
Human and rat skeletal muscle single-nuclei multi-omic integrative analyses nominate causal cell types, regulatory elements, and SNPs for complex traits
- Peter Orchard1,9,
- Nandini Manickam1,9,
- Christa Ventresca1,2,9,
- Swarooparani Vadlamudi3,
- Arushi Varshney1,
- Vivek Rai1,
- Jeremy Kaplan1,
- Claudia Lalancette4,
- Karen L. Mohlke3,
- Katherine Gallagher5,6,
- Charles F. Burant7 and
- Stephen C.J. Parker1,2,8
- 1Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan 48109, USA;
- 2Department of Human Genetics, University of Michigan, Ann Arbor, Michigan 48109, USA;
- 3Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA;
- 4Epigenomics Core, University of Michigan, Ann Arbor, Michigan 48109, USA;
- 5Department of Surgery, University of Michigan, Ann Arbor, Michigan 48109, USA;
- 6Department of Microbiology and Immunology, University of Michigan, Ann Arbor, Michigan 48109, USA;
- 7Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan 48109, USA;
- 8Department of Biostatistics, University of Michigan, Ann Arbor, Michigan 48109, USA
Abstract
Skeletal muscle accounts for the largest proportion of human body mass, on average, and is a key tissue in complex diseases and mobility. It is composed of several different cell and muscle fiber types. Here, we optimize single-nucleus ATAC-seq (snATAC-seq) to map skeletal muscle cell–specific chromatin accessibility landscapes in frozen human and rat samples, and single-nucleus RNA-seq (snRNA-seq) to map cell-specific transcriptomes in human. We additionally perform multi-omics profiling (gene expression and chromatin accessibility) on human and rat muscle samples. We capture type I and type II muscle fiber signatures, which are generally missed by existing single-cell RNA-seq methods. We perform cross-modality and cross-species integrative analyses on 33,862 nuclei and identify seven cell types ranging in abundance from 59.6% to 1.0% of all nuclei. We introduce a regression-based approach to infer cell types by comparing transcription start site–distal ATAC-seq peaks to reference enhancer maps and show consistency with RNA-based marker gene cell type assignments. We find heterogeneity in enrichment of genetic variants linked to complex phenotypes from the UK Biobank and diabetes genome-wide association studies in cell-specific ATAC-seq peaks, with the most striking enrichment patterns in muscle mesenchymal stem cells (∼3.5% of nuclei). Finally, we overlay these chromatin accessibility maps on GWAS data to nominate causal cell types, SNPs, transcription factor motifs, and target genes for type 2 diabetes signals. These chromatin accessibility profiles for human and rat skeletal muscle cell types are a useful resource for nominating causal GWAS SNPs and cell types.
Skeletal muscle tissue accounts for 30%–40% of body mass, which is the largest tissue, on average, in adult humans and is central to basic quality of life and complex diseases (Janssen et al. 2000; Frontera and Ochala 2015). Like other tissues, skeletal muscle is composed of a mixture of different cell types. Most of the tissue is composed of muscle fibers, which may be categorized into different fiber types, each of which display distinct metabolic and molecular phenotypes. The proportion of muscle fibers accounted for by each fiber type varies across individuals (Simoneau and Bouchard 1989). Muscle-related diseases may differentially impact different fiber types, and fiber type proportions are associated with complex phenotypes, including aerobic and anaerobic exercise capacity and type 2 diabetes (T2D) status (Talbot and Maves 2016). Muscle satellite cells are progenitors to muscle fibers, indispensable for the generation and regeneration of muscle (Relaix and Zammit 2012); these cells are present in skeletal muscle tissue, as are several other cell types, such as mesenchymal stem cells, that cooperate in muscle regeneration (Judson et al. 2013; Klimczak et al. 2018). Molecular associations with skeletal muscle tissue/muscle fiber characteristics and muscle-related complex diseases could be mediated in part by these stem cell–like populations; for example, a genetic variant that alters the development of a satellite cell could carry important implications for later muscle function, just as some T2D-associated variants are proposed to impact pancreatic/beta cell development rather than the function of mature beta cells (Travers et al. 2013; Mattis and Gloyn 2020) and facial morphology-associated variants may act through progenitor cell populations (Xiong et al. 2019). Immune cells infiltrate muscle tissue and communicate with muscle cells as well, playing a particularly important role following injury (Pillon et al. 2013). Profiling the transcriptomic and epigenomic landscapes of these cell types and muscle fiber types may therefore contribute to our understanding of the biology of muscle development and muscle-related complex traits.
