
Abstract
Biosynthetic gene clusters (BGCs) are operonic sets of microbial genes that synthesize specialized metabolites with diverse functions, including siderophores and antibiotics, which often require export to the extracellular environment. For this reason, genes for transport across cellular membranes are essential for the production of specialized metabolites and are often genomically colocalized with BGCs. Here, we conducted a comprehensive computational analysis of transporters associated with characterized BGCs. In addition to known exporters, in BGCs we found many importer-specific transmembrane domains that co-occur with substrate binding proteins possibly for uptake of siderophores or metabolic precursors. Machine learning models using transporter gene frequencies were predictive of known siderophore activity, molecular weights, and a measure of lipophilicity (log P) for corresponding BGC-synthesized metabolites. Transporter genes associated with BGCs were often equally or more predictive of metabolite features than biosynthetic genes. Given the importance of siderophores as pathogenicity factors, we used transporters specific for siderophore BGCs to identify both known and uncharacterized siderophore-like BGCs in genomes from metagenomes from the infant and adult gut microbiome. We find that 23% of microbial genomes from premature infant guts have siderophore-like BGCs, but only 3% of those assembled from adult gut microbiomes do. Although siderophore-like BGCs from the infant gut are predominantly associated with Enterobacteriaceae and Staphylococcus, siderophore-like BGCs can be identified from taxa in the adult gut microbiome that have rarely been recognized for siderophore production. Taken together, these results show that consideration of BGC-associated transporter genes can inform predictions of specialized metabolite structure and function.
Microbes produce specialized metabolites with diverse functions, including siderophores, ionophores, antibiotics, antifungals, and signaling molecules (Osbourn 2010). Specialized metabolites therefore often underlie both cooperative and competitive interactions between microbes and microbial interactions with the physiochemical environment (Davies 2013; Sharon et al. 2014; Tyc et al. 2017). The vast majority of specialized metabolites in bacteria are produced by biosynthetic gene clusters (BGCs), which are sets of genomically colocalized genes. Colocalization of genes into BGCs is thought to occur because of selection for coinheritance and coregulation (Fischbach et al. 2008). Although thousands of microbial natural products have been characterized, genomic BGC predictions made using programs such as antiSMASH (Blin et al. 2019b) and ClusterFinder (Cimermancic et al. 2014) suggest that characterized molecules represent just a small fraction of all existing microbial natural products (Medema and Fischbach 2015; Kim et al. 2017). Many of these unknown metabolites may be highly novel owing to enzymatic and combinatorial diversity of genes in BGCs (Jenke-Kodama et al. 2006; Chevrette et al. 2020).
Because of the sheer number of sequenced but otherwise uncharacterized BGCs and the time and costs required for chemical characterization, there is a pressing need for predictions of BGC metabolite structures or functions to enable prioritization of targets for laboratory study (Tran et al. 2019). Prediction of metabolite structure or function for a novel BGC from gene content alone is challenging. For many biosynthetic nonribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs), there is a “colinear” assembly-line regulation in which the order of genes relates to the order of enzymatic modifications on the metabolite during synthesis (Fischbach and Walsh 2006). Using this colinearity rule can help predict some degree of structural detail in NRPSs and PKSs, as is done by antiSMASH and PRISM (Skinnider et al. 2017), but there are many known exceptions to this rule (Wenzel and Müller 2005), and the accuracies of these software predictions have not been formally assessed using a large training data set.
Prediction of BGC metabolite function generally relies on contextual genes associated with BGCs. The observation that genes conferring resistance to the produced metabolite are also colocalized with the BGC motivates investigation of putative resistance genes (self-resistance gene mining) (Mungan et al. 2020; Yan et al. 2020) for functional prediction. For siderophore activity prediction, antiSMASH assigns a functional “siderophore” label for BGCs that contain the IucA/IucC gene family, but this gene is only specific for siderophores with biosynthetic pathways similar to aerobactin (Hider and Kong 2010). More recently, Hannigan et al. (2019) trained neural networks to both identify BGCs in genomes and classify BGCs by known metabolite functions. These networks used protein families from the Pfam database (Pfams) (El-Gebali et al. 2019) found in each BGC as features. They predicted activity labels of antibacterial, antifungal, cytotoxic, and inhibitor, with precisions of 36%, 47%, 61%, and 69% on each class, respectively.
Many specialized metabolites perform their ecological roles extracellularly and thus require transport across cellular membranes. Transporter genes often colocalize in BGCs and have been shown to be compound specific and necessary for export of the product in many cases (Méndez and Salas 2001; Martín et al. 2005; Severi and Thomas 2019). Therefore, transporters may also inform predictions of BGC metabolite structure and function. The distribution of transporters associated with biosynthetic gene clusters has so far been assessed only in characterized BGCs with experimental validation, a small fraction of the total number of BGCs sequenced. At least 40 BGC-associated exporters have been characterized, mostly in the Actinomycetes, with varying degrees of experimental validation (Severi and Thomas 2019).
Transporters associated with BGCs are commonly either ATP-dependent active transporters or ion-gradient-dependent transporters (Martín et al. 2005). ATP-dependent transporters include the ATP-binding cassette (ABC) superfamily of both importers and exporters (Rees et al. 2009) and the MacB tripartite efflux pump (Greene et al. 2018b). Examples of characterized structures of each transporter class and their substrates are shown in Figure 1A. In brief, Type I ABC importers are characterized by the BPD_transp_1 transmembrane (TM) protein family and include MalFGK and MetNI for malate and methionine import in E. coli (ter Beek et al. 2014). Type II ABC importers are characterized by the FecCD TM protein family, and examples include BtuCD and HmuUV (ter Beek et al. 2014) and the FecBCDE system for Iron(III) dicitrate import in Escherichia coli (Staudenmaier et al. 1989). Both types of ABC importers often associate with substrate binding proteins (SBPs), small membrane or periplasmic proteins for substrate uptake (Berntsson et al. 2010; ter Beek et al. 2014). Periplasmic binding proteins, Type II ABC importers, and TonB-dependent receptors are also known to play key roles in siderophore uptake in multiple bacterial species (Ellermann and Arthur 2017).
Distributions of transporter classes in biosynthetic gene clusters. (A) Structures of characterized examples of major transporter classes often found in BGCs, colored and labeled by Pfam domains. The extracellular/periplasmic side of the membrane is shown as a red line, and the intracellular side is in blue. (B) The frequencies of common Pfam transporter domains across the bacterial BGCs in the MIBiG database. (C) The percentages of bacterial BGCs in MIBiG that do and do not contain transporter domains. Each square represents 1% of BGCs. (D) The counts of transporter domains per each bacterial BGC that contains at least one transporter gene across MIBiG.
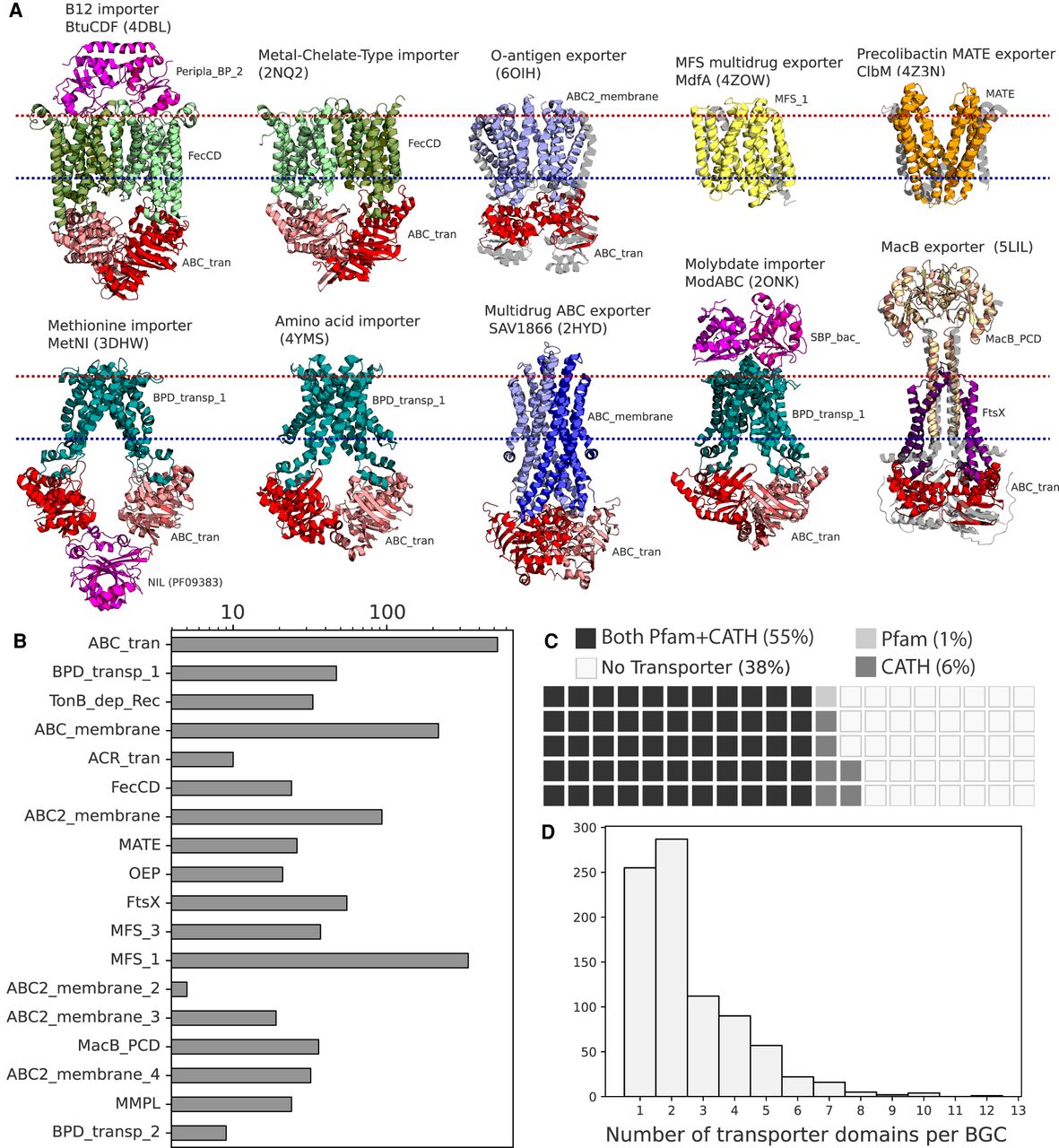
Meanwhile, examples of ABC exporters include McjD for Lasso peptide microcin J25 export (Romano et al. 2018) and the Staphylococcus aureus multidrug exporter Sav1866 (Dawson and Locher 2007), composed of the ABC_membrane TM protein family, whereas the O-antigen polysaccharide exporter is composed of the ABC2_membrane TM protein family (Bi et al. 2018). Export of Nystatin, Doxorubicin, and Mccj25 was found to be dependent on ATP-dependent transporters (Severi and Thomas 2019). The vast majority of ribosomally synthesized and post-translationally modified peptides (RiPPs) and a number of antibiotics from Actinomycetes also rely on characterized ATP-dependent ABC transporters (Méndez and Salas 2001; Gebhard 2012).
