
Different trajectories of polyploidization shape the genomic landscape of the Brettanomyces bruxellensis yeast species
- 1Université de Strasbourg, CNRS, GMGM UMR 7156, 67000 Strasbourg, France;
- 2Université de Bordeaux, ISVV, Unité de Recherche Œnologie EA 4577, USC 1366 INRA, Bordeaux INP, F-33140 Villenave d'Ornon, France;
- 3ENSCBP, Bordeaux INP, 33600 Pessac, France;
- 4Institut Universitaire de France (IUF), 75231 Paris, France
Abstract
Polyploidization events are observed across the tree of life and occur in many fungi, plant, and animal species. During evolution, polyploidy is thought to be an important source of speciation and tumorigenesis. However, the origin of polyploid populations is not always clear, and little is known about the precise nature and structure of their complex genome. Using a long-read sequencing strategy, we sequenced 71 strains from the Brettanomyces bruxellensis yeast species, which is found in anthropized environments (e.g., beer, contaminant of wine, kombucha, and ethanol production) and characterized by several polyploid subpopulations. To reconstruct the polyploid genomes, we phased them by using different strategies and found that each subpopulation had a unique polyploidization history with distinct trajectories. The polyploid genomes contain either genetically closely related (with a genetic divergence <1%) or diverged copies (>3%), indicating auto- as well as allopolyploidization events. These latest events have occurred independently with a specific and unique donor in each of the polyploid subpopulations and exclude the known Brettanomyces sister species as possible donors. Finally, loss of heterozygosity events has shaped the structure of these polyploid genomes and underline their dynamics. Overall, our study highlights the multiplicity of the trajectories leading to polyploid genomes within the same species.
Polyploidy, a state in which organisms carry three or more full sets of chromosomes, is a phenomenon that can be observed throughout plant, animal, and fungi species. Polyploidization has gained interest because of its tremendous effects in the evolution of species or its involvement in cancerogenesis (Adams and Wendel 2005; Gregory and Mable 2005; Gjelsvik et al. 2019). The most obvious and probably well-studied polyploidy events in the tree of life are the whole-genome duplication (WGD) events, which are usually followed by subsequent and massive diversification. One example is the series of two ancient WGD events that occurred in the lineage leading to the ancestor of all vertebrates ∼450 million years ago, and have significantly contributed to the successive evolution of 60,000 extant species (Dehal and Boore 2005; Sacerdot et al. 2018).
Although the effects of polyploidization on the organisms are versatile, there are also different mechanisms to become polyploid (Comai 2005; Fox et al. 2020). The doubling of one's own genome or of a hybrid coming from individuals of the same species would lead to multiple genomic copies of identical or similar descent and defines the mechanisms of autopolyploidization. However, interspecific hybridization would cause the acquisition of additional full chromosomal sets harboring higher genetic variation and defines allopolyploidization. Although it is well observed that a polyploid state causes genomic conflicts, leads to genome instability, or reduces gamete formation, on the contrary, genomic reorganization can ultimately promote diversification from the additional genomic information (Mayer and Aguilera 1990; Wood et al. 2009; Van de Peer et al. 2017). Therefore, polyploidy can play a predominant role in bursts of adaptive divergence and speciation (Leitch and Leitch 2008; Soltis et al. 2015). Polyploidy can be beneficial under certain environmental circumstances and increases the potential for adaptability, taking advantage of evolutionary innovations from neo- and subfunctionalization of duplicated genes (Sanchez-Perez et al. 2008; Eberlein et al. 2017). Environmental changes that require a fast adaptation, for example, can trigger the prevalence of polyploids, which at least for short terms, can have an adaptive advantage from genomic flexibility rather than simply being the “dead-end.”
Some taxa are believed to be more stable to polyploid states than others. These are known to be found frequently among plants, which in contrast to animals are characterized by a development that seems to be more robust to genomic perturbations (Orr 1990). Studies suggest that up to 70% of flowering plants originate from polyploid ancestors, putting it as a major contributor in the evolution of species (Masterson 1994; Levin 2002). It is also suspected in animals that polyploidization plays an even more prevalent role than currently shown, but the analytic tools and effort detecting them are limiting factors. Although animals are characterized by less stable polyploids, it is well acknowledged that most of the vertebrate species originate from ancient polyploidization events too (Gregory and Mable 2005; Wertheim et al. 2013). At the same time, polyploidy is also increasingly observed in single-cell organisms such as yeasts (Bellon et al. 2011; Krogerus et al. 2015; Peter et al. 2018), suggesting that this state can be used as a rapid response to ecologically or human-made changes in anthropogenically used environments, coevolution, or enabled invasions by the acquisition or holding of additional full sets of chromosomes (te Beest et al. 2012; Bertier et al. 2013; Morrow and Fraser 2013; Van de Peer et al. 2017). In the lineage leading to Saccharomyces cerevisiae, a hybridization event between two ancestral species has been followed by subsequent WGD, a means by which, in the subsequent process of extensive genome reorganization, high fertility could be retained (Seoighe and Wolfe 1998; Gerstein and Otto 2009; Gordon et al. 2009; Marcet-Houben and Gabaldón 2015). Moreover, the prevalence of polyploidy, currently observed in S. cerevisiae, is ∼11.5%, as shown in a recent study of 1011 whole-genome sequenced isolates (Peter et al. 2018). Polyploids are particularly enriched in subpopulations associated with the production of beer or bread, highlighting that its domestication most likely triggered the appearance of polyploids to fulfill the desired requirements in industrial settings.
