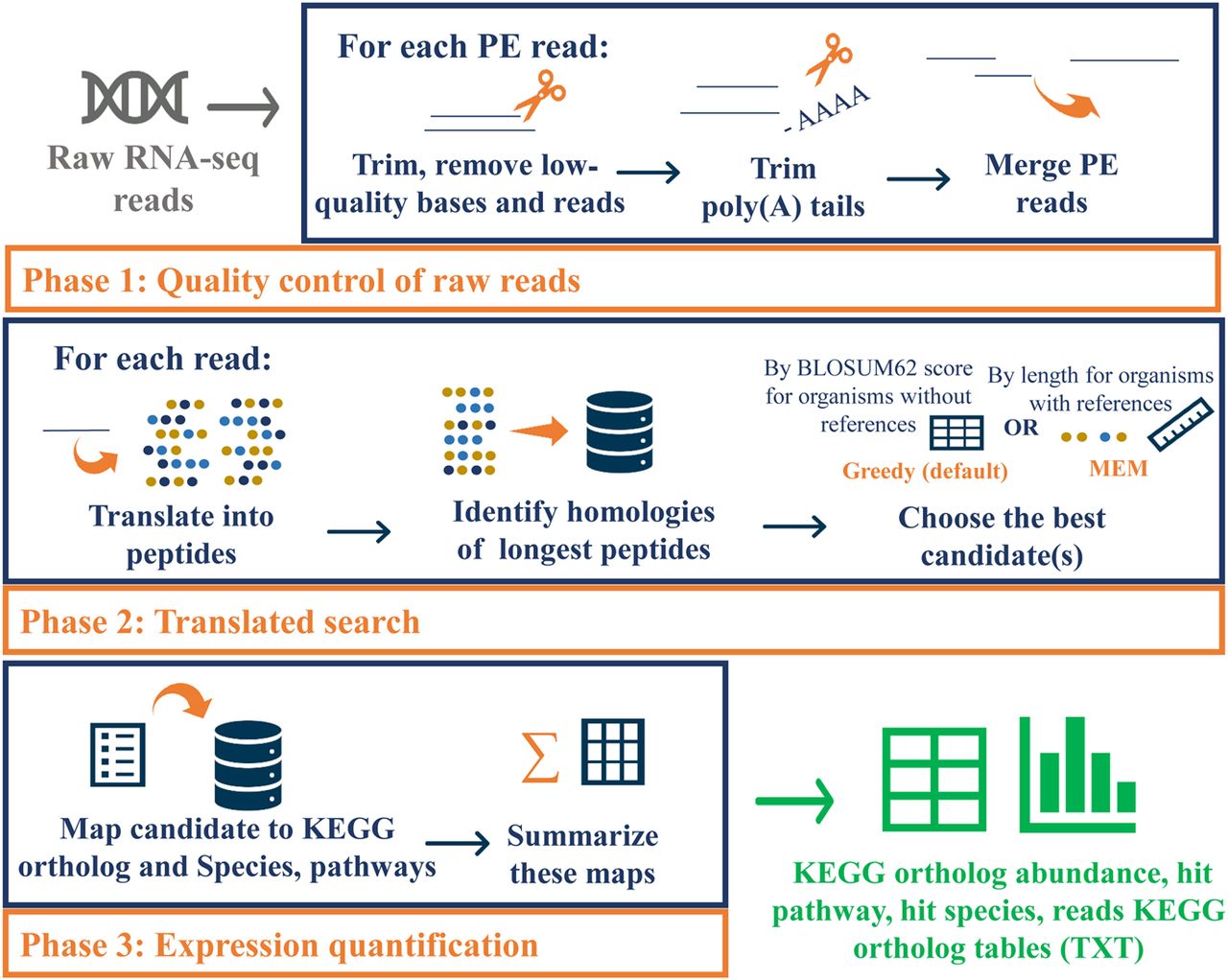
Overview of the Seq2Fun workflow. Seq2Fun accepts raw RNA-seq reads and generates various expression count tables. There are three main phases: quality control; translated search; and expression quantification. Seq2Fun starts by loading read pack (n = 10,000 raw RNA-seq reads), followed by trimming, adaptor and poly(A) tail removal, overlapped paired-end reads merging, and sequence error correction; cleaned reads are translated into all possible amino acid sequences, and the longest fragments are subjected to search in a protein database based on FM-index to identify the most likely functional homologs either by maximum exact match (MEM) or Greedy mode. Each matched read is assigned with protein ID(s), followed by mapping each protein ID with the KEGG ortholog ID, and finally summing each KEGG ortholog to produce a KEGG ortholog abundance table, pathway hit table, species hit table, and KEGG ortholog reads table. An HTML report is also generated to summarize and visualize read qualities and results tables. Cleaned reads labeled with mapped KEGG orthologs are also retrieved.