Ion-gradient-dependent transporters (also known as secondary active transport systems) do not require ATP and facilitate transport of small molecules in response to chemiosmotic gradients (Quistgaard et al. 2016). Those found in BGCs are often examples of the major facilitator superfamily (MFS) and occasionally, the resistance nodulation division (RND) family or the multidrug and toxic compound extrusion (MATE) family. Examples of characterized secondary active transporters for antibiotics include an RND transporter for pyoluteorin, and MFS transporters specific for mitomycin C, virginiamycin S, and landomycin (Severi and Thomas 2019).
Thousands of BGCs with chemically characterized metabolites open up the possibility for a broad genomic and computational analysis of phylogenetically and functionally diverse BGCs. Here, we used a curated version of the Minimum Information about a Biosynthetic Gene cluster database (MIBiG 2.0) of BGCs (Kautsar et al. 2020) and selected transporter-specific protein domain hidden Markov models (HMMs) to perform a wide genomic assessment of the distribution of transporters in BGCs. We found clear correlations between transporter domains and corresponding metabolite features, especially siderophore activity, that indicate underlying logical structure to transporter associations and can inform functional and structural prediction of specialized metabolites from genomics alone.
Results
Genome mining of transporters associated with biosynthetic gene clusters
Using two compiled sets of transporter-specific HMMs (Pfam and CATH) (https://www.cathdb.info/), we cataloged all classes of transporters across the MIBiG 2.0 database of characterized and experimentally validated biosynthetic gene clusters. We found that 56% of the bacterial BGCs in MIBiG contained at least one Pfam transporter hit and an additional 6% contained a CATH transporter hit without a Pfam domain (Fig. 1C). These percentages increased among BGCs that produce antibiotics (71%) and siderophores (78%), indicating that BGCs with these activities are more likely to contain at least one transporter. BGCs with transporters contained 2.5 transporter-associated domains across transport-annotated genes on average (Fig. 1D), which is expected because many ATP-dependent transporter systems have at least two domain complexes.
However, some BGCs contain considerably more transporter ORFs and domains, indicating that sometimes multiple transport systems can be associated with one BGC, although the number of protein domains that function as one transport system can often vary. The number of transporters in a BGC had no association with the number of metabolite structures reported for that BGC. The ATP-binding ABC transporter (ABC_tran) domain and the Major Facilitator Superfamily 1 (MFS_1) domain were the two most common transporter domains found in BGCs (Fig. 1B). A variety of proteins had nucleotide binding domains along with several different transmembrane domains—ABC_membrane and ABC2_membrane domains were most common but ABC2_membrane_2, -_3, and -_4 domains were also represented.
Examining domains specific for export, the ABC_membrane domain is often characteristic of exporters (e.g., Sav1866) (Velamakanni et al. 2008), but recently has been reported in the genes for siderophore uptake (YbtPQ) in Yersinia (Wang et al. 2020) and is therefore not necessarily indicative of export or import alone. The second most common transmembrane domain, ABC2_membrane, has been observed in the O-antigen polysaccharide exporter (Bi et al. 2018). The MacB-FtsX tripartite efflux pump was found in 60 BGCs, whereas the RND (ACR_tran) efflux pump was less common and found in only 10 BGCs. Other known efflux systems, such as SMR, MatE, and the MFS families 2 through 5 were comparatively rare across BGCs.
We next calculated co-occurrence correlations between all transporter protein families across BGCs and observed a strong negative correlation between MFS transporters and ATP-dependent transporters relying on the nucleotide binding domain, and a weaker negative correlation of MatE domains from the ATP-dependent NBD (Fig. 2A). This points toward a dichotomous choice between ATP-dependent and ATP-independent transport associated with a BGC.
Presence of importer-specific domains and co-occurrence between transporters across BGCs. (A) Spearman's correlations between commonly occurring Pfam transporter domains across MIBiG BGCs—only correlations with P < 0.001 are shown. (B) Counts of Pfam transporter substrate binding domain families and corresponding substrate binding protein clusters described by Berntsson et al. (2010). (C) Counts of importer-specific CATH domains across MIBiG BGCs. The CATH functional family “Iron ABC permease” is essentially synonymous with the FecCD Pfam.
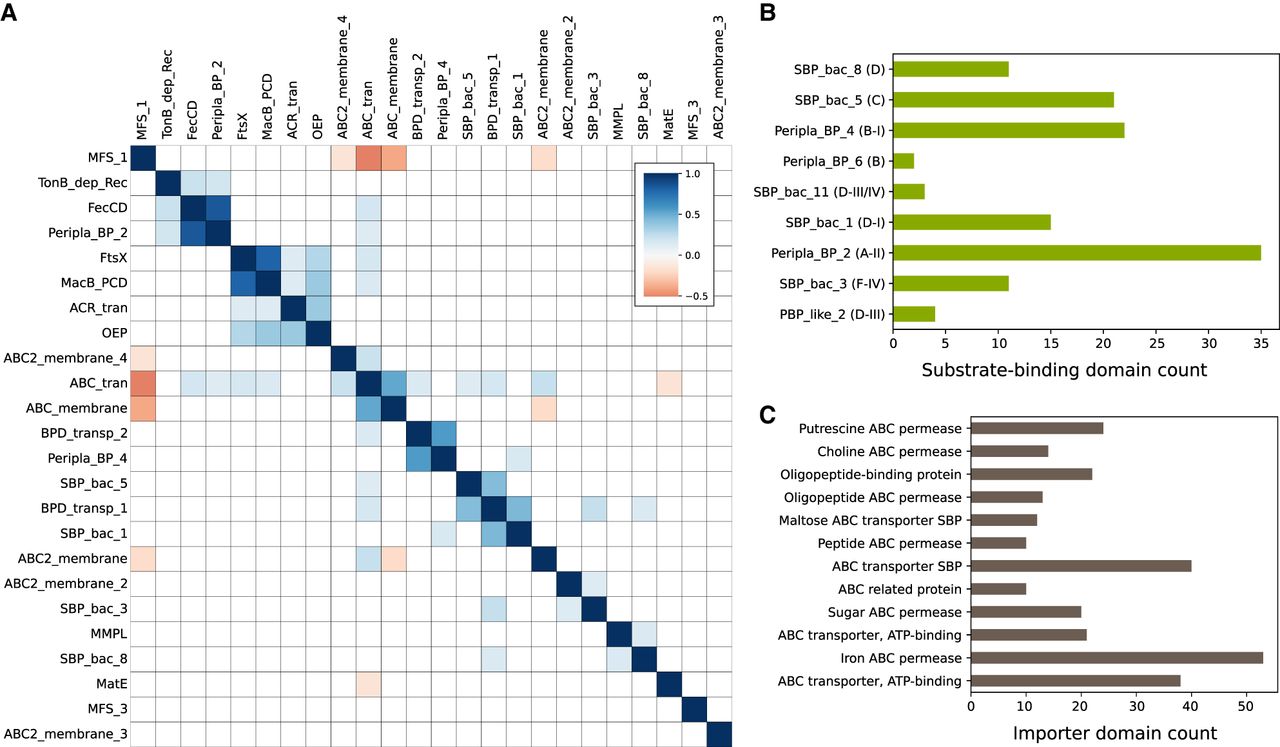
Multiple lines of annotation evidence indicated that many of the transporter genes associated with BGCs were likely to be importers. Importers can be involved in the uptake or re-uptake of molecules like siderophores and may also play roles in importing precursor metabolites for a BGC. The membrane domains specific to Type I importers (BPD_transp_1) and Type II importers (FecCD) were the most often observed ATP-dependent transmembrane domains besides ABC_membrane and ABC2_membrane (Fig. 1B). CATH Protein Structure Classification database HMMs (CATHs) that were specific for importer families in the Transporter Classification Database and found in BGCs included permeases for sugars, oligopeptides, and iron siderophores (Fig. 2C). Eleven percent of BGCs with a transporter also contained a substrate binding protein. Among the substrate binding proteins we searched for, the most common contained domain was Peripla_BP_2, also found in the E. coli B12 importer complex BtuCDF, and variants of this SBP (cluster A-II) are specific for siderophores and cobalamin (Berntsson et al. 2010). We also observed many substrate binding proteins with specificities predicted to include carbohydrates, oligopeptides, and peptide uptake (Fig. 2B; Berntsson et al. 2010). For example, the gene family SBP_bac_1 (SBP cluster D-I) (Berntsson et al. 2010) is specific for uptake of sugars and was found in the BGCs for the glycopeptide Mannopeptimycin and the aminoglycoside spectinomycin and may play a role in sugar precursor uptake (Supplemental Fig. S1). It was also found in the acarbose and acarviostatin BGCs (Supplemental Fig. S1), consistent with their putative roles as carbophors (Guo et al. 2012).