Bulk profiling of skeletal muscle tissue ignores this heterogeneity and is dominated by the most common cell types (muscle fibers), but single-cell/-nucleus methods overcome this and allow profiling of the constituent cell types. In the case of skeletal muscle, the distinction between single-nucleus and single-cell profiling is particularly important as (1) skeletal muscle fibers have an elongated shape that may make them difficult to capture in single-cell suspensions, and (2) muscle fibers are multinucleated, meaning that a single-cell measurement will capture the output of many nuclei. Previous single-cell RNA-seq studies of human (Rubenstein et al. 2020; Xi et al. 2020; De Micheli et al. 2020b), mouse (The Tabula Muris Consortium 2018; Dell'Orso et al. 2019; Giordani et al. 2019; Pawlikowski et al. 2019; Oprescu et al. 2020; De Micheli et al. 2020a), and pig (Qiu et al. 2020) skeletal muscle tissue either capture no muscle fiber nuclei or capture them in unrepresentative proportions. Bulk analysis of pooled, dissected muscle fibers has generated fiber type-specific transcriptional profiles (Chemello et al. 2011, 2019; Raue et al. 2012; Alessio et al. 2019), and analysis of specific isolated muscle resident cell populations (Fukada et al. 2007; Cho and Doles 2017; Liu et al. 2019) has generated insights into targeted cell subpopulations, but these studies are necessarily biased toward specific cell types.
Here, we employ single-nucleus RNA-sequencing (snRNA-seq), ATAC-seq (snATAC-seq), and multiomic profiling (joint snRNA-seq + snATAC-seq) on the 10x Genomics platform to profile gene expression and chromatin accessibility of frozen skeletal muscle cell populations in human and rat. To our knowledge, this represents the first snATAC-seq data from rat skeletal muscle tissue and the first joint single-nucleus gene expression + chromatin accessibility data from human and rat skeletal muscle tissue. First, we examine the influence of fluorescence-activated nucleus sorting (FANS) and nucleus loading concentration on the performance of the 10x Genomics platform. Next, we perform clustering of the snRNA-seq and snATAC-seq nuclei to determine the cell types detected in skeletal muscle tissue samples and map their respective transcriptomes and chromatin landscapes. We then integrate the resulting genomic maps with UK Biobank and T2D-related GWAS results to explore the relationship between these cell types and a broad range of human phenotypes and diseases and nominate causal SNPs at several genomic loci.
Results
FANS negatively impacts 10x snATAC-seq results
Before being loaded onto the 10x platform, nuclei must be isolated from the samples of interest. This process involves cell lysis, which produces viable nuclei as well as substantial cellular debris, some of which remains in the final nuclei suspension. Debris reduces the quality of the resulting libraries. By staining the DNA in live nuclei and using FANS to selectively filter the suspension for stained entities, one can remove cellular debris in the suspension, potentially improving the purity and quality of the suspension loaded onto the 10x platform. Debris must be removed post-lysis, as lysis itself produces much of the debris. Although FANS may result in substantial nucleus loss, this is not a problem when enough input is available. However, the FANS process could stress the nuclei or otherwise alter the snRNA-seq and snATAC-seq results. Comparing quality control metrics and (in the case of snRNA-seq) aggregate gene expression or (in the case of snATAC-seq) aggregate ATAC-seq peaks/signal between snRNA-seq and snATAC-seq libraries generated from nuclei that either did or did not undergo FANS allows one to detect substantial changes that FANS may introduce. Also, because the aggregate of reads from a snATAC-seq library should resemble the profile of an ATAC-seq library on the same biological sample, one can generate bulk and single-nucleus libraries from a single sample and compare quality control metrics and ATAC-seq signal between them. Therefore, to determine the effect of FANS on 10x snRNA-seq and snATAC-seq results, we performed three nuclear isolations from a single human muscle sample, mixed the resulting nuclei together, and performed FANS (using DRAQ7 staining) on one half of the suspension (Fig. 1A). DRAQ7 is a DNA stain and was used to sort positive nuclei. The FANS and non-FANS suspensions were each used to produce two replicate snATAC-seq and two replicate snRNA-seq libraries, resulting in eight total libraries (four snATAC and four snRNA). We also generated two independent bulk ATAC-seq libraries from the same biological sample, allowing us to compare snATAC-seq profiles, with and without FANS, to a comparable bulk ATAC-seq profile.