With polyploidization being recognized as a ubiquitous mechanism in nature with almost unpredictable consequences in terms of genomic conflicts or adaptability, we are just beginning to fully resolve and understand the genomic architecture of natural polyploid populations, their prevalence, and trajectories especially within the same species. The genomic era has accelerated the research on polyploid and hybrid genomes through the access of long-read sequencing data. However, the biggest challenge is still the correct phasing of haplotypes to separate the different sets of chromosomes without any prior knowledge about ploidy and levels of genetic variation between genomic copies (or sometimes referred to as subgenomes). Here, we focused on the Brettanomyces bruxellensis yeast species, a genetically diverse species with different subpopulations of various levels of ploidy that allowed us to shed light on several questions related to polyploidization. As seen for other yeasts of the Saccharomycotina subphylum, the link between ecological origin and genetic differentiation for the different B. bruxellensis clades is primarily supposed to be driven by its anthropogenic influences (de Barros Pita et al. 2011; Avramova et al. 2019). Multiple genetically distinct subpopulations (clusters) correspond to different ecological niches, respectively, wine, beer, tequila/bioethanol, kombucha, and soft drinks (Avramova et al. 2018).
To study their genomic complexity and to allow a detailed view on their genomic architecture for the first time, we sequenced a subset of 71 B. bruxellensis strains from different subpopulations with long- and short-read sequencing strategies. By using two complex phasing strategies, we studied different trajectories of polyploidization in an ecological diverse setting.
Results
Conserved clusters of polyploid isolates
Brettanomyces bruxellensis is known as a diverse species with genetically and ecologically distinct clusters and various levels of ploidy (Avramova et al. 2018; Colomer et al. 2020; Gounot et al. 2020). To dissect the genomic architecture and to further understand the origin as well as the trajectories of recently described polyploid groups, we selected 71 strains with 51 coming from subpopulations defined as polyploids (Fig. 1A; Supplemental Table S1; Avramova et al. 2018). Most of the strains were isolated in Europe and originate from different ecological origins: beer (n = 25), wine (n = 36), tequila/bioethanol (n = 7), and kombucha (n = 3).
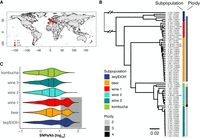
Ploidy and intra-genomic variation. (A) Strain collection. The 71 sequenced strains come from the collection of 1500 isolates (Avramova et al. 2018) and were isolated in different regions worldwide, where they are associated with anthropized environments such as tequila/bioethanol, beer, wine, and kombucha production. (B) Genetic relationship and ploidy level. The sequenced strains, here clustered based on Illumina short-read sequencing data (75PE), separate into six genetically distinct subpopulations—namely, tequila/bioethanol (teq/EtOH), beer, wine (1–3), and kombucha (based on 24,313 genome-wide distributed variants). Forty-eight strains were detected as triploids (69%) coming from five of the six subpopulations: teq/EtOH, beer, wine (1,2), and kombucha (inferred from genome-wide allele frequencies). (C) Genetic diversity within clades inferred from long-read sequencing data. The three subpopulations teq/EtOH (n = 5), beer (n = 22), and wine 1 (n = 7) harbor strains with two clusters of reads bearing low and high genetic variation (underlaid in gray) compared to the reference genome Brettanomyces bruxellensis (Fournier et al. 2017). The subpopulation wine 2 (n = 9), although being polyploid (B), lacks genomic regions with high genetic variation to the reference genome. The three lines within each distribution show the 25%, 50%, and 75% quartiles.
To have a deep insight into the population structure and ploidy levels, we first sequenced the 71 genomes using a whole-genome Illumina short-read sequencing strategy with a 16.9-fold mean coverage. Using this data set, we sampled 24,313 genetic variants evenly distributed across the genome and performed a phylogenetic analysis (Fig. 1B). All the 71 strains were clustered into six well-defined lineages, which correlate with environmental niche, as previously reported (Avramova et al. 2018; Colomer et al. 2020; Gounot et al. 2020). We then assigned ploidy state to each of the strains using SNP frequency distributions within the sequence reads. We categorized their level of ploidy as either being diploid (with SNPs at allele frequencies at 0.5 and 1), triploid (with SNPs at allele frequencies at 0.33 and 0.67), or tetraploid (with SNPs at allele frequencies at 0.25, 0.5, 0.75, and 1) (Supplemental Fig. S1A). We found that the level of ploidy is conserved within but varies across subpopulations (Fig. 1B). The wine 1, wine 2, and beer subpopulations are triploid, whereas the wine 3 and kombucha subpopulations are diploid. The exception is a single kombucha strain being tetraploid (Supplemental Fig. S1A). The teq/EtOH clade harbors one diploid and three triploid strains, whereas the ploidy could not have been assigned to the two other isolates (Supplemental Fig. S1A). For another single strain (I_H06_YJS78889), we could neither identify its ploidy nor assign it to one of the six subpopulations. To exclude the fact that aneuploidies are causing the non-assignment of ploidy levels for the three strains, we looked at read coverage across their genome to identify regions that are absent or present in multiple copies (Supplemental Fig. S1B,C). We showed that the coverage is steady and that these strains do not contain regions with varying coverage, explaining our results.
Overall, we can highlight that the level of ploidy is conserved within genetically diverged subpopulations, but not across them. We showed that the teq/EtOH strains are the most diverse subpopulation and confirmed previous data that additionally suggested this subpopulation as the oldest of the different B. bruxellensis clades (Colomer et al. 2020). The teq/EtOH strains stand in contrast to other subpopulations like the wine 3, which show the lowest degree of genetic variation and therefore might hint toward a single ancestral origin with a recent expansion.
Strategies used to phase the B. bruxellensis polyploid genomes
To resolve the genomic structure of polyploid isolates, we sequenced the genomes of the 71 strains using the Oxford Nanopore sequencing strategy. Long-read sequencing has become the strategy of choice to best resolve structural variation and build high-quality de novo reference assemblies. The difficulty of resolving polyploid genomes, however, lies especially in the attempt to distinguish between the different haplotypes, which are present as independent genomic copies within the same genomes. Although de novo assemblers are not capable of fully differentiating between different haplotypes, seeking instead to provide collapsed haplotypes, several alignment-based algorithms were developed recently to cope with the genomic architecture of polyploid genomes (Schrinner et al. 2020; Shaw and Yu 2020; Abou Saada et al. 2021). They all aim to phase haplotypes into independent entities, but they vary in performance depending on factors such as ploidy, coverage, and the level of genetic divergence between genomic copies of the polyploid genomes.