In the co-occurrence data, we observed pairing of different substrate binding proteins with different transmembrane domains. Peripla_BP_2 positively correlated strongly with FecCD and TonB_dep_Rec, genes known to be involved in siderophore uptake. BPD_transp_1 co-occurred with either SBP_bac_5 (SBP cluster C) or SBP_bac_1, whereas BPD_transp_2 co-occurred with Peripla_BP_4 (SBP cluster B-I). Taken together, these results show a logical organization of importer-specific transporter domains within BGCs that may be involved in either siderophore uptake, precursor uptake, or other roles. Regardless of the substrate specificity of these proteins, care must be taken when assuming that a transporter in a BGC is definitively for export of the matured product.
Prediction of siderophore and antibacterial activity from biosynthetic transporters
Because transporters are required for the ecological functions of biosynthesized specialized metabolites, we used machine learning to test if transporter classes were predictive of BGC-synthesized metabolite structures and functions. We noticed that metabolite activity labels in MIBiG were strongly associated with phylogeny: 83% of antibiotic BGCs were from Gram-positive bacteria, but only 40% of siderophore BGCs were from Gram-positive bacteria in the data set of curated MIBiG BGCs. To reduce the impact of this potential bias, we created separate training and testing data sets for activity prediction for Gram-positive and Gram-negative organisms.
Using our curated set of BGCs with transporters from the MIBiG 2.0 database, we tested for associations between BGC transport genes and metabolite function. We generated two activity classification tasks: (1) distinguishing siderophores (including known ionophores) (n = 16 Gram-positive, n = 24 Gram-negative) from non-siderophores (n = 142 Gram-positive, n = 42 Gram-negative); and (2) distinguishing antibiotics and antifungals (n = 131 Gram-positive, n = 27 Gram-negative) from non-antibiotics (n = 57 Gram-positive, n = 37 Gram-negative). We observed several statistically significant (Fisher's exact test; Q < 0.05) associations in the distribution of transporter types between both the siderophore and other activity classes. Among Gram-positive bacteria, 60% of siderophore BGCs contained the SBP Peripla_BP_2 and 55% contained the FecCD importer, but no BGCs with other activities had either (Fig. 3A; Supplemental Fig. S2). The situation was similar for Gram-negative siderophore BGCs. The TonB-dependent receptor (completely absent from Gram-positive bacteria) was the strongest signal, found in almost 80% of Gram-negative siderophore BGCs with a transporter and never in BGCs with other activities.
Transporter domains are predictive of siderophore BGCs. (A) The frequencies of common transporter Pfam domains across siderophore BGCs and BGCs of other known activities in Gram-positive and Gram-negative bacteria. Bars in green were significantly different in frequency between the two classes (Fisher's exact test; Q < 0.05) (B) Precision-recall curves for two-layer decision trees classifying siderophore BGCs using Pfam transporter, CATH transporter, and Pfam biosynthetic gene features in Gram-negative and Gram-positive bacteria. (C) Examples of three siderophore BGCs without activity labels in MIBiG 2.0, which could be identified using transporter frequencies. Transporter genes are blue, core biosynthetic genes (NRPS and PKS) are dark red, accessory biosynthetic genes are light red, and regulatory genes are green.
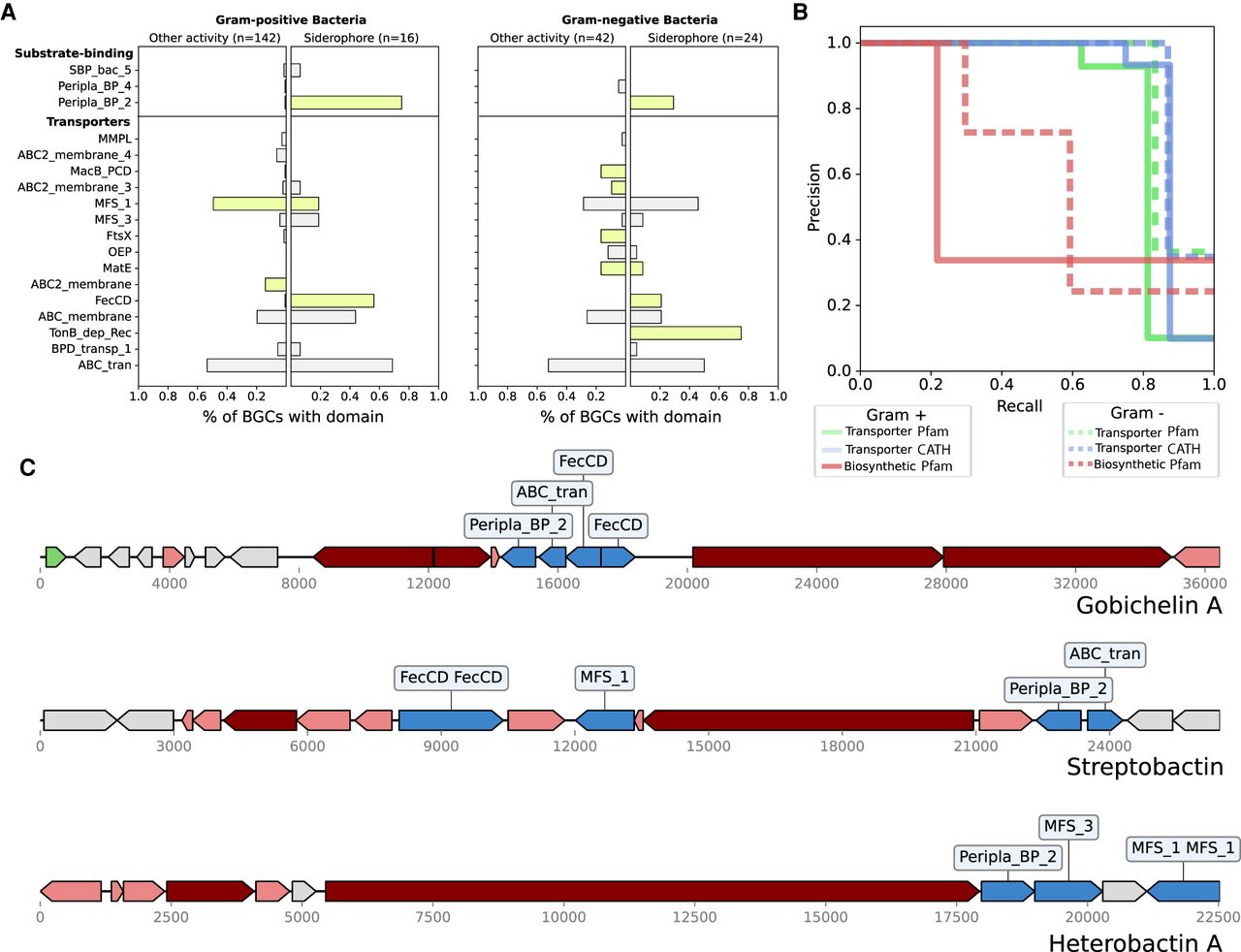
To assess siderophore predictability from BGC gene content, we used decision trees with only two layers applied to different feature sets of protein domain annotations, transport-affiliated Pfams, transport-affiliated CATH HMMs, and biosynthetic Pfams. To avoid issues with class imbalance, we report precision and recall on the siderophore class, because siderophore prediction requires searching for a minority class (siderophores) within a background of mostly non-siderophores. With the transport-only features, we found that just with two gene decisions, it is possible to achieve 100% precision with <80% recall using either Pfam or CATH transporter annotations for Gram-negative siderophores, and 100% precision with <80% recall using CATH transporter annotations for Gram-positive siderophores (Fig. 3B; Supplemental Table S5). On the other hand, when using all biosynthetic annotations within BGCs, we found that two-layer decision trees trained on biosynthetic genes performed substantially worse at predicting siderophore activity than those trained on transporter genes (Fig. 3B; Supplemental Table S5). We further validated our results by training LASSO linearized regression models, which do not model interactions between features. These models obtained a slightly improved area under the precision-recall curve (AUPRC), indicating that very simple transporter patterns are highly predictive of whether a BGC is siderophore producing or not in our data set (Fig. 3B). Transporter features predictive of siderophores were consistently selected by LASSO across stratified cross-validation repeats, giving evidence that these patterns are robust (Supplemental Fig. S3). The top predictive biosynthetic features of siderophores were the IucA/IucC protein family (used by antiSMASH to label siderophores and which is known to be involved in aerobactin biosynthesis) and condensation domains (likely to capture nonribosomal peptide siderophores), but predictive effect sizes were smaller than those for transporters (Supplemental Fig. S3).
Using siderophore-specific transporter genes, we attempted to predict siderophore classes for any remaining gene clusters that have no annotated function in MIBiG2 (that we had not already hand curated). We searched for gene clusters containing the siderophore-predictive genes FecCD and Peripla_BP_2 and found six additional BGCs with no annotated activity, three of which were experimentally validated by the literature to be siderophores (Fig. 3C; Carran et al. 2001; Matsuo et al. 2011; Chen et al. 2013). The remaining three identified BGCs were false positives. One of them, the BGC for the antibiotic Ficellomycin, only contained these transport genes in flanking regions not known to be involved in biosynthesis (Liu et al. 2017), whereas the herbimycin A BGC contains the transporters genes in the reverse reading frame from the BGC, separated by an unusual 16-kb intergenic region and the genes appear to be fragmented (Rascher et al. 2005). The other false positive was Lividomycin, an antibiotic that does seem to be a rare non-siderophore BGC with FecCD and Peripla_BP_2 transporters in the MIBiG2.0 database.
To understand the extent of transporter specificity for particular classes of BGCs, we expanded our analysis to 95,293 BGCs in the antiSMASH database, which is a set of predicted BGCs in microbial genomes from the RefSeq database. Frequencies of general transporters and transporter genes that were specific for siderophore biosynthesis in MIBiG (FecCD and the TonB-dependent Receptor) were calculated across all antiSMASH BGCs by bacterial genus (Fig. 4; Supplemental Fig. S4). The general ABC transporter ATP-binding domain and MFS superfamily transporter genes varied in their frequencies across different classes of BGCs, but there were some consistent patterns. Thiopeptides, NRPS-independent siderophores, and arylpolyene BGCs tended to have an MFS exporter, but ribosomally synthesized products (e.g., Lasso peptides and Lantipeptides) consistently had ATP-dependent transport mechanisms (Fig. 4A).