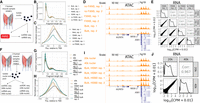
Impact of FANS and loading concentration on 10x Genomics snATAC-seq and snRNA-seq results. (A) Study design to determine the effect of FANS on snRNA-seq and snATAC-seq results. Muscle cartoon adapted from Scott et al. 2016. HSM1 refers to one specific skeletal muscle sample (“human skeletal muscle 1”). Bulk ATAC-seq was performed on HSM1 as well (two replicates, each separate nuclei isolations). (B) Fragment length distribution and (C) TSS enrichment for two snATAC-seq libraries that did not undergo FANS and two that did, as well as two bulk ATAC-seq replicates from the same sample (“Bulk”). (D) ATAC-seq signal at the ANK1 locus for FANS or non-FANS input snATAC-seq libraries, and the two bulk ATAC-seq libraries. All tracks are normalized to 1-M reads and have the same y-axis range corresponding to zero reads per million to three reads per million. Gene model (GENCODE v19 basic) displays protein coding genes only. (E) Correlation between FANS and non-FANS snRNA-seq libraries; each point represents one gene. (F) Study design to determine the effect of loading 20k versus 40k nuclei into the 10x platform, utilizing HSM1 as well as a second sample, HSM2 (“human skeletal muscle 2”). Bulk ATAC-seq was performed on HSM1 (same libraries as in A) and on HSM2 (two replicates, each separate nuclei isolations). (G) Fragment length distribution and (H) TSS enrichment for snATAC-seq libraries after loading 20k versus 40k nuclei, as well as for the four bulk ATAC-seq libraries (two each from the two muscle samples, “HSM1 bulk” and “HSM2 bulk”). (I) ATAC-seq signal at the ANK1 locus for the 20k and 40k libraries and the four bulk ATAC-seq libraries. All tracks are normalized as in D. Gene model (GENCODE v19 basic) displays protein coding genes only. (J) Correlation between snRNA-seq libraries resulting from loading 20k versus 40k nuclei.
First, we examined the four snATAC-seq libraries, comparing the aggregate signal for each library to bulk ATAC-seq libraries from the same biological sample. We called peaks for the four libraries and ran the ataqv quality control software package (Orchard et al. 2020) on the aggregated data to examine the overall transcription start site (TSS) enrichment and fragment length distributions. The fragment length distributions for each library resembled the expected stereotypical ATAC-seq fragment length distribution, showing an abundance of short fragments as well as mononucleosomal fragments (Fig. 1B; Buenrostro et al. 2013); however, the TSS enrichment was lower in the FANS libraries (Fig. 1C), indicating that the FANS libraries had a lower signal-to-noise ratio. This difference in signal-to-noise ratio is demonstrated when visualizing the ATAC-seq signal at genomic regions active in muscle, such as the ANK1 locus (Fig. 1D; Scott et al. 2016). We additionally overlapped TSS-distal ATAC-seq peaks from each of the libraries with existing chromatin states from diverse tissues and cell types (Roadmap Epigenomics Consortium et al. 2015) and found that the peaks from the non-FANS libraries showed considerable overlap with skeletal muscle enhancers, whereas the peaks from the FANS libraries showed poor overlap (Supplemental Fig. S1). ATAC-seq signal across FANS libraries showed poor correlation with the two bulk ATAC-seq libraries from the same sample (Supplemental Fig. S2). We therefore concluded that FANS, at least as it was performed here, has a clear negative impact on 10x snATAC-seq results.
Next, we examined the four snRNA-seq libraries. All four libraries showed high correlation, indicating that FANS does not substantially alter snRNA-seq results at the pseudobulk gene expression level (Fig. 1E). In order to determine if FANS altered the yield of quality nuclei, we used read counts and mitochondrial contamination to select quality nuclei from each library (Supplemental Fig. S3). We found that FANS substantially increased the number of quality nuclei obtained (2264 and 2343 for non-FANS libraries; 8423 and 7779 for FANS libraries). We therefore concluded that FANS has little effect on pseudobulk gene expression measurements but may alter nucleus yield.
snATAC-seq and snRNA-seq results are robust to nucleus loading concentrations
The concentration at which nuclei are loaded onto the 10x platform is an important parameter affecting data quality and the number of nuclei available for downstream analysis. Increasing the loading concentration increases the maximum number of nuclei from which data can be obtained; however, it also increases the probability that multiple nuclei end up with the same gel bead, thereby increasing the doublet rate. Balancing these outcomes is important to maximize the amount of quality data and number of nuclei available for downstream analysis. To evaluate the effect of increasing the number of nuclei loaded onto the platform, we performed a separate experiment in which we isolated nuclei from two muscle samples, mixed them together, and then loaded either 20k or 40k nuclei (as quantified by a Countess II FL Automated Cell Counter) into a 10x well for snRNA-seq and for snATAC-seq (Fig. 1F). We also generated two independent bulk ATAC-seq libraries from the biological sample for which bulk ATAC-seq profiles were not already available, allowing us to compare snATAC-seq profiles to comparable bulk ATAC-seq profiles.