To properly phase our polyploid genomes, we sought to apply different strategies depending on the level of divergence of the copies constituting these genomes. Reasons to believe that there are different levels of variation have been given by previous studies, which indicated that at least two single polyploid strains from the wine 1 and beer subpopulations have likely experienced polyploidization events by having an additional genomic copy of high genetic variation (Borneman et al. 2014).
To estimate the genetic divergence, we hence aligned the long reads of each strain to the B. bruxellensis reference genome (Supplemental Fig. S2A; Fournier et al. 2017). We identified three subpopulations (teq/EtOH, beer, and wine 1) for which the genetic variation resolves in a bimodal distribution with a low as well as a high genetic variation level cluster of reads (Fig. 1C). These subpopulations stand in contrast to the other three subpopulations (wine 2, wine 3, and kombucha), which solely comprise low genetic divergent reads. Moreover, wine 2 is the only polyploid subpopulation that bears reads with only low genetic diversity.
With the two types of polyploid subpopulations exhibiting either low or high genomic variation, we further applied two different phasing strategies to study their genomic architecture:
-
To resolve the origin of the genetic diversity, and to determine if the cluster with reads of high genetic variation complements an additional genomic copy, we separated the long reads into distinct clusters based on their diversity level. We clustered reads with peaks of low genetic variation at 2 SNPs per kb and high genetic variation showing 24.4 SNPs per kb (Supplemental Fig. S2A,B). Reads between the two distributions (i.e., with a variation between 10 and 14 SNPs per kb) were ignored to avoid the assignment of reads to the wrong cluster (Supplemental Fig. S2B). Based on these sets of reads, we generated de novo assemblies in order to have the phased copies of the polyploid genomes.
-
The low genetic variation observed in the polyploid wine 2 subpopulation did not allow us to separate reads based on their genetic divergence (Fig. 1C). Consequently, we used nPhase, a phasing algorithm that we recently developed (Abou Saada et al. 2021). Briefly, nPhase resolves the genome into distinct haplotypes and provides accurate and contiguous haplotype predictions using short- and long-read sequencing data without any prior information of the true ploidy. It accurately identifies heterozygous positions using highly accurate short reads and clusters long reads into haplotypes based on the presence of similar heterozygous SNP profiles (Abou Saada et al. 2021).
Genomic architecture of the polyploid wine 2 subpopulation
We applied the nPhase phasing algorithm to the sequenced genomes of the wine 2 subpopulation, showing a low intra-genomic variation. We focused on six of the 10 strains for which we had high-quality long- and short-read sequencing data, allowing us to phase their genome properly into independent haplotigs (Fig. 2; Supplemental Fig. S3A).
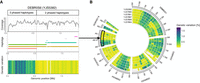
Autopolyploidization for the wine 2 subpopulation. (A) Separation of haplotypes. Phasing the genomes of strains from the polyploid wine 2 subpopulation resolves the generally low intra-genomic variation into haplotigs along the genome. The presence of two haplotypes resolves in lower genetic variation as it does when three haplotigs are present at a given position. Maximal genetic variation between haplotypes increases from 0.93% to 1.79% with the presence of a third phased haplotype. To control that the variations in genetic difference are not artifacts coming from a variable coverage along these regions, the genome-wide coverage was calculated. The coverage is consistent across regions that harbor either two or three phased haplotypes. (B) Conserved patterns of phased haplotypes along the genomes of six strains of the wine 2 subpopulation. Having either two or three phased haplotypes at a site is conserved among different strains from the same subpopulation.
We observed that the chromosomes are phased into regions underlying in most cases two or three haplotigs (Supplemental Fig. S3A). Some regions bear multiple and often small haplotigs and underline the complexity in phasing polyploid genomes with haplotypes that reflect high genetic similarity. In addition, the level of genetic divergence varies along the genomes of the six strains. Whenever nPhase resolves a region into two haplotigs, the genetic variation in these regions is lower compared to regions where it distinguishes between three haplotigs (Fig. 2A). Here, the highest genetic variation in the presence of two haplotigs is 0.93%, whereas on average it is as low as 0.09% (Fig. 2B; Supplemental Fig. S3B). In the presence of a third phased haplotig, the genetic variation can be as high as 1.79% with an average genetic variation of 0.54% (Fig. 2B; Supplemental Fig. S3C). Consistent coverage levels support the hypothesis that the prediction of only two haplotypes is not a result of the absence of a third copy for part of the chromosome (Fig. 2A). Therefore, although the differentiation of three haplotigs underlines the existence of three genetically different genomic copies at that site, the phasing resolving into two haplotigs represents a region with two identical haplotypes plus the existence of genetically different copy.
Further, we can show the presence of conserved regions in all six strains that are characterized by the presence/absence of a third phased haplotype (Fig. 2B; Supplemental Fig. S3B,C). Some regions, for example the first 1 Mb on Chromosome 1, are characterized by two identical copies and a non-identical copy, resolving into two phased haplotypes. This region is followed by another 1-Mb region that resolved into three haplotypes in all six strains. An explanation for the alternation of such regions phased into two or three haplotypes is the occurrence of loss of heterozygosity (LOH) events. LOH events are characterized by the removal of polymorphic markers that distinguish different genomic copies in diploid or polyploid individuals and consequently reduce the genetic variation. nPhase outputs only unique haplotypes present in the data, irrespective if one haplotype contains twice the number of reads as the other, and therefore indicates the occurrence of LOH events in the genome.
