
Improved redox homeostasis owing to the up-regulation of one-carbon metabolism and related pathways is crucial for yeast heterosis at high temperature
- Liang Song1,2,3,
- Jun-Yan Shi1,2,3,
- Shou-Fu Duan1,
- Da-Yong Han1,2,
- Kuan Li1,
- Ri-Peng Zhang1,2,
- Peng-Yu He1,2,
- Pei-Jie Han1,
- Qi-Ming Wang1 and
- Feng-Yan Bai1,2
- 1State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China;
- 2College of Life Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
-
↵3 These authors contributed equally to this work.
Abstract
Heterosis or hybrid vigor is a common phenomenon in plants and animals; however, the molecular mechanisms underlying heterosis remain elusive, despite extensive studies on the phenomenon for more than a century. Here we constructed a large collection of F1 hybrids of Saccharomyces cerevisiae by spore-to-spore mating between homozygous wild strains of the species with different genetic distances and compared growth performance of the F1 hybrids with their parents. We found that heterosis was prevalent in the F1 hybrids at 40°C. A hump-shaped relationship between heterosis and parental genetic distance was observed. We then analyzed transcriptomes of selected heterotic and depressed F1 hybrids and their parents growing at 40°C and found that genes associated with one-carbon metabolism and related pathways were generally up-regulated in the heterotic F1 hybrids, leading to improved cellular redox homeostasis at high temperature. Consistently, genes related with DNA repair, stress responses, and ion homeostasis were generally down-regulated in the heterotic F1 hybrids. Furthermore, genes associated with protein quality control systems were also generally down-regulated in the heterotic F1 hybrids, suggesting a lower level of protein turnover and thus higher energy use efficiency in these strains. In contrast, the depressed F1 hybrids, which were limited in number and mostly shared a common aneuploid parental strain, showed a largely opposite gene expression pattern to the heterotic F1 hybrids. We provide new insights into molecular mechanisms underlying heterosis and thermotolerance of yeast and new clues for a better understanding of the molecular basis of heterosis in plants and animals.
Hybrid vigor, or heterosis, refers to the phenomenon that hybrid offspring are more vigorous than their genetically different homozygous parents. Hybrid vigor was first described in plants by Charles Darwin in 1876, who observed that cross-pollinated plants usually show better performances in height, weight, and fertility than their self-pollinated counterparts (Darwin 1876). This phenomenon was rediscovered in maize in 1908 (Shull 1908), and the term “heterosis” was introduced by Shull (1914) for this concept. Since then, heterosis has been widely observed in plants and animals and extensively exploited in agriculture, resulting in a great and constant increase in agricultural productivity worldwide (Lippman et al. 2008; Schnable and Springer 2013; Hochholdinger and Baldauf 2018).
The question of why hybrids show better performances in different traits has fascinated scientists for more than a century. Despite extensive studies on the question that have been performed since the early days of Darwin, little consensus has yet been reached about the genetic basis and molecular mechanisms of heterosis (Hochholdinger and Hoecker 2007; Lippman and Zamir 2007; Hochholdinger and Baldauf 2018; Govindaraju 2019). Different genetic hypotheses have been proposed to explain the phenomenon, including the three classic models: dominance (Davenport 1908; Bruce 1910; Jones 1917), overdominance (Shull 1911; East 1936), and epistasis (Richey 1942; Powers 1944). According to the dominance model, the complementation of slightly deleterious recessive alleles in heterozygotes leads to heterosis (Shull 1908; Charlesworth and Willis 2009). The overdominance model suggests that interactions occur between the alleles in the progeny and result in better performance than either homozygous parents (Shull 1911; Li et al. 2008; Chen 2013). The epistasis hypothesis, however, believes that interactions between nonallelic genes in hybrids result in heterosis (Yu et al. 1997; Fiévet et al. 2010; Hochholdinger and Baldauf 2018). There are experimental evidences that support each of these hypotheses. However, none of the classic models that were proposed before the era of molecular biology can fully explain the molecular mechanism of heterosis (Fiévet et al. 2018; Hochholdinger and Baldauf 2018).
In recent years, genomics, transcriptomics, proteomics, and systemic tools have been used in the studies of heterosis, resulting in improved understanding of heterosis at the molecular level (Hochholdinger and Hoecker 2007; Birchler et al. 2010; Schnable and Springer 2013; Fiévet et al. 2018; Fujimoto et al. 2018; Hochholdinger and Baldauf 2018; Vasseur et al. 2019). The contribution of different gene expression models to heterosis has been observed. These models include nonadditive or allele-specific gene expression, single-gene overdominance or underdominance, and single-parent expression complementation (Hoecker et al. 2008; Li et al. 2008; Wei et al. 2009; Guo et al. 2010; Krieger et al. 2010; Riddle et al. 2010; Paschold et al. 2012; Baldauf et al. 2018; Gonzalez-Bayon et al. 2019; Zhao et al. 2019). However, no consensus set or enriched functional categories related to heterosis have been identified (Fujimoto et al. 2018; Hochholdinger and Baldauf 2018). Other studies ascribe heterosis to a systemic property resulting from nonlinear concave genotype–phenotype relationships of living systems (Fiévet et al. 2018; Vasseur et al. 2019). Aiming to unify the theories for heterosis, a metabolic or energy-use efficiency hypothesis has been proposed (Ginn 2010, 2017; Goff 2011). Previous studies have shown that inbred organisms usually have increased rates of protein turnover relative to noninbred organisms (Hawkins et al. 1986; Hedgecock et al. 1996; Bayne 2004). Based on theoretical biophysics calculations, Ginn (2010, 2017) postulated that the higher levels of protein turnover among inbred organisms can be attributed to the accumulations of misfolded and aggregated proteins that require degradation by the inbred organisms’ protein quality control systems. Both protein synthesis and degradation are energy-consuming processes; inbred organisms must consume more energy to sustain a given biomass and are thus less “metabolically efficient.” Goff (2011) speculated that hybrids achieve greater energy efficiency via selective protein synthesis and metabolism. The model describes that cells distinguish between parental alleles based on the relative stability of the encoded proteins and use allele-specific gene expression to conserve energy and promote growth. Outcrossing provides more opportunity for allele selection and thereby increases the potential for enhanced vigor (Goff 2011). However, solid molecular evidence for the energy-use efficiency hypothesis has not been shown.