Presence of general and siderophore-specific transporters by biosynthetic class and bacterial genus across the antiSMASH database. (A) ATP-dependent and ATP-independent (MFS) transporters are commonly associated with a variety of BGCs in the antiSMASH database across a wide range of genera. Each point is the percentage of a BGC class with a transporter within a particular genus. Each genus is colored by its Gram status, and genera with fewer than 20 BGCs of a particular class are excluded. (B) Siderophore-specific transporters are associated with few BGC classes in the antiSMASH database. Each point is the percentage of a BGC class with a transporter within a particular genus. Each genus is colored by its Gram status, and genera with fewer than 20 BGCs of a particular class are excluded.
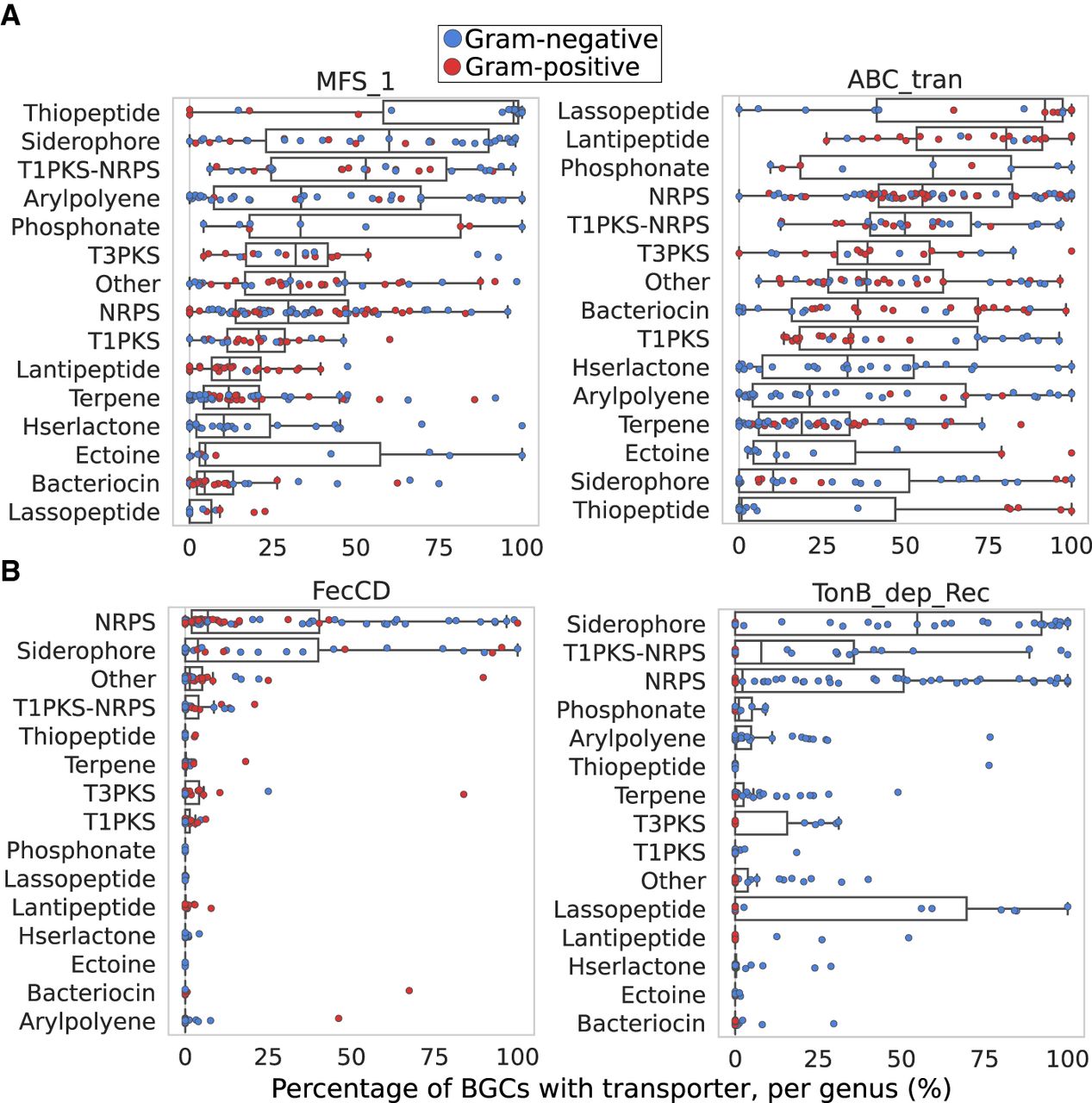
Alternatively, the TonB-dependent receptor and the FecCD Type II importer were highly restricted to specific classes of BGCs. They only consistently appeared in NRPS-independent siderophore and NRPS clusters and were nearly absent from entire other classes of BGCs (Fig. 4B). This is consistent with the known NRPS-independent and NRPS biosynthetic pathways for siderophores. The TonB-dependent receptor was also found associated with Lasso peptides (possibly functioning as a resistance gene) (Mathavan et al. 2014), but FecCD was not. Twenty-one percent of NRPS clusters contained a FecCD gene, and 28% contained a TonB-dependent receptor, possibly indicating that at least one in five uncharacterized NRPSs may function as siderophores. Thus, even in an uncurated data set of thousands of BGCs, it appears as though siderophore-specific transporters only rarely occur in biosynthetic gene clusters that are unlikely to have siderophore functions.
Classifying antibiotics and antifungals from either transporters or biosynthetic genes proved more challenging than classifying siderophores (Supplemental Table S5; Supplemental Fig. S5). In Gram-negative bacteria, we observed positive associations between the MacB-FtsX tripartite efflux pump with antibacterial or antifungal activity (Fisher's exact test; Q < 0.05). MacB and associated components (FtsX and OEP) were positively associated with antibiotic activity by LASSO logistic regression. This result was stable across both cross-validation folds and repeats of cross-validation (Supplemental Fig. S5). We found that seven of 27 Gram-negative antibacterial BGCs contained a MacB, and no BGCs in our classes of other activities contained MacB. Although MacB is involved in export of the siderophore Pyoverdine (for which there is not an accurate BGC in MIBiG) in Pseudomonas (Greene et al. 2018a), in general, MacB may be a strong indicator of antibacterial activity for a BGC. Previously, we identified a number of MacB-FtsX exporters in BGCs from novel Acidobacteria (Crits-Christoph et al. 2018), possibly indicating a role in antibacterial activity for these BGCs. LASSO effect sizes for individual biosynthetic genes were substantially lower (Supplemental Fig. S5).
Association of biosynthetic transporter classes with the molecular weights and lipophilicity of their putative substrates
We next hypothesized that transporter classes could be predictive of other molecular features beyond functional activity. There was no strong correlation between the molecular size of the metabolite produced and Gram status of the bacteria encoding each corresponding BGC in the MIBiG data set. Thus, we tested for differences in transporter classes in BGCs producing metabolites that were (1) less than and (2) greater than 1000 Da in size across all bacteria. There was a significant difference in the frequencies of some transporters between BGCs with different metabolite molecular weights (Fig. 5A). The strongest difference was in the distribution of MFS transporters—found in 57% of BGCs with products <1000 Da, but only 14% of those >1000 Da (Fisher's exact test; Q < 0.05). The 95th percentile of metabolite molecular weights for clusters with MFS_1 and without ABC_tran was 1082 Da, which may be approaching a biological limit for the molecular weights of substrates for these transporters. Conversely, the ATP-dependent ABC_tran domain was found in 89% of BGCs producing high molecular weight compounds but only 42% of those producing low molecular weight compounds (Fisher's exact test; Q < 0.05). MacB/FtsX and the rarer transmembrane domains were also associated with higher molecular weight compounds.
Transporter domains associated with molecular size and partition coefficient. (A) The frequencies of common transporter Pfam domains in BGCs that synthesize metabolites >1000 Da (left) and <1000 Da (right). Bars in green were significantly different in frequency between the two classes (Fisher's exact test; Q < 0.05). (B) Precision-recall curves for two-layer decision trees and LASSO logistic regression models classifying BGCs producing metabolites >1000 Da using Pfam transporter, CATH transporter, and Pfam biosynthetic gene features. (C) The distribution of metabolite molecular weights synthesized by BGCs with at least one NBD-binding ABC transporter domain, at least one MFS domain, and the ABC2_membrane_3 transmembrane domain. (D) Predicted partition coefficients (log P) for metabolites synthesized by BGCs that contain at least one variant of two different ABC transporter transmembrane domains.