Moreover, the existence of the conserved regions of LOH events among the six strains can hint at hotspots for LOH events. Such hotspots have been shown in other species like S. cerevisiae (Peter et al. 2018), in which they frequently cause the removal of genetic variation. Alternatively, this conserved pattern could also hint toward a recent common ancestor. But with these strains being isolated in different countries of two continents (Supplemental Table S1), this explanation is less likely.
Overall, the utilization of long- and short-read sequences in combination with complex phasing strategies enables deciphering of the genomic structure of polyploid genomes of low genetic variation and allowing study of its dynamic. Further, for the wine 2 subpopulation, the only polyploid clade with a low intra-genomic variation, the genomes of six strains revealed conserved regions having undergone LOH events.
Three polyploid clades contain a genetically diverged genomic copy
Next, we focused on the triploid genomes of the teq/EtOH, beer, and wine 1 subpopulations, which show genetically very heterogeneous genomes. To enable comparative analyses, we first separated long reads based on their genetic divergence compared to the reference genome (Supplemental Fig. S2A). We clustered long reads from the bimodal distribution with reads bearing low genetic variation (peak at 2 SNPs per kb) and reads with high genetic variation (peak at 24.4 SNPs per kb) (Supplemental Fig. S2B). As previously mentioned, reads with a variation between 10 and 14 SNPs per kb were ignored to avoid assigning reads to the wrong cluster. The determination of the ratio between the number of reads with a low genetic variation and the total coverage (reads with low and high genetic variation) within 10-kb windows across the genome allowed us to determine the average genomic ploidy level of each strain at a given genomic position (Supplemental Fig. S4A). We identified that the three groups (teq/EtOH, beer, and wine 1) contained two genomic copies with low genetic variation and a single genomic copy that shows a high genetic divergence (or vice versa), which on average complemented to 3n genome-wide (Supplemental Fig. S4B).
The fact that the beer and wine 1 subpopulations contain isolates with higher genetic variation compared to the reference genome was already shown previously for a single strain from each subpopulation (Borneman et al. 2014). The authors claimed the possibility of interspecific hybridization events having taken place. We can, for the first time, highlight that this phenomenon of having a genetically different genomic copy within these subpopulations is frequent and conserved. Additionally, although previous analyses have underpinned the prevalence of polyploid strains in the teq/EtOH subpopulation, we can also show that teq/EtOH strains contain a genomic copy as genetically different to the reference genome of B. bruxellensis as in beer and wine 1 isolates.
To ultimately allow comparative studies between the different genomic copies among these genomes as well as with genomes from the other subpopulations, we first performed de novo genome assemblies using SMARTdenovo (Liu et al. 2021). We generated independent de novo assemblies. First, we used only the long reads that contained low genetic variation for all strains from the three subpopulations (teq/EtOH, beer, and wine 1). We repeated this step independently for the long reads that were exclusively bearing high genetic variation to the reference genome to prepare de novo assemblies (Supplemental Fig. S5; Supplemental Table S2). Then, we created group-specific reference genomes by concatenating the de novo assemblies created from low and high genomic variation, respectively (Methods). This was performed for a representative strain from each group. By performing a competitive mapping approach using these group-specific reference genomes with scaffolds made from low and high genetic variation, we separated the short reads for each strain from the three groups, respectively, into short reads with low or high genetic variation (teq/EtOH, beer, and wine 1) (Supplemental Fig. S5). Then, we aligned the short reads independently back to the B. bruxellensis reference genome. For strains that were either diploid, or polyploid without reads with high genetic variation compared to the reference genome, we aligned short-read sequences directly to the reference genome B. bruxellensis (wine 2, wine 3, and kombucha subpopulations).
First, we determined if there was any bias in mapping rates of the short reads to the reference genome/assemblies between the high diversity strains aligned to the de novo assemblies compared to the low diversity strains aligned to the B. bruxellensis reference genome. Both showed similar alignment rates of 94% and 92.5%, respectively, indicating no bias in the alignment owing to the applied phasing strategy. Then, we determined the genetic diversity of the 71 strains by performing a principal component analysis (Fig. 3A). By looking at the first two principal components explaining 53.7% of the variation from 24,110 sampled genome-wide distributed SNPs, we can show that the genomic copies with high genetic variation (“High”) of 40 strains from the teq/EtOH, beer, and wine 1 subpopulations are clearly distinct from the genomic copies with low genetic variation (“Low”) and cluster group-specific.