With the advantages of clear genetic and genomic background and being amenable to large-scale laboratory experiments, Saccharomyces cerevisiae and related yeast species have been used in the study on heterosis (Steinmetz et al. 2002; Marullo et al. 2006; Naumov et al. 2006; Sinha et al. 2006; Timberlake et al. 2011; Shapira et al. 2014; Blein-Nicolas et al. 2015; Shapira and David 2016; Bernardes et al. 2017; Herbst et al. 2017; Martí-Raga et al. 2017; Fiévet et al. 2018; Jansen et al. 2018). However, it is also difficult to find consensus from these studies about the mechanism of heterosis. Steinmetz et al. (2002) and Sinha et al. (2006) showed that both dominance and epistasis at three quantitative trait genes—MKT1, END3, and RHO2—contributed to growth heterosis at high temperatures in some yeast hybrids but were not conserved in others. Plech et al. (2014) reported that heterosis was prevalent among domesticated but not wild strains of S. cerevisiae. Based on the study on an interspecific hybrid between S. cerevisiae and Saccharomyces paradoxus strains, Herbst et al. (2017) ascribed heterosis to the impairment of growth-limiting pathways caused by regulatory incompatibility in hybrids. According to this hypothesis, the regulatory mechanisms related to growth-rate-limiting and safe-guard processes in hybrids were perturbed because of genomic incompatibility, allowing hybrid cells to grow faster and finally leading to heterosis (Bar-Zvi et al. 2017; Herbst et al. 2017).
Our recent population genomic study on S. cerevisiae showed a hallmark difference in heterozygosity between the wild and domesticated populations of the species. Wild isolates are exclusively homozygous, whereas domesticated isolates are generally heterozygous (Duan et al. 2018). The domesticated populations appear to have originated from ancestors formed by outcrossing between diverse wild isolates, and heterosis is probably responsible for the improved high temperature tolerance ability of domesticated isolates (Duan et al. 2018). The goal of this study is to check if heterosis is prevalent in wild S. cerevisiae at high temperature, to investigate correlation of heterosis with genetic distances, and to illuminate molecular mechanisms underlying thermotolerant heterosis of yeast. We provide new insights into molecular mechanisms underlying heterosis and thermotolerance of yeast and new clues for a better understanding of the molecular basis of heterosis in plants and animals.
Results
Wild yeast hybrids showed growth heterosis at high temperature
A total of 53 wild strains of S. cerevisiae (Supplemental Table S1) were selected from different wild lineages of the species recognized in our previous studies (Wang et al. 2012; Duan et al. 2018). These strains represent the largest genetic diversity of the wild population of S. cerevisiae documented so far (Duan et al. 2018). We generated 641 F1 hybrids without any genetic marker by spore-to-spore mating between pairs of the wild S. cerevisiae strains with genetic distance ranging from 0.323 × 10−3 to 15.337 × 10−3 (Supplemental Table S1). The F1 hybrids showing increased fitness (i.e., heterosis) and decreased fitness (i.e., outbreeding depression) were determined based on the fitness variables including maximum growth rate and growth efficiency compared with their parents at 30°C and 40°C, respectively. The F1 hybrids with different degrees of heterosis were then identified. If a hybrid shows a fitness value being significantly higher than the average value of its parents (mid-parent value [MPV]) or than the fitness value of the better parent, it is defined as a hybrid with mid-parent heterosis (MPH) or better-parent heterosis (BPH) (Zörgö et al. 2012). If the fitness of a F1 hybrid is significantly lower than that of the worst parent, the hybrid is classified in the group of outbreeding depression or worst parent heterosis (WPH), as defined in Zörgö et al. (2012). We found that at 30°C, 39.7% and 27.4% of the F1 hybrids showed MPH and BPH, respectively, in maximum growth rate, and 64.0% and 34.1% showed MPH and BPH, respectively, in growth efficiency (Supplemental Fig. S1A). However, the growth difference between the F1 hybrids and their parents was generally unremarkable, and the mean fitness of the F1 hybrids did not deviate from the MPV significantly in maximum growth rate (Mann–Whitney U test, P = 0.153) and slightly deviated from MPV (P = 0.007) in growth efficiency (Supplemental Fig. S1B). In contrast, the F1 hybrids showed evident heterosis at 40°C. In average, the fitness of the F1 hybrids was significantly higher than the MPV in both growth rate and efficiency (Mann–Whitney U test, P < 0.001) (Fig. 1A; Supplemental Table S1). A total of 95.8% and 75.9% of the F1 hybrids showed MPH and BPH, respectively, in growth rate, and 92.4% and 69.5% of the F1 hybrids showed MPH and BPH, respectively, in growth efficiency (Fig. 1B). Only 16 (2.5%) and 14 (2.2%) out of the 641 F1 hybrids tested showed outbreeding depression in growth rate and efficiency, respectively (Fig. 1B). Notably, 81.3% (13/16) and 57.1% (8/14) of the depressed F1 hybrids in growth rate and efficiency, respectively, share a common parental strain BJ22 which is aneuploid (Duan et al. 2018). These results show that heterosis is prevalent in F1 hybrids of wild S. cerevisiae strains at high temperature.
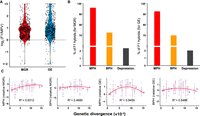
Heterosis of F1 hybrids of wild S. cerevisiae, and correlation between heterosis and the genetic distance of parental strains at 40°C. (A) Maximum growth rate (MGR) and growth efficiency (GE) of F1 hybrids relative to the average values of their parents (mid-parent value [MPV]). (B) Proportions of F1 hybrids showing mid-parent heterosis (MPH), better-parent heterosis (BPH), and outbreeding depression. (C) Correlation between genetic distances of parental strains and growth performance of F1 hybrids relative to the MPV (MPH) or the better parent value (BPH) in terms of MGR or GE.