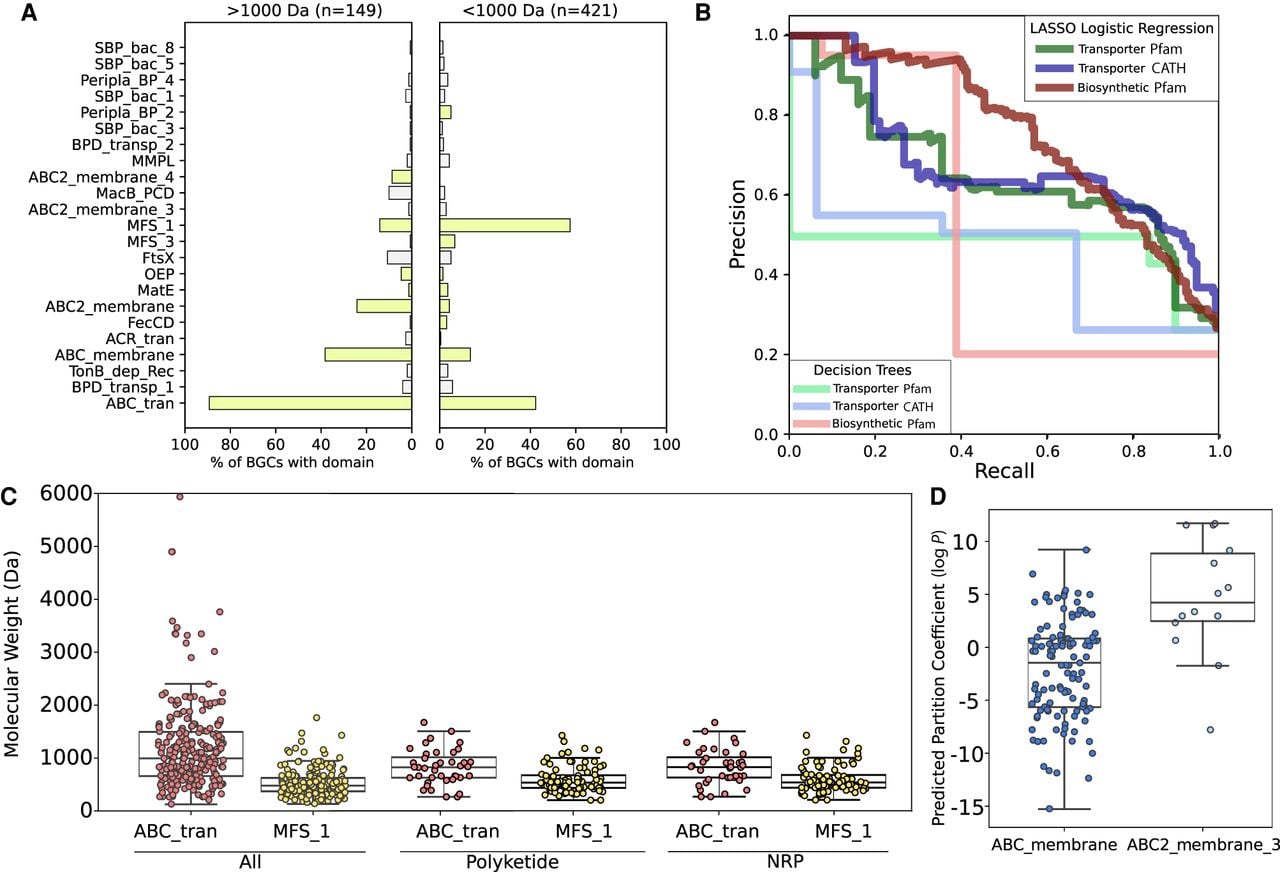
We tested for an association within the two largest chemical classes in the data set, PKs and NRPs, and found that also within the PKS and NRPS biosynthetic classes ATP-dependent transporters were associated with larger metabolite molecular weights than the MFS family (Fig. 5C). We also observed that the ABC2_membrane_4 was associated almost exclusively with large RiPPs, with almost all of the BGCs in which it is found in producing compounds >1500 Da in size. After training both LASSO logistic regression and two-layer decision tree models to classify whether produced molecules are >1000 Da, we found that transporter genes were able to distinguish large from small metabolites with moderate precision and recall (Supplemental Table S5) and an AUPRC of up to 42%, and biosynthetic genes performed similarly (Fig. 5B). Top transporter features consistently had larger effect sizes than top biosynthetic features, indicating that transporter-based features provided clearer signals. This result was stable across both cross-validation folds and repeats (Supplemental Fig. S6). The biosynthetic protein family most associated with high molecular weight metabolites was Glycos_transf_2, likely owing to the addition of sugar groups to metabolites by these enzymes in BGCs.
It has previously been reported that transporters can be specific for compounds with similar hydrophilicity (Rempel et al. 2020). The lipophilicity of a metabolite is often considered critical for its success in clinical development for human therapeutics (Arnott and Planey 2012). With LASSO logistic regression, we predicted partition coefficients (log P), a measure of lipophilicity, for all of the metabolites in MIBiG, and tested how well metabolite partition coefficients could be predicted by gene content. We found that the presence of five transporter classes was significantly associated with increased lipophilicity (Fisher's exact test; Q < 0.05). In particular, we observed an association between varying ATP-dependent transmembrane domains and log P, with ABC2_ membrane_3 domain co-occurring with BGC-metabolites with a high log P (median 4.4) (Fig. 5D) and ABC2_membrane_4 domain co-occurring with BGC-metabolites with a low log P (median −6.2). Although the ABC2_membrane_4 association is likely a result of its exclusive association with large RiPP products, the ABC2_membrane_3 domain occurred in multiple BGC classes, mostly polyketides, and was still associated with a decrease in log P just within the polyketide class. LASSO logistic regression distinguished log P > 0 from log P < 0 with 77% AUPRC (Supplemental Table S5; Supplemental Fig. S7). On this task biosynthetic genes were distinctly superior over transporters at prediction of log P, obtaining a 83% AUPRC with a LASSO logistic regression trained on biosynthetic genes (Supplemental Table S5).
Identifying novel siderophore-like biosynthetic gene clusters in the human microbiome
To show the predictive utility of BGC-associated transporters, we mined BGCs with siderophore-specific transporters in metagenomic genomes (dereplicated per species) from (1) the gut microbiomes of neonatal infants in the intensive care unit (Olm et al. 2019), and (2) a cross-study collation of genomes assembled from multiple human gut studies (Nayfach et al. 2019). Identified “siderophore-like” BGCs putatively produce siderophores, as they contained the transporter classes that can achieve near 100% siderophore specificity in the MIBiG database: (1) Peripla_BP_2 and FecCD in Gram-positive bacteria, and (2) Peripla_BP_2, FecCD, and TonB_dep_Rec in Gram-negative bacteria. We identified 1442 BGCs with siderophore-like transporter classes (Fig. 6A; Supplemental Table S6) and then grouped them into novel gene cluster families using BiG-SCAPE, resulting in 75 siderophore-like gene cluster families (Fig. 6B).
BGCs with siderophore-like transporters from human gut microbiomes. (A) Concatenated ribosomal protein tree (collapsed to the genus level) for high-quality genomes from the infant and adult gut microbiomes that encode siderophore-like BGCs. On the right are counts of siderophore BGCs from infant gut genomes (blue) and adult gut genomes (red). (B) Gene Cluster Families of BGCs containing known siderophores (bright red) and human microbiome–derived BGCs with siderophore-like transporters. BGCs are connected by similarity to other BGCs in the same gene cluster family, calculated using BiG-SCAPE. (C) Families of siderophore-like BGCs without any similarity to existing known BGCs. BGCs in the network are colored by the taxonomy of the genome of origin and are grouped and labeled by the antiSMASH reported biosynthetic class: for NRPS gene clusters, the adenylation domain specificities are reported.
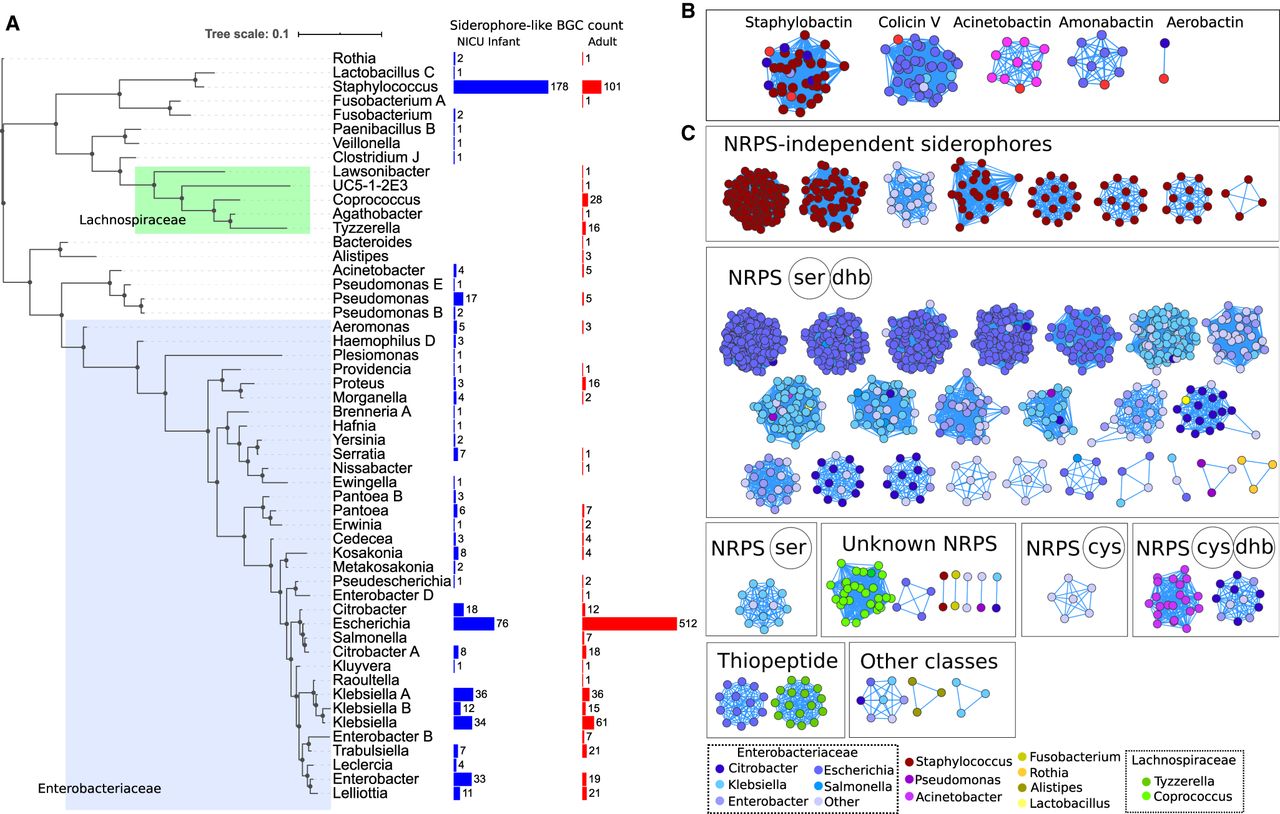
Most siderophore-like BGCs were in large gene cluster families with other BGCs that also contained the same set of transporter hits. Twenty-three percent of microbial genomes from the neonatal infant gut microbiomes had siderophore-like BGCs, but only 3% of those assembled from adult gut microbiomes did. Siderophore-like BGCs were identified across a range of bacterial genera, but the majority were from the Staphylococcus or Enterobacteriaceae, which are known to be in high abundance in the neonatal gut microbiome. The genera with the most siderophore-like BGCs were Staphylococcus and Klebsiella, common hospital-acquired pathogens of neonates. Five of the 75 identified siderophore-like BGC families included a known representative gene cluster in MIBiG, and four of the known representatives were siderophores, again pointing to the specificity of these transporter classes.