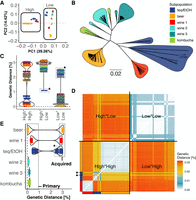
Three independent interspecific hybridization events. (A) Three distinct clusters of genomic copies with high genetic variation. A principal component analysis shows that the genomic copies with high genetic variation to the reference genome of strains in the subpopulations teq/EtOH, beer, and wine 1 are not only different to the genomic copy with low genetic variation but are also genetically distinct between subpopulations (based on 24,110 genome-wide distributed SNPs). (B) Phylogenetic relationship from reads with low genetic variation to the reference genome. The genomic copies with low genetic variation are different between the six subpopulations teq/EtOH, beer, wine 1–3, and kombucha, which group according to their ecological origin (based on 24,110 genome-wide distributed SNPs). (C–E) Pairwise genetic diversity between genomic copies from imputed whole-genome sequences. (C) (Left [High*High]) Pairwise comparison of the genomic copies with high levels of intra-genomic variation between strains of the same subpopulations (single-colored dots) show a genetic diversity of <1% (average of 0.13%). Between strains from different subpopulations (two-colored dots indicate the subpopulation dependency of the compared strains), this diversity varies between 1.76% and 3.05%. (Middle [High*Low]) The genetic distance between the genomic copies with low and high levels of variation, irrespective if it is within the same or between strains, is on average 2.92% (two-colored dots indicate the subpopulation dependency of the compared strains). The strain III_F09_YJS8068 is an outlier (black triangle) as it appears to be an admixed diploid with only 1.1% divergence between the highly and lowly diverged parts of its genome. (Right [Low*Low]) Genetic distances between genomic copies of low intra-genomic variation is generally below (0.9%) between strains of the same or different subpopulations. The admixed strain III_F09_YJS8068 is the exception, because it also has the highest variation between the low intra-genomic regions of its genome and the low intra-genomic copies of other strains (>2%). (D) Heatmap showing the genetic distance between genomic copies of 40 polyploid individuals. The only strains whose genomic copies (low intra-genomic variation and high intra-genomic variation) are similar is the admixed III_F09_YJS8068 (black triangle). Here, both genomic copies cluster together with all other genomic copies of high intra-genomic variation. (E) Acquired genomic copy of unknown origin. The genomic copies with low genetic variation were assigned as the primary genomic copies present in all individuals (2n-4n), whereas the genomic copies with high genetic variation were assigned as acquired genomic copies, only present in 40 polyploids of the subpopulations teq/EtOH, beer, and wine 1. Pairwise genetic analysis with the primary genome of the beer subpopulation as a reference shows a clear gap between the genetic variation that defines the primary and the acquired genome. The primary genome of the beer subpopulation is similarly distant to its own acquired genomic copy as well as to the acquired genomic copies of the other two polyploid groups wine 1 and teq/EtOH. The two genetic clusters beyond the 0.9% for the teq/EtOH do not only comprise the pairwise comparison with the acquired genomic copies. The dotted rectangle corresponds to comparisons with the admixed diploid, for which both copies are equally distinct. Genetic distances were calculated pairwise per chromosome and then average per genome (JC69).
We then checked for the genetic relationship of the genomic copies with only low genomic variation, because such genomic copies were present in all strains of all 71 strains (Fig. 3B). We can show that the strains cluster in the six subpopulations as previously seen using the raw Illumina data (Fig. 1B). The single strain I_H06_YJS7889, initially unable to be associated with a subpopulation, now clusters with other teq/EtOH strains.
The acquired divergent copies highlight clade-specific allopolyploidy events
To study the origin of the genetically divergent copies present in the three subpopulations, we imputed whole-genome FASTA-alignment files for every individual. First, we compared the genomic copies with high genetic variation (High*High) within and between groups. We calculated pairwise genetic distances and found that the divergence between these copies within the subpopulations was 0.13% on average (Fig. 3C, single-colored dots). In contrast, the genetic divergence of these copies across the subpopulations was 2.59% on average, ranging from 1.76% to 3.05% (two-colored dots).
When comparing the genetic distance between the low and highly (High*Low) diverged genomic copies across all the genomes, we observed that the genetic distance is 2.92% on average (Fig. 3C, High*Low). With 3.16%, the largest genetic distance can be seen between the wine 1 and kombucha subpopulations. The single outlier strain is III_F09_YJS8068 (teq/EtOH) and has the closest genetic distance between its own two genomic copies, with about 1.1% (Fig. 3C, black triangle). With more than 2, III_F09_YJS8068's low variation genome is also the most distant one to all other low variation genomes (Fig. 3C, Low*Low). This observation points to III_F09_YJS8068 being an admixed diploid whose two genomic copies are mixtures of lowly and highly diverged sequences, placing it between the highly diverged and lowly diverged genomes. The other genomes bearing low genetic variation are <1% diverged (Low*Low). In fact, using the representation of pairwise distances in the heatmap format reasserts the three genetically distinct entities of the genomic copies with high genetic variation (Fig. 3D, High*High), whereas the genomic copies with low genetic variation are more similar (Fig. 3D, Low*Low). Pairwise comparison using the low variable genomic copies of the beer clade as a reference confirm that inter-clade transfer of genomic copies can be excluded as a potential cause in the acquisition of additional genomic copies with high genetic variation in the three polyploid subpopulations (Fig. 3E) and underpin the presence of additional and unrelated copies in these three groups.
Because there is a conservation of a closely related diploid genome across the isolates of the species, we define this part as the primary genome of B. bruxellensis (Fig. 3E). It is present in all the strains and harbors a genetic variation of <1% to the reference genome. The exception is the admixed strain III_F09_YJS8068, which groups within the teq/EtOH subpopulation and has as the only strain a minimum genetic distance of 2.01% and maximum genetic distance of 2.53% to the other primary genomes. In addition to these primary genomic copies, a highly divergent copy is present in three groups (teq/EtOH, beer, and wine 1 subpopulations) and was defined as a new or “acquired” genomic copy (Fig. 3E). Although it clearly exceeds the genetic variation of the primary genome, the acquired genomic copies open the discussion about where they originate from and if they have been acquired as a result of interspecies hybridization.
To test whether the additional copies have been acquired from sister species as part of the genus Brettanomyces, we sequenced and generated de novo genome assemblies for four of the sister species: B. anomala, B. nanus, B. custerianus, and B. acidodurans (Supplemental Table S3; Supplemental Fig. S6A–D). Although we were able to show collinearity between the acquired copies and the reference genome B. bruxellensis (Supplemental Fig. S6E,F), the genomes of the sister species to B. bruxellensis were too dissimilar to retain any correlation using the same parameters. Only by lowering the parameters, we were able to show a correlation, suggesting less synteny paired with high genetic differentiation (Supplemental Fig. S6G), as already shown by Roach and Borneman (2020). With a genetic divergence of 2.5%–3% between the acquired and the primary genomic copies, however, it seems unlikely that sister species with a genetic similarity of <77% have been involved in the acquisition of the additional genomic copies (Roach and Borneman 2020).
Overall, we have shown that the triploid genomes of the wine 1, teq/EtOH, and beer subpopulations are composed of a part, which is common to every B. bruxellensis isolate, as well as a newly acquired divergent copy. These results strongly suggest that these events must have occurred independently with closer, so far unknown, and far related isolates that we would, according to the genetic distance of ∼3%, define as different species to B. bruxellensis.