The genetic distances between the parental strains (Supplemental Table S1) were calculated based on their genome sequences determined previously (Duan et al. 2018). The correlation between the degree of heterosis and the parental genetic distance was analyzed using linear and nonlinear models (Wei and Zhang 2018; Vasseur et al. 2019). We found that the quadratic model was favored in explaining the data observed (Supplemental Table S2). We found a hump-shaped relationship between MPH and parental genetic distance in both growth rate (R2 = 0.9312, P < 0.05) and efficiency (R2 = 0.9459, P < 0.05). A relatively weaker hump-shaped relationship between BPH and parental genetic distance was also observed in growth rate (R2 = 0.4685, P < 0.05) and efficiency (R2 = 0.5488, P < 0.05) (Fig. 1C). The optimal mating distances were estimated to be 7.42 × 10−3 to 10.56 × 10−3 for different types of heterosis (Supplemental Table S2).
Additive and nonadditive gene expression in F1 hybrids
To uncover the molecular mechanisms of heterosis of wild yeast at high temperature, we compared the gene expression profiles of selected F1 hybrids and their parents in the logarithmic growth phase at 40°C using RNA sequencing (RNA-seq) analysis. The strains were selected based on the considerations that the hybrids showed clear growth performance differences from their parents and that the parental strains represented different lineages with different genetic distances. A total of 94 strains, including 59 F1 hybrids and their parents (34 strains), were used in the RNA-seq analysis (Supplemental Table S3). Among the F1 hybrids, 50 showed BPH and the other nine showed outbreeding depression in both growth rate and efficiency, except two of the depressed hybrids showed outbreeding depression only in growth rate (Supplemental Table S3). We obtained high-quality RNA-seq data from two biological repeats for each strain (Supplemental Table S4). The reliability of the RNA-seq data was validated by quantitative real-time PCR (qRT-PCR) analysis. Eighteen genes were randomly selected from the actively expressed genes, and their expression levels in 18 of the RNA samples subjected to RNA-seq analysis were determined by qRT-PCR analysis (Supplemental Table S5). A strong correlation between the expression levels of the 18 genes determined by RNA-seq and qRT-PCR was observed in each sample (Supplemental Fig. S2).
The expression of a gene in a F1 hybrid is expected to occur in one of the two modes: additive and nonadditive. The additive expression occurs when the expression level of a gene in a F1 hybrid is equal to the average expression level of the gene in its parents (MPV), indicating the same expression levels of the alleles in the hybrid and the corresponding parents. Nonadditive expression occurs when the expression level of a gene in a F1 hybrid significantly deviates from the MPV, suggesting altered expression levels of the corresponding alleles in the hybrid (Hochholdinger and Hoecker 2007; Wei et al. 2009). A correlation of nonadditive gene expression with heterosis has been observed in plant (Li et al. 2009; Birchler et al. 2010; Riddle et al. 2010; Zhao et al. 2019). We found that additive expression was prevalent in the F1 hybrids of S. cerevisiae analyzed (Fig. 2A). Only a limited number (0–7.9%) of the expressed genes showed a nonadditive expression pattern in the F1 hybrids (Fig. 2A; Supplemental Table S6). However, we found a clear difference in the numbers and proportions of nonadditive genes between the heterotic and depressed F1 hybrids (Fig. 2B), although the limited number of the depressed hybrids compared did not allow a convincing statistical comparison.
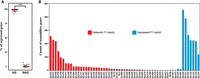
Gene expression variations of F1 hybrids and their parents at 40°C. (A) Proportions of additive genes (AGs) and nonadditive genes (NAGs) in every F1 hybrid. (***) P < 0.001 (Mann–Whitney U test). (B) Counts of nonadditive genes in every heterotic and depressed F1 hybrid.
We then compared the expression levels of the nonadditive genes relative to the MPV. The heatmap showed that a considerable number of the nonadditive genes showed a largely opposite expression level between the heterotic and depressed F1 hybrids (Supplemental Fig. S3A). Statistical analysis showed that the number of the nonadditive genes with expression levels below the MPV was much higher than that of the nonadditive genes with expression levels above the MPV in the heterotic F1 hybrids. However, an opposite trend was observed in the depressed F1 hybrids (Supplemental Fig. S3B). The result implies possible roles of nonadditive genes in the manifestation of heterosis or outbreeding depression in S. cerevisiae.
Nonadditive genes are enriched in specific pathways
The nonadditive genes recognized from all the F1 hybrids used in RNA-seq analysis were then subjected to correlation analysis using the weighted gene coexpression network analysis (WGCNA) tool (Langfelder and Horvath 2008). Briefly, significantly coexpressed nonadditive genes were clustered in separate modules. The genes in the modules with expression levels significantly correlated with growth rate or efficiency were selected for further analyses (Supplemental Fig. S4). A total of 523 coexpressed nonadditive genes with expression levels significantly correlated with the fitness of the hybrids (R ≥ 0.5 or ≤−0.5, P < 0.05) were identified (Supplemental Table S7). The nonadditive genes in the heterotic F1 hybrids were clustered into two major groups (I and II) with expression levels generally lower (down-regulated) and higher (up-regulated) than the MPV, respectively (Fig. 3A). A largely opposite expression pattern of these nonadditive genes was observed in the depressed F1 hybrids. That is, the nonadditive genes that were up-regulated in the heterotic F1 hybrids were generally down-regulated in the depressed F1 hybrids and vice versa (Fig. 3A).
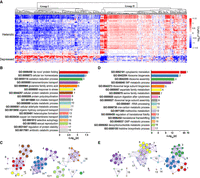
Functional categories of nonadditive genes in F1 hybrids of wild S. cerevisiae. (A) Heatmap shows the clustering of nonadditive genes based on their expression levels in each F1 hybrid relative to the average expression levels of the genes in the parents (MPV) of the hybrid according to the scale on the right, which depicts the values of log2 (F1/MPV). (B,D) Nonredundant Gene Ontology (GO) terms significantly enriched with the nonadditive genes in groups I and II, respectively. P-values represent statistical significance. (C,E) Enrichment networks show the intra- and inter-cluster functional similarities of the enriched GO terms shown in B and D, respectively. Enrichment networks are created by representing each enriched term as a node and connecting pairs of nodes with kappa similarities above 0.3. Up to 10 terms represented by colored nodes are included per cluster. The color code is respectively the same with that in B and D. The sizes of the nodes are proportional to the numbers of input genes falling into the terms.