The rest of the siderophore-like gene cluster families that were identified had no closely characterized representative in MIBiG, indicating that there is likely capacity for production of multiple novel siderophores in the human gut microbiome (Fig. 6C). Almost all siderophore-like BGCs were either NRPS or NRPS-independent siderophore classes, the latter of which is based on the presence of the IuA/IuC gene family, as in aerobactin biosynthesis. Of the NRPS siderophore-like gene clusters, the majority had adenylation domain specificities for serine (ser) and 2,3-dihydroxybenzoate (dhb), indicating similar catechol-containing nonribosomal biosynthetic pathways to siderophores like enterobactin and salmochelin (Supplemental Table S7). We observed substantial genetic diversity between gene cluster families containing similar NRPS domains, which may indicate the existence of possible unknown derivatives of these siderophores in the human microbiome. In adult gut microbiome samples, one large novel NRPS siderophore-like gene cluster with unknown adenylation specificity was identified in Coprococcus, members of the Lachnospiraceae, often considered to be important commensals in the human gut (Chen et al. 2017; Duvallet et al. 2017). Thus, although a majority of siderophore-like BGCs in the human microbiome contained core enzymes similar to known siderophore biosynthetic pathways, there was substantial genetic diversity that could indicate further unexplored structural variation.
Discussion
We uncovered several strong associations between transporters within characterized BGCs and features of the corresponding BGC-synthesized metabolites. With regard to prediction of metabolite activity, we quantified the specificity of TonB-dependent receptors, FecCD, and Periplasmic binding protein 2 for siderophore-producing BGCs. This complements existing literature indicating that genes in these families are specific for siderophore import in both Gram-positive and Gram-negative bacteria (Chu et al. 2010). We also identified a putative association between the MacB tripartite efflux pump and antibacterial/antifungal activity. Based on these findings, a strategy of targeting novel BGCs containing MacB for characterization may be useful for antibiotic prospecting. In addition to activity prediction, we used metabolite structural information in MIBiG to predict metabolite molecular weight and lipophilicity from BGC gene content. We discovered a strong relationship between the transporters in characterized BGCs and the molecular weight of their synthesized metabolites. The strong dichotomy between ATP-dependent transporters (using the ABC_tran nucleotide binding domain) and MFS family transporters points toward required ATP-dependence for transporting metabolites >1000 Da. We also identified relationships between two understudied membrane components (ABC2_membrane_3 and ABC2_membrane_4) and substrate log P, possibly indicating trade-off in membrane domains for molecules of different chemical properties. Future phylogeny-based subdivision of these families may improve on general protein family annotations to increase the predictive power of transporter substrate characteristics.
There are multiple caveats to our work. Molecular activity of specialized metabolites based on functions proven in the laboratory may be very different from the ecological roles that metabolites play in natural settings (Behnsen and Raffatellu 2016; van der Meij et al. 2017; Kramer et al. 2020). Further, many BGCs may produce multiple variants of a metabolite (Fischbach and Clardy 2007), only some of which may be reported. There may also be reference-database biases in our gene searches—although they are sensitive, it is possible that phylogenetically divergent microbes use transporter genes that are not hit by our sequence models. Finally, as reported, a significant proportion of BGCs contain no transporter at all or a transporter gene genomically adjacent to a BGC may not be functionally linked. There are both technical and biological reasons why a BGC might not contain a transporter gene. First, the transporter(s) for the metabolite produced may be encoded elsewhere in the genome. Second, the BGC's genomic boundaries may be misannotated, and the transporter may be downstream from annotated genes. Third, it is possible that the metabolite being produced performs its primary function intracellularly and does not require a transporter for export. It is also possible that there are unannotated transport systems in BGCs: To further investigate this, we identified unannotated proteins with transmembrane domains in BGCs and found that 18% of BGCs in MIBiG without a transporter contained one unknown membrane protein. Despite these caveats, it appears as though transporter genes provide simple and strong signals for inferring both activity and chemical properties of metabolites produced by BGCs.
Siderophores are both considered critical pathogenicity factors for many human-associated microbes (Weakland et al. 2020) and are also known to facilitate interactions with other microbes and the innate immune system in the human gut microbiome (Behnsen and Raffatellu 2016; Holden et al. 2016; Lam et al. 2018; Zhu et al. 2020). Therefore, being able to annotate genes for the production of siderophores across diverse bacterial species may be critical for understanding the distribution of virulence factors, yet it is difficult to do using traditional annotation pipelines alone. We observed a high prevalence of siderophore-like BGCs in bacterial genomes from NICU premature infant guts, suggesting that the premature infant gut could be more prone to invasion by pathogens with siderophore virulence factors. Potentially novel siderophore-like BGCs were most consistently found to be encoded in the genomes of members of the Enterobacteriaceae and Staphylococcus in the premature infant microbiome. Only in the adult microbiome data sets did we identify siderophore-like BGCs in the Lachnospiraceae, that are often considered important commensals, indicating that there may also be commensal siderophore production in adult gut microbiomes. Importantly, we identified siderophore-like BGCs in these taxa that are not homologous to known siderophore clusters, indicating that there is still substantial unknown chemical diversity of siderophores, even within well-studied lineages.
In general, here we showed that consideration of transporter genes can aid holistic functional prediction of BGC products. A transporter-guided approach could be especially useful for identification of siderophore targets for medical (Nagoba and Vedpathak 2011) and biotechnological applications (Ahmed and Holmström 2014). Given the large diversity of BGCs and that chemical characterization of their products can be time and resource intensive, better functional prediction of BGCs for targeted study can improve selection of targets for antimicrobial discovery and downstream activity tests.
Methods
Curation and selection of BGCs and transporter annotations
We parsed the MIBiG 2.0 database of biosynthetic gene clusters metadata and extracted information including host genus, compound count, chemical structures of the metabolite product, known metabolite activities, and the number of open reading frames for each BGC. Using Entrez and NCBI, we assigned the expected Gram status for each BGC based on phylum, coded 0 = Gram-negative, 1 = Gram-positive, 2 = Fungal, 3 = other. For the purpose of this manuscript, we only analyzed BGCs from Gram-positive and Gram-negative bacteria. We noticed that the activity labels in MIBiG 2.0 were often incomplete, and manually added a set of antibacterial, siderophore, and antifungal labels derived from the literature (Supplemental Table S1). We found that 28 BGCs (1.8%) in MIBiG were unusually large in length, and comparisons to published papers on these BGCs showed that their MIBiG counterparts were overextended in comparison to the validated BGC. For this reason, we eliminated the 28 BGCs over 60 ORFs in length. Using Python and RDKit, we calculated molecular weights and partition coefficients (log P) using the algorithm described in Wildman and Crippen (1999) for all 1042 MIBiG BGCs with a single associated compound structure. Two hundred thirty-eight BGCs have more than one associated metabolite structure, and these multistructure BGCs were not used in our structural association analyses. We annotated biosynthetic genes in MIBiG with HMMER hmmsearch (Eddy 1998) and cath-resolve-hits (Lewis et al. 2019) on a set of the 99 most commonly represented biosynthetic Pfams in antiSMASH BGCs obtained from Cimermancic et al. (2014) to generate a counts table of biosynthetic protein families for each BGC (Supplemental Table S4).
To obtain a comprehensive overview of the distribution of transport-associated protein domains in biosynthetic gene clusters, we generated two separate feature tables: (1) using CATH (Sillitoe et al. 2019) HMMs, and (2) using Pfam (El-Gebali et al. 2019) HMMs. For the first, we downloaded all proteins in the Transporter Classification Database (TCDB) (Saier et al. 2016) and annotated them with hmmsearch and cath-resolve-hits, using all CATH Functional Family HMMs. We then selected all CATH HMMs that were represented at least five times. We then manually curated this list down to 180 final CATH HMMs that were transport specific. We then calculated the specificities of each CATH HMM for TCDB families; 80 were specific for exactly one TCDB family.
For the second set of features, we took all protein sequences with an annotation including “Transport” in the antiSMASH database v2 (Blin et al. 2019a) and annotated these proteins with the Pfam-A set of HMMs using hmmsearch and the option‐‐cut_ga. We then selected highly represented HMMs and manually curated this list to be transporter specific and representative of the major transporter classes in TCDB. We then also selected the Pfam Substrate Binding Protein and Periplasmic Binding Protein HMMs that were represented more than five times in MIBiG. When comparing both the Pfam and CATH set of HMMs, we found substantial overlap, but the CATH set is composed of 166 domain features, whereas the Pfam set only contains 18 (Supplemental Tables S2, S3).
Machine learning to predict metabolite structural and functional characteristics
To identify associations between metabolite functional classes and structural properties with BGC gene content, we used traditional statistical tests and different machine learning models. Our classification tasks were (1) siderophore (n = 16 Gram-positive; n = 24 Gram-negative) versus other activity (n = 142 Gram-positive; n = 42 Gram-negative), (2) antibiotic and antifungal (n = 131 Gram-positive; n = 27 Gram-negative) versus other activity (n = 57 Gram-positive; n = 37 Gram-negative), (3) metabolite molecular weight > 1000 Daltons (n = 149) versus metabolite molecular weight < 1000 Daltons (n = 421), and (4) predicted partition coefficient log P < 0 (n = 220) versus log P ≥ 0 (n = 350). For the functional classification tasks, we noticed a strong class imbalance with Gram status, so we performed functional classification separately for BGCs from Gram-positive and Gram-negative bacteria. We first tested for univariate differences in proportional representation of each transporter BGC between classes for classification tests using Fisher's exact test in Python (Q < 0.05, Benjamini–Hochberg correction) (Hochberg and Benjamini 1990).