LOH events shaping the genomic landscape of interspecific hybrids
Hybrid genomes are dynamic entities with LOH events playing an important role in their evolution (Smukowski Heil et al. 2017; Lancaster et al. 2019). As already seen for the triploid genomes of the wine 2 subpopulation, these events can cause the removal of genetic variation along the genomes in a conserved manner (Fig. 2B). Moreover, these events would resolve in a difference of genomic content from the parental genomes. When preparing de novo assemblies from reads with either high or low intra-genomic variation, we observed significantly shorter assemblies (median 9.1 Mb) for the genomic copies harboring high intra-genomic variation in comparison to de novo assemblies from reads with high intra-genomic variation or the reference genome of B. bruxellensis (P-value = 1.3 × 10−10) (Supplemental Fig. S7A; Fournier et al. 2017). In fact, strains from different subpopulations showed a trend in which even assembly size seemed to be not only different but also conserved between subpopulations (Supplemental Fig. S7B). Therefore, with the significantly shorter de novo assemblies for the acquired genomic copies, we hypothesized that these polyploid genomes with heterogenous levels of genetic variation have undergone LOH events as well.
To check for LOH events along the polyploid genomes, we looked at the coverage from reads belonging to the primary and acquired genome, determined if they are complementary to the total coverage, and analyzed their proportion to the total coverage. Here, we used the coverage from the short reads aligned to the B. bruxellensis reference and previously separated genomes using competitive mapping (Supplemental Fig. S5) along the chromosomes to check for reciprocal shifts in coverage (Supplemental Fig. S8A,B). We can show that regions that lack reads aligned to the acquired genomic copy reveal an increase in coverage at the primary genome complementing the total coverage. On the other hand, this also appears to be the case for several regions of the primary genome, where aligned reads represent only a single genomic copy (one-third of the total coverage), whereas the acquired genomic copy appears to be present in two copies (two-thirds of the total coverage). These results confirm the reorganization of the polyploid genome through LOH events in the subpopulations teq/EtOH, beer, and wine 1.
Then, we used the primary genome as a reference and determined how many copies are present throughout the genome within the polyploid strains. We calculated its ratio per 10 kb nonsliding windows to the total coverage to assess its proportion. Our results show that the polyploid genomes have undergone massive LOH events (Fig. 4). Most regions appear to have been lost/gained within a subpopulation-specific pattern. On Chromosome 1 for example, the beer and wine 1 subpopulations lack a significant part of the acquired genomic copy (1.7 Mb for beer strains and 1.2 Mb for wine 1 strains). Other (often small) events are private to single strains.
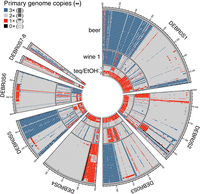
Dynamic genomic landscape of polyploid strains. The polyploid genomes of the three subpopulations beer, wine 1, and teq/EtOH underlie massive modifications through LOH events. The primary genome (low genetic variation) was used as a reference. Conserved patterns of modified regions for the primary genome were identified by determining the gain or loss of its copies in each strain, here varying between three (3×) and zero (0×). Only a few modified regions are unique to single or few strains. There are no common regions that show the same patterns across subpopulations. The teq/EtOH subpopulation shows a division into two clusters, each consisting of three individuals. The ploidy level was estimated in 10-kb windows.
Next, we determined if the parts absent from the acquired genomic copy in the three subpopulations can complement the smaller de novo genome assemblies (Supplemental Fig. S7). For the beer strains, we calculated that LOH events have caused the loss of 26.6% of regions on average from the acquired copy (Supplemental Fig. S8A). This makes 9.54 Mb of the acquired genomic copy still kept, which is similar in size to the de novo assembly of 8.9 Mb (Supplemental Fig. S7B). For the wine 1 subpopulation, 22.3% of the acquired genomic copy is lost on average, making 10.1 Mb still being present (Supplemental Fig. S8B). Here, the de novo assembly size (10.1 Mb) matches exactly the number of retained regions in our analysis (Supplemental Fig. S7B).
The teq/EtOH strains, however, show a pattern of loss/gain of genomic regions from the primary genome that enables the distinction of two subgroups, denoted as teq/EtOH 1 and teq/EtOH 2. The teq/EtOH 2 has almost entirely lost the second copy of the primary genome, being replaced by a second copy of the acquired genome (Fig. 4). Both subclades have lost 10.5% of the acquired genomic copy on average (12.1% for teq/EtOH 1 and 8.8% for teq/EtOH 2). The average of both (11.64 Mb) is comparable to the average de novo assembly size of 10.7 Mb.
The conserved patterns of LOH within each subpopulation opens the discussion of whether these patterns are the consequence of adaptation, random processes, or point at a recent shared ancestry. In the evolution of species, polyploidy has been shown to potentially play an important role in the acquisition of new traits or the amplification of already existing traits in the context of the acquisition of resistances (Jackson and Tinsley 2003; Augustine et al. 2013), interactions (Thompson et al. 2004; Těšitelová et al. 2013), coping with changing environments (Selmecki et al. 2015), or the occupation of novel ecological niches (Wani et al. 2018). Further, the different environments where B. bruxellensis’ polyploid subpopulations are associated with bioethanol production, wine, or beer fermentation are harsh environments and require different characteristics from the strains as a high tolerance against alcohol and acidity, for example.