The two groups of the nonadditive genes were respectively subjected to Gene Ontology (GO) enrichment analysis using the Metascape tool (Zhou et al. 2019). The terms resulting from GO analysis were then connected to form different enrichment networks based on quantitative measurements of the functional similarities of the terms (Huang et al. 2009). The group I nonadditive genes were significantly enriched in 18 GO terms (P < 0.01) (Fig. 3B; Supplemental Table S8A). The GO terms associated with protein quality control formed one main network (Fig. 3C). This network includes the GO terms of protein folding, cellular protein catabolic process, response to stress, and regulation of protein stability. The GO terms associated with cellular ion homeostasis, iron chelate transport, and ion transmembrane transport formed the second network. The GO terms of glutamine family amino acid biosynthetic process, which is associated with oxidative stress response (Altman et al. 2016), formed the third network, together with GO terms of cellular aldehyde metabolic, lactate metabolic, and antibiotic catabolic processes (including response to toxic substance) (Fig. 3C). The genes associated with DNA repair in this group, including RAD51, RAD54, and RDH54 (Pâques and Haber 1999), were enriched in a separate GO term named heteroduplex formation (Fig. 3C; Supplemental Table S8A).
The group II nonadditive genes were significantly overrepresented in 17 GO terms (P < 0.05) (Fig. 3D; Supplemental Table S8B). These GO terms were mainly clustered into two main networks (Fig. 3E). The GO terms associated with translation and ribosome biogenesis and assembly formed one major network. The GO terms of one-carbon (1C) metabolism, inosine monophosphate (IMP) metabolism, and methionine, serine, and aspartate family amino acid metabolism formed another main network (Fig. 3E).
The largely opposite expression levels of key nonadditive genes in the heterotic and depressed F1 hybrids are shown in Supplemental Figures S5 and S6. These genes are associated with protein quality control, DNA repair, or stress response (Supplemental Fig. S5) or with 1C, IMP, or serine and methionine metabolism (Supplemental Fig. S6). They were further classified into five categories based on their expression levels compared with those of their parents (Stupar et al. 2008; Schnable and Springer 2013). The expression levels of the nonadditive genes in group I were generally low parent (LP)-like (equal to the LP value) or below LP (lower than the LP value) in the heterotic F1 hybrids but high parent (HP)-like (equal to the HP value) or above HP (higher than the HP) in the depressed F1 hybrids (Fig. 4). The expression levels of the nonadditive genes in group II were generally HP-like or above HP in the heterotic F1 hybrids but LP-like or below LP in the depressed F1 hybrids (Fig. 4).
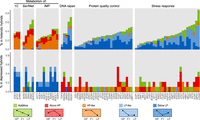
Proportions of key nonadditive genes showing different expression models in heterotic and depressed F1 hybrids of wild S. cerevisiae growing at 40°C. Five expression models are defined as illustrated at the bottom. (HP) High parent value; (LP) low parent value. The nonadditive genes are associated with six different functional categories as indicated. (1C) One-carbon; (Ser/Met) serine and methionine; (IMP) inosine monophosphate.
Lower ROS level in the cells of heterotic F1 hybrids
High temperature usually triggers oxidative stress on the cells owing to the overproduction of reactive oxygen species (ROS) (Morano et al. 2012; Foyer and Noctor 2016). Genes related with oxidative-reduction processes and other stress responses were generally up-regulated in the depressed F1 hybrids but down-regulated in the heterotic F1 hybrids (Fig. 3A; Supplemental Fig. S5). Genes enriched in the glutamine metabolism process, which is crucial for cellular ROS homeostasis (Altman et al. 2016), were also up-regulated in the depressed F1 hybrids (Fig. 3A; Supplemental Fig. S5). These data suggest higher oxidative stress level in the depressed F1 hybrids. Consistently, the up-regulation of genes related with protein quality control and DNA repair systems (Fig. 3A; Supplemental Fig. S5) suggests more protein and DNA damage probably owing to a higher level of ROS in the depressed F1 hybrids. We therefore measured the ROS levels in the F1 hybrids and their parental strains when growing at 40°C (Supplemental Table S9). The average ROS levels in the heterotic F1 hybrids were significantly lower than the average ROS levels of their parents (MPV; P = 0.005). However, the average ROS level in the depressed F1 hybrids did not significantly differ from the MPV of their parents (P = 0.094) (Fig. 5A). The majority (74.0%) of the heterotic F1 hybrids had ROS levels lower than the MPV of their parents, whereas the majority (77.8%) of the depressed F1 hybrids had ROS levels higher than the MPV of their parents (Fig. 5B).
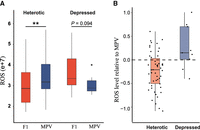
Levels of reactive oxygen species (ROS) within cells of F1 hybrids of wild S. cerevisiae and their parents growing at 40°C. (A) Cellular ROS levels of F1 hybrids and average cellular ROS levels of their parents (MPV) in the groups of heterotic and depressed F1 hybrids, respectively. (**) P < 0.01 (Mann–Whitney U test). (B) Cellular ROS levels of heterotic and depressed F1 hybrids relative to the MPV. The relative ROS level in a F1 hybrid was calculated according to the formula log2 (F1/MPV).
1C metabolism plays a key role in thermotolerant heterosis in yeast
The nonadditive genes that were up-regulated in the heterotic F1 hybrids but down-regulated in the depressed F1 hybrids are mostly enriched in the GO terms 1C metabolism, IMP metabolism, and methionine and serine biosynthetic process (Fig. 3D,E). IMP metabolism requires 10-formyl-THF, which is produced by 1C metabolism (Ducker et al. 2016). Serine is an indispensable donator to the 1C unit that contributes to the folate cycle and the methionine cycle (Locasale 2013; Rosenzweig et al. 2018). Therefore, these processes are centered by 1C metabolism as shown in the GO enrichment network analysis (Fig. 3E). 1C metabolism is a universal metabolic process supporting multiple physiological processes as well as redox defense (Locasale 2013; Fan et al. 2014; Ducker and Rabinowitz 2017; Rosenzweig et al. 2018). To maintain the redox homeostasis, cells evolved a series of regulatory systems. The cofactors NADH and NADPH are key components in these systems (Rosenzweig et al. 2018). One of the major pathways contributing to NADH and NADPH production is 1C metabolism (Fan et al. 2014; Rosenzweig et al. 2018).