We then assessed the predictive power of the three sets of features for each BGC: transporter Pfam HMMs, transporter CATH HMMs, and biosynthetic Pfam HMMs. Features were counts of protein families, which were standardized using the StandardScaler function in the scikit-learn package. Given the nature of our study, we used simple models to ensure reliability and interpretability of our results. We fit two classes of machine learning models: (1) LASSO-penalized logistic regression, which fits a linear model with a sparsity penalty on weights; and (2) shallow decision trees (of depth one or two), which can classify based on splitting at most two features (Franklin 2005). All models were trained using the Python package scikit-learn. Because of data size and class imbalance, we fit models using repeated, stratified k-fold cross-validation (“RepeatedStratifiedKFold” in scikit-learn) with five repeats and five folds. On each cross-validation split of our data, we computed the area under the precision-recall curve (AUPRC) to evaluate performance for our class-imbalanced tasks. Thus for the final output of this procedure, we reported the mean of accuracy, precision, recall, and AUPRC each generated from five repeats of different random fivefold partitions of the data. We further use these repeated cross-validation splits to see which features are consistently used for classification across repeats.
Annotating metagenomic siderophore-like BGCs from the human microbiome
To assess the distribution of siderophore-like BGCs in the human microbiome, we downloaded two sets of genomes assembled from metagenomes obtained from the human gut microbiome: 2425 genomes from a neonatal intensive care unit (NICU) premature infant microbiome (Olm et al. 2019) and 24,345 genomes from a diverse set of mostly adult human cohorts (Nayfach et al. 2019). We ran antiSMASH 5.0 on these genomes and then scanned predicted BGCs for at least two of the Pfams that were found to be specific for siderophore BGCs (FecCD, Peripla_BP_2, and Ton_dep_Rec). We dereplicated these BGCs and compared them to known MIBiG siderophore BGCs using the software BiG-SCAPE (Navarro-Muñoz et al. 2020) run with default settings. We then considered BGCs with either set of hits and reported their genomic taxonomic distribution based on the closest BLAST hit representatives of genomic ribosomal proteins to taxonomic genera defined by GTDB (minimum percent identity of hits >80%) (Parks et al. 2018).
Data access
All Python code used in this paper, along with the data analyzed, antiSMASH BGCs, and additional data tables, is available at GitHub (https://github.com/nickbhat/bgc_tran) and as Supplemental Code and Data.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This research was supported, in part, by the National Institutes of Health under awards RAI092531A, R01-GM109454, and R35-GM134922. J.F.B. and Y.S.S. are Chan Zuckerberg Biohub Investigators.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.268169.120.
[3] Freely available online through the Genome Research Open Access option.
References
- ↵Ahmed E, Holmström SJM. 2014. Siderophores in environmental research: roles and applications. Microb Biotechnol 7: 196–208. 10.1111/1751-7915.12117
- ↵Arnott JA, Planey SL. 2012. The influence of lipophilicity in drug discovery and design. Expert Opin Drug Discov 7: 863–875. 10.1517/17460441.2012.714363
- ↵Behnsen J, Raffatellu M. 2016. Siderophores: more than stealing iron. mBio 7: e01906-16. 10.1128/mBio.01906-16
- ↵Berntsson RPA, Smits SHJ, Schmitt L, Slotboom DJ, Poolman B. 2010. A structural classification of substrate-binding proteins. FEBS Lett 584: 2606–2617. 10.1016/j.febslet.2010.04.043
- ↵Bi Y, Mann E, Whitfield C, Zimmer J. 2018. Architecture of a channel-forming O-antigen polysaccharide ABC transporter. Nature 553: 361–365. 10.1038/nature25190
- ↵Blin K, Pascal Andreu V, de Los Santos ELC, Del Carratore F, Lee SY, Medema MH, Weber T. 2019a. The antiSMASH database version 2: a comprehensive resource on secondary metabolite biosynthetic gene clusters. Nucleic Acids Res 47: D625–D630. 10.1093/nar/gky1060
- ↵Blin K, Shaw S, Steinke K, Villebro R, Ziemert N, Lee SY, Medema MH, Weber T. 2019b. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res 47: W81–W87. 10.1093/nar/gkz310
- ↵Carran CJ, Jordan M, Drechsel H, Schmid DG, Winkelmann G. 2001. Heterobactins: a new class of siderophores from Rhodococcus erythropolis IGTS8 containing both hydroxamate and catecholate donor groups. Biometals 14: 119–125. 10.1023/A:1016633529461
- ↵Chen Y, Unger M, Ntai I, McClure RA, Albright JC, Thomson RJ, Kelleher NL. 2013. Gobichelin A and B: mixed-ligand siderophores discovered using proteomics. Medchemcomm 4: 233–238. 10.1039/c2md20232h
- ↵Chen L, Wilson JE, Koenigsknecht MJ, Chou W-C, Montgomery SA, Truax AD, June Brickey W, Packey CD, Maharshak N, Matsushima GK, 2017. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat Immunol 18: 541–551. 10.1038/ni.3690
- ↵Chevrette MG, Gutiérrez-García K, Selem-Mojica N, Aguilar-Martínez C, Yañez-Olvera A, Ramos-Aboites HE, Hoskisson PA, Barona-Gómez F. 2020. Evolutionary dynamics of natural product biosynthesis in bacteria. Nat Prod Rep 37: 566–599. 10.1039/c9np00048h
- ↵Chu BC, Garcia-Herrero A, Johanson TH, Krewulak KD, Lau CK, Sean Peacock R, Slavinskaya Z, Vogel HJ. 2010. Siderophore uptake in bacteria and the battle for iron with the host; a bird's eye view. Biometals 23: 601–611. 10.1007/s10534-010-9361-x
- ↵Cimermancic P, Medema MH, Claesen J, Kurita K, Wieland Brown LC, Mavrommatis K, Pati A, Godfrey PA, Koehrsen M, Clardy J 2014. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 158: 412–421. 10.1016/j.cell.2014.06.034
- ↵Crits-Christoph A, Diamond S, Butterfield CN, Thomas BC, Banfield JF. 2018. Novel soil bacteria possess diverse genes for secondary metabolite biosynthesis. Nature 558: 440–444. 10.1038/s41586-018-0207-y
- ↵Davies J. 2013. Specialized microbial metabolites: functions and origins. J Antibiot 66: 361–364. 10.1038/ja.2013.61
- ↵Dawson RJP, Locher KP. 2007. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett 581: 935–938. 10.1016/j.febslet.2007.01.073
- ↵Duvallet C, Gibbons SM, Gurry T, Irizarry RA, Alm EJ. 2017. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat Commun 8: 1784. 10.1038/s41467-017-01973-8
- ↵Eddy SR. 1998. Profile hidden Markov models. Bioinformatics 14: 755–763. 10.1093/bioinformatics/14.9.755
- ↵El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, 2019. The Pfam protein families database in 2019. Nucleic Acids Res 47: D427–D432. 10.1093/nar/gky995
- ↵Ellermann M, Arthur JC. 2017. Siderophore-mediated iron acquisition and modulation of host-bacterial interactions. Free Radic Biol Med 105: 68–78. 10.1016/j.freeradbiomed.2016.10.489
- ↵Fischbach MA, Clardy J. 2007. One pathway, many products. Nat Chem Biol 3: 353–355. 10.1038/nchembio0707-353
- ↵Fischbach MA, Walsh CT. 2006. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem Rev 106: 3468–3496. 10.1021/cr0503097
- ↵Fischbach MA, Walsh CT, Clardy J. 2008. The evolution of gene collectives: how natural selection drives chemical innovation. Proc Natl Acad Sci 105: 4601–4608. 10.1073/pnas.0709132105
- ↵Franklin J. 2005. The elements of statistical learning: data mining, inference and prediction. Math Intelligencer 27: 83–85. 10.1007/bf02985802
- ↵Gebhard S. 2012. ABC transporters of antimicrobial peptides in firmicutes bacteria—phylogeny, function and regulation. Mol Microbiol 86: 1295–1317. 10.1111/mmi.12078
- ↵Greene NP, Kaplan E, Crow A, Koronakis V. 2018a. Antibiotic resistance mediated by the macB ABC transporter family: a structural and functional perspective. Front Microbiol 9: 950. 10.3389/fmicb.2018.00950
- ↵Greene NP, Kaplan E, Crow A, Koronakis V. 2018b. Corrigendum: Antibiotic resistance mediated by the macB ABC transporter family: a structural and functional perspective. Front Microbiol 9: 2318. 10.3389/fmicb.2018.02318
- ↵Guo X, Geng P, Bai F, Bai G, Sun T, Li X, Shi L, Zhong Q. 2012. Draft genome sequence of Streptomyces coelicoflavus ZG0656 reveals the putative biosynthetic gene cluster of acarviostatin family α-amylase inhibitors. Lett Appl Microbiol 55: 162–169. 10.1111/j.1472-765X.2012.03274.x
- ↵Hannigan GD, Prihoda D, Palicka A, Soukup J, Klempir O, Rampula L, Durcak J, Wurst M, Kotowski J, Chang D, 2019. A deep learning genome-mining strategy for biosynthetic gene cluster prediction. Nucleic Acids Res 47: e110. 10.1093/nar/gkz654
- ↵Hider RC, Kong X. 2010. Chemistry and biology of siderophores. Nat Prod Rep 27: 637. 10.1039/b906679a
- ↵Hochberg Y, Benjamini Y. 1990. More powerful procedures for multiple significance testing. Stat Med 9: 811–818. 10.1002/sim.4780090710