To justify the conserved pattern per group from an adaptive evolutionary perspective, we checked if the regions, either gained or lost from the primary genome, are enriched for genes with particular functions. We used the genome annotation from Gounot et al. (2020) and checked for Gene Ontology (GO) terms in regions that have gained or lost a copy of the primary genome. Additionally, we checked regions that are different in ploidy between the beer and the wine 1 subpopulation. Both approaches, however, revealed no enrichment (Supplemental Table S4). Further, we focused on the set of 56 candidate genes described in Colomer et al. (2020) (Supplemental Table S5). These genes are associated with particular functions and phenotypes, such as maltose assimilation, ethanol production, or sulphite tolerance and play important (positive and negative) roles in different industrial applications. We could not find a pattern in which particular gene groups are linked to regions that are characterized by a conserved number of genomic copies for the primary genome (Supplemental Fig. S9). Either these patterns of conservation have been acquired through adaptive processes for which we could not find any proof, or alternatively they have occurred through random processes. As already seen for the polyploid wine 2 subpopulation, LOH events are shared among strains and, with LOH events at similar positions (e.g., DEBR0S1), hotspots for LOH might be involved. However, we have no evidence for these at this point. Further, we would conclude that a recent ancestry could be also an explanation for the underlying pattern of LOH events, but because of the different origins of isolation in space and time, we believe it is highly unlikely to argue that the observed and conserved pattern within the subpopulations is caused by a recently shared common ancestry.
Overall, the three subpopulations with polyploid genomes coming from interspecific hybridization events are highly dynamic where LOH events have caused conserved patterns of low genetic diversity regions within each subpopulation. How these variations, especially on the gene level, are finally expressed at a phenotypic level will have to be the goal of following studies investigating the phenotypic landscape of the different subpopulations.
Expanded mitochondrial genomes and large inversions for the teq/EtOH subpopulation
With the polyploid subpopulations having undergone massive and independent modifications of their nuclear genome, we checked if this also accounts for their mitochondrial (Mt) genome. For this, we generated de novo assemblies from short-read sequencing data. We were able to prepare single circularized scaffolds for 48 of the 71 strains (Supplemental Table S6). Overall, de novo assemblies revealed an increase in size from 75.3 to 89 kb for all subpopulations, except teq/EtOH, whose Mt genome size was >100 kb (Fig. 5A).

De novo assemblies for the mitochondria reveal its expansion in all subpopulations and high level of reorganization for the teq/EtOH strains. (A) De novo assembly size difference. All subpopulations increased their mitochondrial genome compared to the reference genome (accession number GQ354526.1), from 75.3 to 88–90 kb. The teq/EtOH subpopulation stands out with de novo assembly sizes >100 kb. Red dotted line denotes the mitochondrial size of the reference genome. (B) Mitochondrial synteny. Synteny is conserved among subpopulations (compared to the reference), with the exception for the teq/EtOH subpopulation. Additionally, synteny within teq/EtOH was not congruent among strains. Here, the strain I_H06_YJS7889 harbors a large inversion. (C) Intron content contributes to genome expansion of teq/EtOH subpopulation. For teq/EtOH strains, the two protein-coding genes COB and COX1 have increased in intron content, whereas other subpopulations have the expected intron sizes compared to the reference genome (accession number GQ354526.1).
We then extracted gene positions and prepared a synteny approach to look for the organization of the different mitochondrial genomes. We can show that the teq/EtOH strains, apart from their extended sequence length, harbor several large inversions, whereas the organization of the mitochondria from the other subpopulation are aligned with the organization as found for the reference genome (Fig. 5B).
By calculating pairwise genetic distances from concatenated gene sequences, teq/EtOH strains are clearly distinct from all other strains (Supplemental Fig. S10). Highest genetic distance of 3% was detected between two wine strains (wine 2: I_B01_YJS7812; wine 3: II_B03_YJS7914) with the teq/EtOH strains.
By looking at intronic content, we can show that two of the intron-carrying and protein-coding mitochondrial genes, COB and COX1, are partially involved in the overall size increase of the mitochondria in teq/EtOH strains. The other strains show no difference in intronic regions (Fig. 5C).
Discussion
The Brettanomyces bruxellensis yeast species is known to harbor subpopulations with various levels of ploidy (Avramova et al. 2018; Colomer et al. 2020; Gounot et al. 2020). For the first time, we provide a detailed insight into the complex genomic architecture of these polyploid subpopulations. We noticed that there is a high conservation of ploidy in each subpopulation and four of them, associated with three different ecological environments (tequila/ethanol production, wine making, and beer brewing) are exclusively characterized by triploids.
Because polyploidy can be achieved in different ways (allopolyploidization or autopolyploidization), the final genomic composition might vary by distinct levels of intra-genomic information. At the same time, the intra-genomic variation will define the boundaries of genomic flexibility and therefore drive evolution in almost unpredictable and different ways (Ng et al. 2012; Selmecki et al. 2015).
By using two different phasing strategies, we elucidated the genomic architecture of polyploid subpopulations of B. bruxellensis with various levels of intra-genomic variation. We highlighted that all six populations harbor a primary genome irrespective of their ploidy, which is defined as the genetic variation that does not exceed the 1% compared to the reference genome B. bruxellensis (Fournier et al. 2017). This is lower but in accordance with previous papers elucidating the genetic variation of B. bruxellensis, because they did not phase the genomes into distinct haplotypes (1.2%) (Gounot et al. 2020). Further, we can show the existence of three allopolyploid subpopulations (teq/EtOH, beer, and wine 1) with an acquired genomic copy with a genetic divergence of about 3% compared to the reference genome. They clearly exceed the average intra-genomic variation of the primary genome, undermining the occurrence of interspecific hybridization events in these subpopulations. The known sister species within the same genus are rejected as donors for the interspecific hybrids owing to too high genetic divergence of at least 23% (Roach and Borneman 2020).