In S. cerevisiae, ADE3 (C1-tetrahydrofolate synthase gene) is a key gene in the cytosolic 1C metabolism, which contributes to NADPH production in the pathway. MTD1 (NAD-dependent 5,10-methylenetetrahydrafolate dehydrogenase) contributes to the production of NADH in this pathway (Piper et al. 2000). These two genes were up-regulated in most heterotic but down-regulated in most depressed F1 hybrids (Supplemental Fig. S6). We randomly selected four heterotic F1 hybrids and knocked out ADE3 or MTD1 from them. The null mutants (ade3Δ/Δ or mtd1Δ/Δ) of the hybrids showed a remarkable decrease in growth performance at 40°C (Fig. 6A,B; Supplemental Fig. S7A,B). The addition of N-acetyl-L-cysteine, which is a well-characterized antioxidant able to scavenge ROS (Zafarullah et al. 2003), rescued or improved the growth of the ade3Δ/Δ and mtd1Δ/Δ mutants at 40°C but did not influence or even compromise the growth of the wild-type strains (Fig. 6A,B; Supplemental Fig. S7A,B). Both or at least one of the mutants showed significantly increased cellular ROS levels and NADP+/NADPH ratios compared with the wild-type strains at 40°C (Fig. 6C; Supplemental Fig. S7C). These results suggest that the up-regulation of the cytosolic 1C metabolism pathway contributes to the heterosis of F1 hybrids of yeast at high temperature by providing more power for redox defense.
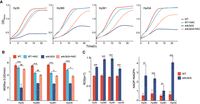
Effects of ADE3 gene deletion on the growth and cellular oxidative stress of F1 hybrids of wild S. cerevisiae at 40°C. (A) Growth curves of wild types (WTs) and ade3Δ/Δ mutants of four F1 hybrids without or with 15 mM N-acetyl-L-cysteine (NAC) in the medium. (B) MGRs of wild types and ade3Δ/Δ mutants of four F1 hybrids without or with 15 mM NAC in the medium. (C) Comparisons of cellular ROS levels (left) and NADP+/NADPH ratios (right) between the wild types and ade3Δ/Δ mutants of four F1 hybrids. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (NS) not significant, P > 0.05 (Mann–Whitney U test). Error bars, SDs (n ≥ 3).
To test if the up-regulation of ADE3 could improve the fitness of depressed hybrids, we expressed multiple copies of the gene in three depressed hybrids and their parents using an expression plasmid. However, we did not observe growth improvement of the mutants at 40°C (Supplemental Fig. S8). On the other hand, we found that the growth of the depressed hybrids and their parents at 40°C was significantly improved by the addition of N-acetyl-L-cysteine (Supplemental Fig. S8). The data suggest that elevated oxidative stress contributes to the decreased fitness of depressed F1 hybrids at high temperature. However, artificially up-regulating only one gene associated with 1C metabolism is unable to improve the outcome of the whole pathway. It is also possible that other pathways are required for the improvement of redox homeostasis or for the manifestation of thermotolerant heterosis in yeast.
Discussion
We show here that heterosis is prevalent in F1 hybrids of wild S. cerevisiae strains at high temperature. The degree of heterosis and parental genetic distance show a hump-shaped relationship. Improved redox homeostasis and energy-use efficiency caused by the up-regulation (HP-like or over HP level) of a limited number of genes associated with metabolism pathways centered by 1C metabolism probably play a key role in heterosis of yeast at high temperature. We provide new insights into molecular mechanisms underlying heterosis and thermotolerance of yeast and new clues for a better understanding of the molecular basis of heterosis in plants and animals.
The result of our study does not agree with that of Plech et al. (2014), who did not detect heterosis in hybrids formed by wild S. cerevisiae strains. The following factors are probably responsible for the inconsistency. First, the growth performance was generally tested at 30°C in the previous study. In our study, when average fitness of the parental strains and F1 hybrids was compared at 30°C, the latter also did not show heterosis (Supplemental Fig. S1). However, evident heterosis was detected in the F1 hybrids growing at 40°C (Fig. 1). Second, the strains used by Plech et al. (2014) were not genetically intact. Both homozygous and heterozygous diploid strains contained one or two of the hphMX4, kanMX4, and natMX4 cassettes. Another study has shown that the genetic markers have a significant cost on the growth of Saccharomyces strains, and unmarked strains usually grow better than the marked versions of the same strains (Bernardes et al. 2017). In our study, we used original homozygous wild strains, and the hybrids were generated by spore-to-spore mating without any genetic markers. Although the F1 hybrids did not show evident heterosis in optimum growth conditions at 30°C, they most likely will have an adaptive advantage in food and ethanol fermentation environments, in which yeast cells encounter various stresses, including high osmolarity, elevated temperature, and increased ethanol concentration. All these stresses can cause an elevated ROS level in the cells (Auesukaree 2017). This might explain the prevalence of heterozygosity in domesticated strains of S. cerevisiae (Duan et al. 2018).
A correlation between heterosis and genetic distance between inbreeding parental lines was generally observed in plants (East 1936; Chen 2010; Pandey et al. 2018) but was not observed in previous studies on Saccharomyces yeasts (Shapira et al. 2014; Bernardes et al. 2017). Shapira et al. (2014) did not observe a correlation between heterosis in hybrids and parental genetic distances in S. cerevisiae. Although Bernardes et al. (2017) showed a significant increase in MPH with increasing genetic distance, the relationship was driven entirely by the interspecific (S. cerevisiae × S. paradoxus) hybrids. Our result implies that the degree of thermotolerant heterosis in yeast increases along with the increase of parental genetic distance but decrease when the distance exceeds an optimal mating distance. The result is consistent with the theory prediction that the fitness of an individual hybrid is maximized when the genetic distance between its parents (i.e., mating distance) is neither too small nor too large (Wei and Zhang 2018). The nucleotide diversity and the maximal intraspecific genetic distance of S. cerevisiae are 6.63 × 10−3 and 16.39 × 10−3, respectively (Duan et al. 2018). The estimated optimal mating distances (7.42 × 10−3 to 10.56 × 10−3), as shown in Supplemental Table S2, also agree with the prediction of Wei and Zhang (2018) that the optimal mating distances are generally slightly greater than the nucleotide diversities of the species concerned but smaller than the observed maximal intraspecific genetic distances.