- ↵Holden VI, Breen P, Houle S, Dozois CM, Bachman MA. 2016. Klebsiella pneumoniae siderophores induce inflammation, bacterial dissemination, and HIF-1α stabilization during pneumonia. mBio 7: e01397-16. 10.1128/mBio.01397-16
- ↵Jenke-Kodama H, Börner T, Dittmann E. 2006. Natural biocombinatorics in the polyketide synthase genes of the actinobacterium Streptomyces avermitilis. PLoS Comput Biol 2: e132. 10.1371/journal.pcbi.0020132
- ↵Kautsar SA, Blin K, Shaw S, Navarro-Muñoz JC, Terlouw BR, van der Hooft JJJ, van Santen JA, Tracanna V, Suarez Duran H, Pascal Andreu V, 2020. MIBiG 2.0: a repository for biosynthetic gene clusters of known function. Nucleic Acids Res 48: D454–D458. 10.1093/nar/gkz882
- ↵Kim HU, Blin K, Lee SY, Weber T. 2017. Recent development of computational resources for new antibiotics discovery. Curr Opin Microbiol 39: 113–120. 10.1016/j.mib.2017.10.027
- ↵Kramer J, Özkaya Ö, Kümmerli R. 2020. Bacterial siderophores in community and host interactions. Nat Rev Microbiol 18: 152–163. 10.1038/s41579-019-0284-4
- ↵Lam MMC, Wyres KL, Judd LM, Wick RR, Jenney A, Brisse S, Holt KE. 2018. Tracking key virulence loci encoding aerobactin and salmochelin siderophore synthesis in Klebsiella pneumoniae. Genome Med 10: 77. 10.1186/s13073-018-0587-5
- ↵Lewis TE, Sillitoe I, Lees JG. 2019. cath-resolve-hits: a new tool that resolves domain matches suspiciously quickly. Bioinformatics 35: 1766–1767. 10.1093/bioinformatics/bty863
- ↵Liu Y, Li M, Mu H, Song S, Zhang Y, Chen K, He X, Wang H, Dai Y, Lu F, 2017. Identification and characterization of the ficellomycin biosynthesis gene cluster from Streptomyces ficellus. Appl Microbiol Biotechnol 101: 7589–7602. 10.1007/s00253-017-8465-4
- ↵Martín JF, Casqueiro J, Liras P. 2005. Secretion systems for secondary metabolites: how producer cells send out messages of intercellular communication. Curr Opin Microbiol 8: 282–293. 10.1016/j.mib.2005.04.009
- ↵Mathavan I, Zirah S, Mehmood S, Choudhury HG, Goulard C, Li Y, Robinson CV, Rebuffat S, Beis K. 2014. Structural basis for hijacking siderophore receptors by antimicrobial lasso peptides. Nat Chem Biol 10: 340–342. 10.1038/nchembio.1499
- ↵Matsuo Y, Kanoh K, Jang J-H, Adachi K, Matsuda S, Miki O, Kato T, Shizuri Y. 2011. Streptobactin, a tricatechol-type siderophore from marine-derived Streptomyces sp. YM5-799. J Nat Prod 74: 2371–2376. 10.1021/np200290j
- ↵Medema MH, Fischbach MA. 2015. Computational approaches to natural product discovery. Nat Chem Biol 11: 639–648 10.1038/nchembio.1884
- ↵Méndez C, Salas JA. 2001. The role of ABC transporters in antibiotic-producing organisms: drug secretion and resistance mechanisms. Res Microbiol 152: 341–350. 10.1016/S0923-2508(01)01205-0
- ↵Mungan MD, Alanjary M, Blin K, Weber T, Medema MH, Ziemert N. 2020. ARTS 2.0: feature updates and expansion of the antibiotic resistant target seeker for comparative genome mining. Nucleic Acids Res 48: W546–W552. 10.1093/nar/gkaa374
- ↵Nagoba B, Vedpathak D. 2011. Medical applications of siderophores. Electron J Gen Med 8: 229–235. 10.29333/ejgm/82743
- ↵Navarro-Muñoz JC, Selem-Mojica N, Mullowney MW, Kautsar SA, Tryon JH, Parkinson EI, De Los Santos ELC, Yeong M, Cruz-Morales P, Abubucker S, 2020. A computational framework to explore large-scale biosynthetic diversity. Nat Chem Biol 16: 60–68. 10.1038/s41589-019-0400-9
- ↵Nayfach S, Shi ZJ, Seshadri R, Pollard KS, Kyrpides NC. 2019. New insights from uncultivated genomes of the global human gut microbiome. Nature 568: 505–510. 10.1038/s41586-019-1058-x
- ↵Olm MR, Bhattacharya N, Crits-Christoph A, Firek BA, Baker R, Song YS, Morowitz MJ, Banfield JF. 2019. Necrotizing enterocolitis is preceded by increased gut bacterial replication, Klebsiella, and fimbriae-encoding bacteria. Sci Adv 5: eaax5727. 10.1126/sciadv.aax5727
- ↵Osbourn A. 2010. Secondary metabolic gene clusters: evolutionary toolkits for chemical innovation. Trends Genet 26: 449–457. 10.1016/j.tig.2010.07.001
- ↵Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil P-A, Hugenholtz P. 2018. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol 36: 996–1004. 10.1038/nbt.4229
- ↵Quistgaard EM, Löw C, Guettou F, Nordlund P. 2016. Understanding transport by the major facilitator superfamily (MFS): structures pave the way. Nat Rev Mol Cell Biol 17: 123–132. 10.1038/nrm.2015.25
- ↵Rascher A, Hu Z, Buchanan GO, Reid R, Richard Hutchinson C. 2005. Insights into the biosynthesis of the benzoquinone ansamycins geldanamycin and herbimycin, obtained by gene sequencing and disruption. Appl Environ Microbiol 71: 4862–4871. 10.1128/AEM.71.8.4862-4871.2005
- ↵Rees DC, Johnson E, Lewinson O. 2009. ABC transporters: the power to change. Nat Rev Mol Cell Biol 10: 218–227. 10.1038/nrm2646
- ↵Rempel S, Gati C, Nijland M, Thangaratnarajah C, Karyolaimos A, de Gier JW, Guskov A, Slotboom DJ. 2020. A mycobacterial ABC transporter mediates the uptake of hydrophilic compounds. Nature 580: 409–412. 10.1038/s41586-020-2072-8
- ↵Romano M, Fusco G, Choudhury HG, Mehmood S, Robinson CV, Zirah S, Hegemann JD, Lescop E, Marahiel MA, Rebuffat S, 2018. Structural basis for natural product selection and export by bacterial ABC transporters. ACS Chem Biol 13: 1598–1609. 10.1021/acschembio.8b00226
- ↵Saier MH, Reddy VS, Tsu BV, Ahmed MS, Li C, Moreno-Hagelsieb G. 2016. The transporter classification database (TCDB): recent advances. Nucleic Acids Res 44: D372–D379. 10.1093/nar/gkv1103
- ↵Severi E, Thomas GH. 2019. Antibiotic export: transporters involved in the final step of natural product production. Microbiology 165: 805–818. 10.1099/mic.0.000794
- ↵Sharon G, Garg N, Debelius J, Knight R, Dorrestein PC, Mazmanian SK. 2014. Specialized metabolites from the microbiome in health and disease. Cell Metab 20: 719–730. 10.1016/j.cmet.2014.10.016
- ↵Sillitoe I, Dawson N, Lewis TE, Das S, Lees JG, Ashford P, Tolulope A, Scholes HM, Senatorov I, Bujan A, 2019. CATH: expanding the horizons of structure-based functional annotations for genome sequences. Nucleic Acids Res 47: D280–D284. 10.1093/nar/gky1097
- ↵Skinnider MA, Merwin NJ, Johnston CW, Magarvey NA. 2017. PRISM 3: expanded prediction of natural product chemical structures from microbial genomes. Nucleic Acids Res 45: W49–W54. 10.1093/nar/gkx320
- ↵Staudenmaier H, Van Hove B, Yaraghi Z, Braun V. 1989. Nucleotide sequences of the fecBCDE genes and locations of the proteins suggest a periplasmic-binding-protein-dependent transport mechanism for iron(III) dicitrate in Escherichia coli. J Bacteriol 171: 2626–2633. 10.1128/JB.171.5.2626-2633.1989
- ↵ter Beek J, Guskov A, Slotboom DJ. 2014. Structural diversity of ABC transporters. J Gen Physiol 143: 419–435. 10.1085/jgp.201411164
- ↵Tran PN, Yen MR, Chiang CY, Lin HC, Chen PY. 2019. Detecting and prioritizing biosynthetic gene clusters for bioactive compounds in bacteria and fungi. Appl Microbiol Biotechnol 103: 3277–3287. 10.1007/s00253-019-09708-z
- ↵Tyc O, Song C, Dickschat JS, Vos M, Garbeva P. 2017. The ecological role of volatile and soluble secondary metabolites produced by soil bacteria. Trends Microbiol 25: 280–292. 10.1016/j.tim.2016.12.002
- ↵van der Meij A, Worsley SF, Hutchings MI, van Wezel GP. 2017. Chemical ecology of antibiotic production by actinomycetes. FEMS Microbiol Rev 41: 392–416. 10.1093/femsre/fux005
- ↵Velamakanni S, Yao Y, Gutmann DAP, van Veen HW. 2008. Multidrug transport by the ABC transporter Sav1866 from Staphylococcus aureus. Biochemistry 47: 9300–9308. 10.1021/bi8006737
- ↵Wang Z, Hu W, Zheng H. 2020. Pathogenic siderophore ABC importer YbtPQ adopts a surprising fold of exporter. Sci Adv 6: eaay7997. 10.1126/sciadv.aay7997
- ↵Weakland DR, Smith SN, Bell B, Tripathi A, Mobley HLT. 2020. The Serratia marcescens siderophore, serratiochelin, is necessary for full virulence during bloodstream infection. Infect Immun 88: e00117-20. 10.1128/iai.00117-20
- ↵Wenzel SC, Müller R. 2005. Formation of novel secondary metabolites by bacterial multimodular assembly lines: deviations from textbook biosynthetic logic. Curr Opin Chem Biol 9: 447–458. 10.1016/j.cbpa.2005.08.001
- ↵Wildman SA, Crippen GM. 1999. Prediction of physicochemical parameters by atomic contributions. J Chem Inf Comput Sci 39: 868–873. 10.1021/ci990307l
- ↵Yan Y, Liu N, Tang Y. 2020. Recent developments in self-resistance gene directed natural product discovery. Nat Prod Rep 37: 879–892. 10.1039/c9np00050j
- ↵Zhu W, Winter MG, Spiga L, Hughes ER, Chanin R, Mulgaonkar A, Pennington J, Maas M, Behrendt CL, Kim J, 2020. Xenosiderophore utilization promotes Bacteroides thetaiotaomicron resilience during colitis. Cell Host Microbe 27: 376–388.e8. 10.1016/j.chom.2020.01.010