We further highlight that to our knowledge, the B. bruxellensis species is one (or the) rare case, in which these different scenarios, allo- and autopolyploidy, respectively, can be observed in close related subpopulations. We observed different trajectories for strains not only associated with different environments (teq/EtOH, beer, and wine) but also associated with the same environment while being part of a genetically distinct cluster. The “wine”-associated strains fall into three genetically diverged subpopulations. With the two subpopulations wine 1 and wine 2 being triploid compared to wine 3 (diploid), only wine 1 has acquired a third genomic copy from interspecific hybridization, whereas wine 2 has solely genetically similar haplotypes.
Different trajectories of polyploidizations in nature were mostly studied (and observed) in plants, which give no clues about the importance as well as prevalence of polyploidization and their trajectories in animal or fungal systems (Leggatt and Iwama 2003; Gregory and Mable 2005; Barker et al. 2016). At the same time, these mechanisms, when observed and studied in extant polyploids, have mostly (when not exclusively) been determined between species systems rather than within. We highlight that future studies, especially in the animal and fungi kingdom, are required that screen individuals on a large scale to study prevalence and trajectories of polyploids across ecologically diverged, naturally occurring subpopulations. Indications that polyploidy could be a more common state were shown by two recent studies that genotyped more than 1000 individuals from the S. cerevisiae and B. bruxellensis yeast species, with a prevalence of polyploids of 11.4% (Peter et al. 2018) and 54% (Avramova et al. 2018), respectively.
Finally, we speculate that the different trajectories of polyploids in the subpopulations of B. bruxellensis are linked to the adaptation to the different anthropized environments. Polyploids in general have been given a lot of attention in the context of adaptivity and diversification, in which many extant species originate from ancient polyploid states (Seoighe and Wolfe 1998; Gerstein and Otto 2009; Gordon et al. 2009; Marcet-Houben and Gabaldón 2015). Although a polyploid state itself can allow adaptability, it is often seen as a transient state, which is followed by massive modifications thereafter to cope with genetic incompatibilities and to regain fertility in the long term. Evidence for this process has been gained through the detection of paralogous gene sets with different historical trajectories in many naturally diploid taxa, undermining the process of genomic modifications after polyploidization. With a prevalence of 54% polyploids, plus evidence for three independent interspecific hybridization events, polyploidy is very abundant and most likely does not underlie random effects for B. bruxellensis. The genomes of the allo- and autopolyploid subpopulations are characterized by massive genomic modifications that have established conserved pattern of rearranged blocks. These underline on the one hand the independent acquisition of genetically diverse genomic copies for the allopolyploid subpopulations, but most likely they reflect the regain of fitness and overcome of genomic incompatibilities while being able to adapt to harsh and changing conditions in their anthropized environments. The high tolerance against sulfur dioxide, for example, a treatment to prevent wine fermentation from spoilage by B. bruxellensis, could be the response of this yeast to the recently increased usage of this agent from the industry and have been mostly observed for the wine 1 subpopulation (Avramova et al. 2019).
Our study clearly highlights for the first time the coexistence of a large repertoire of evolution punctuated by various independent polyploidization events within a species and addresses the need to further resolve the genomic architecture of polyploid species complexes from diverse ecological settings.
Methods
Strain selection and sequencing
Seventy-one strains, coming from the previously defined clades of Brettanomyces bruxellensis were chosen for this project (Avramova et al. 2018). All strains were sequenced using the long- and short-read sequencing strategy, Oxford Nanopore and Illumina sequencing, respectively. Long-read sequencing was performed and processed as described (Fournier et al. 2017; Istace et al. 2017). Short-read sequencing was conducted and analyzed with respect to Gounot et al. (2020). The reference genome of B. bruxellensis for the subsequent analysis was chosen from Fournier et al. (2017). A full description of the sample preparation can be found in the Supplemental Material (see “Selection of strains and DNA extraction” and “Library preparation and sequencing” sections).
Short-read (Illumina) analyses
Short reads were either aligned directly to the reference genome or in a competitive mapping approach to concatenated de novo assemblies from long-read sequences. The latter enabled the differentiation between short reads with low or high genetic divergence to the reference genome of polyploid genomes, respectively, to the genomic copy with high or low genetic variation. Calculation of pairwise distances and regions underlying variation in copy numbers were performed based on the genetic variation and its coverage (allele frequency) to the reference genome. A full description of the analysis performed with this Illumina sequence data set can be found in the Supplemental Material (see “Short reads [Illumina] sequences analysis” section).
Long-read (Oxford Nanopore) analyses
Low and high intra-genomic variation clusters were determined in genomes to define the degree of ploidy per strains based on their SNP density to the reference. De novo assemblies were prepared from polyploid strains with divergent clusters. The algorithm published in Abou Saada et al. (2021) was used to decipher genomic variation of genomes of strains with only a low degree of genetic variation to the reference genome. A full description of the analysis performed with this Oxford Nanopore sequence data set can be found in the Supplemental Material (see “Long reads [Oxford Nanopore] sequences analysis” section).
Data access
All raw sequencing data generated in this study have been submitted to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser/home) under accession number PRJEB41126 (for the 71 Brettanomyces bruxellensis isolates) and PRJEB41125 (for the B. anomala, B. nanus, B. custerianus, and B. acidodurans species).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the Agence Nationale de la Recherche (ANR-18-CE20-0003-02 and ANR-18-CE12-0013-02) and the European Research Council (ERC Consolidator Grant 772505). This work of the Interdisciplinary Thematic Institute IMCBio, as part of the ITI 2021-2028 program of the University of Strasbourg, CNRS, and Inserm, was supported by IdEx Unistra (ANR-10-IDEX-0002) and by SFRI-STRAT'US project (ANR-20-SFRI-0012) and EUR IMCBio (ANR-17-EURE-0023) under the framework of the French Investments for the Future Program. J.S. is a Fellow of the University of Strasbourg Institute for Advanced Study (USIAS) and a member of the Institut Universitaire de France.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275380.121.
-
Freely available online through the Genome Research Open Access option.
- Received February 11, 2021.
- Accepted October 25, 2021.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