The conclusion of our study is apparently not in accordance with the regulatory incompatibility model, which attributes heterosis to the impairment of growth-limiting pathways in the hybrid (Bar-Zvi et al. 2017; Herbst et al. 2017). This hypothesis was proposed based on the study of one interspecific hybrid created by mating haploid strains of S. cerevisiae and S. paradoxus, with their HO genes being replaced by different drug-resistant genetic markers (Herbst et al. 2017). Because only one hybrid was used in the study of Herbst et al. (2017), it is not sure if the phenomenon observed is common in yeast hybrids. The increased DNA damages observed in the hybrid suggest that such hybrids will not have an adaptive advantage when living in nature. Indeed, although interspecific hybrids of different Saccharomyces species commonly occur in fermentation environments (Hittinger 2013; Gallone et al. 2019; Langdon et al. 2019), interspecific hybrids of S. cerevisiae and S. paradoxus have rarely been found (Pontes et al. 2019). Nevertheless, Herbst et al. (2017) showed the importance of gene regulation in heterosis of yeast hybrids.
On the other hand, the result of our study is in agreement with the energy-use efficiency hypothesis (Ginn 2010, 2017; Goff 2011). This hypothesis was proposed mainly based on the observation that inbred organisms usually have increased rates of protein turnover relative to noninbred organisms (Hawkins et al. 1986; Hedgecock et al. 1996; Bayne 2004). According to this model, inbred organisms consume more energy in the degradation of misfolded and aggregated proteins by the protein quality control systems (Ginn 2010), whereas hybrids achieve greater energy efficiency via selective expression of parental alleles encoding more stable proteins (Goff 2011), or reduce the rate of toxic soluble oligomer formation (Ginn 2017). A weakness of this hypothesis is that it was proposed only on the basis of theoretical analyses. Here we provide substantial evidence at the transcriptome level and an alternative more reasonable explanation to the model. The down-regulation of genes associated with protein quality control systems in the heterotic F1 hybrids of S. cerevisiae (Figs. 3, 4; Supplemental Fig. S5) suggests a lower protein turnover rate in the hybrids than in the parental strains and depressed hybrids. The result of our RNA-seq analysis suggests that the heterotic F1 hybrids achieve lower protein turnover rate through the up-regulation of genes in the 1C metabolism and related pathways (Figs. 3, 4; Supplemental Fig. S6), leading to improved redox homeostasis in the hybrids (Fig. 5). The reduced level of oxidative stress results in reduced errors in protein synthesis and folding and subsequently to reduced expression levels of genes associated with protein quality control systems, including protein folding, chaperons, proteolysis, and autophagy (Figs. 3, 4; Supplemental Fig. S5). In addition, the improved redox homeostasis also leads to the reduced expression levels of genes associated with DNA repair, stress responses, and ion homeostasis, as well as many other genes (Figs. 3, 4; Supplemental Fig. S5). Therefore, the heterotic F1 hybrids achieve improved energy-use efficiency by the down-regulation of more genes at the cost of the up-regulation of only a limited number of genes associated with key pathways for stress defense.
Gene expression profiling has been extensively used to illuminate molecular mechanisms of heterosis in plant hybrids, and some studies have shown that specific genes or pathways might contribute to heterosis for a number of traits in maize, rice, the tomato, and Arabidopsis (Hoecker et al. 2008; Wei et al. 2009; Krieger et al. 2010; Gonzalez-Bayon et al. 2019). However, the working models of these genes are still unclear, and whether such genes contribute to general molecular mechanisms of heterosis remains to be determined. From the genes and pathways with expression levels significantly correlated with heterosis of the F1 hybrids of S. cerevisiae, we infer that the up-regulation (overdominance or HP dominance) of genes associated with 1C metabolism and related pathways might be causative for the manifestation of heterosis of the yeast. As mentioned above, 1C metabolism as a universal metabolic process provides cells with the building blocks, as well as the reducing power, necessary to maintain high rates of proliferation (Rosenzweig et al. 2018). The up-regulation of these genes endows yeast cells with an improved ability to scavenge ROS triggered by high temperature or other stresses, contributing to the manifestation of growth heterosis of the hybrids. Because the growth rate is proportional to the total translation rate and to the number of ribosomes per cell (Lin and Amir 2018), the higher expression level of genes responsible for ribosome biogenesis and other pathways associated with translation observed in the heterotic F1 hybrids (Fig. 3) is probably the effect, not the cause, of heterosis. The observed association between decreased rates of protein metabolism and heterosis in animals and plants (Hawkins et al. 1986; Hedgecock et al. 1996; Bayne 2004) implies a possible role of 1C metabolism process in the manifestation of heterosis of these species, for reduced ROS level in the cell contributed by 1C metabolism will decrease protein damages, thus leading to decreased protein turnover rate.
Why genes in 1C metabolism and related processes are up-regulated in the F1 hybrids remains to be illuminated. Previous studies have shown that additive expression of a gene is attributable mostly to cis-regulation, whereas nonadditive expression is attributable mostly to trans-regulation (Lemos 2008; Wei et al. 2009; McManus et al. 2010). Genes with antagonistic cis-trans interactions are more likely to be overdominant or underdominant in hybrids (Hochholdinger and Hoecker 2007; Lemos 2008; McManus et al. 2010; Schaefke et al. 2013). The expression patterns of genes in the 1C metabolism and related processes in the F1 hybrids suggest that they are most likely subjected to trans-regulation or antagonistic cis-trans regulation. Trans-acting regulators (e.g., transcription factors and chromatin modifiers) are diffusible, and thus, the genes of the F1 hybrids are subjected to the regulation of these regulators from both parents. The trans-acting regulators with slightly deleterious mutations in one parent can probably be complemented by those from the other parent in the hybrid cells, resulting in HP dominance if the genes are subjected to only trans-regulation or in over- or underdominance if the genes are subjected to cis-trans regulation (Hochholdinger and Hoecker 2007; Lemos 2008; McManus et al. 2010; Schaefke et al. 2013). These mechanisms are in accordance with the dominance model (complementation of slightly deleterious recessive alleles of trans-acting regulator genes) and with the epistasis model (interactions between cis- and trans-acting elements), respectively. The largely positive correlation between heterosis and parental genetic distance until an optimal mating distance observed in this study implies that the complementation model may play a role. However, the benefit of the complementation will probably be offset by the harm of genetic incompatibility when the parental genetic distance is beyond the optimal mating distance, as shown in this study (Fig. 1; Wei and Zhang 2018).
To our knowledge, we are probably the first to use outbreeding depressed F1 hybrids as contrast in the study on the molecular mechanisms of heterosis. Although the number of depressed F1 hybrids generated and used in this study is limited, they are helpful to figure out the genes responsible for heterosis and manifest the contribution of these genes. As for the heterosis, genes associated with 1C metabolism and the related processes are apparently also responsible for the manifestation of the depression. The down-regulation of these genes might compromise redox homeostasis, leading to increased level of ROS (Fig. 5), which in turn causes more DNA and protein damages. We therefore observed elevated expression levels of genes associated with DNA repair and protein quality control systems in the depressed hybrids (Figs. 3, 4; Supplemental Fig. S5). The increased level of oxidative stress might also compromise homeostasis of many other metabolism processes, thus triggering the up-regulation of genes responsible for cellular homeostasis and stress responses (Figs. 3, 4; Supplemental Fig. S5). Therefore, the depressed F1 hybrids showed an opposite gene expression pattern to the heterotic F1 hybrids and reduced growth rate and efficiency at high temperature. The down-regulation (LP-like or below LP level) of genes in 1C metabolism and the related processes is probably caused by regulatory incompatibility or antagonistic cis-trans interactions. As discussed above, antagonistic cis-trans interactions are more likely to be not only overdominant (above HP) but also underdominant (below LP) in hybrids (Hochholdinger and Hoecker 2007; Lemos 2008; McManus et al. 2010; Schaefke et al. 2013).
Aneuploidy is probably another cause of outbreeding depression in S. cerevisiae. The majority (7/9, 77.8%) of the depressed F1 hybrids used in our RNA-seq analysis share a common parental strain BJ22 (Supplemental Table S2). We found that only 22.7% (4/15) of the F1 hybrids generated by crossing BJ22 with other strains showed MPH, much lower than the overall proportion (96.2%) of the F1 hybrids showing MPH (Fig. 1; Supplemental Table S1). The result implies the responsibility of this specific strain for the depression. Our previous study has shown that strain BJ22 is aneuploid (2N + 5), which has two extra copies of Chromosome I, two extra copies of Chromosome III, and one extra copy of Chromosome VI (Duan et al. 2018). The hybrids with BJ22 as one parent are most likely also aneuploid. Chromosomal differences may generate outbreeding depression (Frankham et al. 2011). Investigations in S. cerevisiae have revealed growth defects of aneuploid cells resulting from cell cycle delays, DNA and protein damage, protein folding errors, and elevated ROS levels (Torres et al. 2007; Tsai and Nelliat 2019). The result of RNA-seq analysis of the depressed F1 hybrids obtained in our study is consistent with the phenomena observed in the previous studies. Illumination of the mechanisms causing outbreeding depression is certainly helpful for a better understanding of the mechanisms underlying heterosis.
Finally, it is worth noting that the physiological and molecular interpretations of the mechanisms underlying heterosis presented in this study are only valid for S. cerevisiae hybrids growing at high temperature. Whether the yeast hybrids show similar degrees of heterosis and gene expression variations under other stressful conditions remains to be tested.
Methods
Parental yeast strains and generation of F1 hybrids
A total of 53 parental strains (Supplemental Table S1) were selected from different wild lineages of S. cerevisiae, which were shown to be homozygous by genome analysis (Duan et al. 2018). Parental genetic distance as indicated by the percentage of SNP difference between two parental strains were determined using the Python software package EggLib (De Mita and Siol 2012).
The parental strains were inoculated onto YPD agar (w/v, 1% yeast extract, 2% peptone, 2% D-glucose, and 2% agar) plates for 24 h at 30°C and then transferred onto sporulation plates (1% potassium acetate and 2% agar) and incubated for 3–4 d at 30°C. Asci were collected and digested in 10 mg/mL Zymolyase (20T) solution for 15 min at 30°C. Tetrad dissection and spore-to-spore mating were performed under an MSM 400 micromanipulator microscope (Singer Instruments) on YPD plates. The colonies formed by zygotes that were judged by the mating structure of paired spores were selected. Finally, a total of 641 F1 hybrids were created.
Growth performance test
The mitotic proliferative abilities of parental strains and F1 hybrids in YPD broth at 30°C and 40°C, respectively, were tested in duplicates in microplates using a Bioscreen analyser C (Thermic Labsystems) as described by Warringer and Blomberg (2003). For testing the effect of N-acetyl-L-cysteine on the growth of yeast strains, the compound was added to YPD broth to a final concentration of 15 mM (Topf et al. 2018). The fitness variables, including maximum growth rate and growth efficiency, were extracted from high-density growth curves as described previously (Duan et al. 2019).
Correlation analyses between heterosis and parental genetic distance
The relationship between heterosis and parental genetic distance was analyzed using linear and nonlinear models as described by Wei and Zhang (2018) with minor modifications. Specifically, growth performance was determined at 40°C as described above. We binned the hybrids with similar parental genetic distance (G) under a window size of 1 × 10−3, computed the average G for each bin, and then calculated the average performance of the hybrids for each window. The quadratic equations were fitted by the nonlinear least-squares method using the lm function in R following the formula performance ∼G/2 + G, and the linear model was computed following the formula performance ∼G.
RNA-seq, gene expression profiling, and GO enrichment analyses
The F1 hybrids (59 strains) and their parents (35 strains) selected for RNA-seq analysis are listed in Supplemental Table S2. Yeast cells were incubated in YPD broth at 40°C and harvested at the log-phase. Total RNA was extracted and processed using a commercial RNA purification kit (Thermo Fisher Scientific) according to the manufacturer's protocols. RNA samples were sequenced on an Illumina HiSeq X Ten platform, and at least two Gb clear reads were obtained for each strain.
The FASTX toolkit (https://github.com/agordon/fastx_toolkit/) was used to evaluate the raw data and discard low-quality reads. The clean reads were aligned to the reference genome of strain S288C using HISAT2 (Pertea et al. 2016), and the numbers of reads mapped to each gene were counted by HTSeq (Anders et al. 2015). The Q30 was >89%, and >85% of the reads were mapped to the reference genome (Supplemental Table S3). The gene expression levels were calculated based on the values of fragments per kilobases per million mapped reads (FPKM). The genes with an expression level >1 FPKM were identified as actively expressed genes and subjected for further analyses. The genes with expression levels in a F1 hybrid that significantly deviated from the average expression levels of the genes in its parents (MPV) determined using the algorithm DESeq (P ≤ 0.01, FDR ≤ 0.1) (Groszmann et al. 2015) were identified as nonadditive genes. GO enrichment analysis of nonadditive genes was implemented using Metascape (Zhou et al. 2019), and the GO terms with corrected P < 0.01 were considered as significantly enriched. Enrichment networks were then constructed based on functional similarities of the terms that were measured using an algorithm adopts kappa statistics (Huang et al. 2009). The algorithm quantitatively measures the degree of the agreement of how genes share the similar annotation terms, resulting in kappa similarities ranging from zero to one. Enrichment networks were created by representing each enriched term as a node and connecting pairs of nodes with kappa similarities above 0.3 (Zhou et al. 2019).
Gene expression pattern analysis of nonadditive genes
Based on the expression level of a gene in a F1 hybrid relative to the expression level of the gene in the HP and LP, a nonadditive gene was classified further into one of the five patterns: namely, additive (HP > F1 > LP), HP-like (F1 = HP), LP-like (F1 = LP), above HP (F1 > HP) and below LP (F1 < LP) as defined previously (Stupar et al. 2008; Rapp et al. 2009; Tian et al. 2018). For each nonadditive gene, its FPKM counts in a F1 hybrid, and in its parents were log-transformed and tested for significant deviations between the F1 hybrid and its parents and between the two parents using one-way analysis of variance (ANOVA). Subsequently, we assessed pairwise contrasts using the TukeyHSD post hoc test (P < 0.05).
Validation of RNA-seq data by qRT-PCR
qRT-PCR was used to validate the gene expression levels determined by RNA-seq analysis. Nineteen RNA samples were randomly selected from the samples subjected to RNA-seq analysis, and cDNA libraries were constructed using the first-strand cDNA synthesis with the PrimeScript RT reagent kit with gDNA Eraser (TaKaRa). Seventeen genes were selected, and primers were designed using the Primer Express 3.0 software (Applied Biosystems) (Supplemental Table S4). The qRT-PCR was performed using a Roche LightCycler480 II real-time PCR system (Roche). Each reaction contained 10 μL of 2 × FastStart universal SYBR Green master (Roche), 2.0 μL diluted cDNA, and 0.8 μL each of the forward and reverse primers in a final volume of 20 μL. The PCR conditions consisted of predenaturation for 5 min at 95°C, followed by 40 cycles of 15 sec at 95°C, 25 sec at 60°C, and 25 sec at 72°C. At the end of the PCR cycles, a melting curve analysis was performed to validate the specificity of the PCR product. The housekeeping gene LYS14 was used as a reference for normalization, and three replicates were performed for each cDNA sample. Data analysis was performed as reported previously (Livak and Schmittgen 2001).
ROS and NADP+/NADPH ratio measurement
ROS levels in yeast cells were assayed with 2,7′-dichlorodihydrofluorescein diacetate (Sigma-Aldrich H2DCF-DA). Briefly, strains were incubated in YPD broth for 4 h at 40°C, and then H2DCF-DA was added to a final concentration of 10 μM and incubated for 30 min in the dark at 40°C. Yeast cells were collected by centrifuge and washed three times with phosphate buffered saline (PBS; 135 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, and 8 mM K2HPO4 at pH 7.2). Fluorescence intensity was measured by spectrophotometry with excitation of 488 nm and emission of 525 nm.
NADP+ and NADPH were extracted and measured using the NADP+/NADPH quantitation kit (Sigma-Aldrich) as described by Zhang et al. (2016).
Knocking out ADE3 and MTD1 genes
Gene deletion was performed using the protocol as described in our previous study (Duan et al. 2019). Shortly, for deleting the first copy of the target gene, the plasmid pKAN-ADE3 or pKAN-MTD1 was generated by inserting two DNA fragments containing sequences homologous to the 5′- and 3′-terminals of ADE3 or MTD1 of strain BJ23 into the plasmid pKAN. The plasmids pAG32-ADE3 and pAG32-MTD1 were constructed in the same way for deleting the second copy of the genes. We finally constructed mutant strains with ade3::hphMX/ade3::kanMX or mtd1::hphMX/ mtd1::kanMX. The concentration of G418 and hygromycin used in the YPD plates for mutant selection is 300 μg/mL and 400 μg/mL, respectively.
Overexpression of ADE3
To overexpress the gene ADE3, an expression vector was constructed and transformed into target strains using the protocol as described by Duan et al. (2019) with minor modifications. In brief, the hphMX marker was amplified from the plasmid pAG32, and the DNA segment including the promoter, open reading frame, and terminator of ADE3 was amplified from the wild S. cerevisiae strain BJ23. The marker and the DNA segment were fused into the expression plasmid pRS423, which was then transformed into targeted strains.
Statistical analysis
Standard statistical analyses were performed in R project (v3.3.1) (R Core Team 2016). The statistical significance was obtained using the Student's t-test or Mann–Whitney U test executed by the t.test or wilcox.test function of R project with a two-sided alternative hypothesis. ANOVA was used in gene-expression-level variation analysis.
Data access
The raw RNA-sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA659808. The information of the RNA-sequencing data obtained from every sample is available in Supplemental Table S4.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This study was supported by the Chinese Academy of Sciences (grants no. QYZDJ-SSW-SMC013 and no. 153211KYSB20160029) and the Ministry of Science and Technology of China (KY201701011, the Science and Technology Partnership Program).
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.262055.120.
- Received February 6, 2020.
- Accepted February 12, 2021.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