The snATAC-seq libraries displayed the expected fragment length distributions and comparable TSS enrichments (Fig. 1G,H). We examined the aggregate signal of the snATAC-seq libraries next to bulk ATAC-seq libraries from the same samples and confirmed that both libraries showed strong signal, comparable to that of bulk data (Fig. 1I). Overlap between TSS-distal ATAC-seq peaks called on both libraries and chromatin states were likewise similar, showing relatively high overlap with skeletal muscle enhancers (Supplemental Fig. S4), and the ATAC-seq signal in the libraries correlated with bulk ATAC-seq signal to an extent comparable to the correlation between two bulk ATAC-seq libraries (Supplemental Fig. S5). After selecting quality nuclei (Supplemental Fig. S6), we found that the higher loading concentration yielded 2104 nuclei whereas the lower concentration yielded 830 nuclei (after doublet removal).
Correlation between the snRNA-seq libraries was high, indicating that the loading concentration could be changed substantially without compromising data quality (Fig. 1J). We again found the higher loading concentration yielded more quality nuclei than the lower concentration (3475 vs. 2056) (Supplemental Fig. S7) after doublet removal. As the aggregate gene expression/ATAC-seq signal profile was comparable between loading concentrations, we concluded that snRNA-seq/snATAC-seq results are robust to the number of nuclei loaded. One caveat to these conclusions is that the actual number of nuclei loaded into the well may differ from our estimated numbers, as debris in the nuclei preps may affect the accuracy of the nuclei counts.
Clustering of human and rat snATAC-seq and snRNA-seq identifies skeletal muscle cell types
To determine cell types present in skeletal muscle samples, we selected high-quality ATAC and RNA nuclei from the FANS/non-FANS libraries and the 20k/40k nuclei libraries generated above and performed joint clustering. snATAC-seq libraries that underwent FANS were excluded as they failed to provide quality data. We generated and included a snATAC-seq library containing a mix of human and rat nuclei (Supplemental Figs. S8, S9; Supplemental Tables S1, S2), as well as a single-nucleus multi-omics library (gene expression + chromatin accessibility from the same nucleus) containing a mix of human and rat nuclei (Supplemental Figs. S10, S11). Information about the biological samples and post-QC nucleus summary statistics for each library is provided in Supplemental Table S3. In total, we obtained 26,340 human snRNA-seq (mean UMIs = 8159), 5274 human snATAC-seq (mean reads = 97,173), 643 rat snATAC-seq (mean reads = 138,705), 1265 human multiome (mean RNA UMIs = 2638; mean ATAC reads = 66,470), and 722 rat multiome (mean RNA UMIs = 2346; mean ATAC reads = 74,544) nuclei. We used Seurat (Butler et al. 2018; Stuart et al. 2019) to jointly cluster the snRNA-seq, snATAC-seq, and multi-omics nuclei and identified seven cell type clusters (Fig. 2A). Nuclei from different modalities, species, and libraries integrated well, indicating that clustering was not driven by technical factors (Fig. 2B). To ensure that the clusters were robust, we additionally used integrative nonnegative matrix factorization (iNMF) as implemented in the LIGER (linked inference of genomic experimental relationships) software package (Welch et al. 2019) to jointly cluster the nuclei and found that cluster assignments were largely concordant (95.9% of nuclei were assigned to the same cluster) (Supplemental Fig. S12).
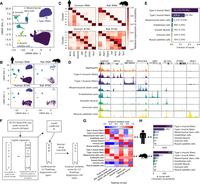
Joint clustering of human and rat snATAC-seq and snRNA-seq identifies skeletal muscle cell types. (A) UMAP after clustering human snATAC-seq, human snRNA-seq, rat snATAC-seq, and human and rat dual modality (snATAC-seq + snRNA-seq) nuclei with Seurat. (B) UMAP facetted by species and modality. Dual modality nuclei were clustered using RNA and are displayed within the “RNA” facets. (C) Gene expression (snRNA-seq, including dual modality nuclei RNA) or accessibility (snATAC-seq; gene promoter + gene body) of marker genes. Values are column-normalized. (D) ATAC-seq signal for human snATAC-seq (+ dual modality) nuclei in each cluster. All tracks are normalized to 1M reads. (E) Fraction of nuclei assigned to each cell type. (F) Logistic regression-based approach to score similarity between TSS-distal ATAC-seq peaks (>5 kb from TSS) and Roadmap Epigenomics enhancer states. For all TSS-distal ATAC-seq peaks across all cell types, we scored the accessibility of the peak (0/1) in each of the muscle cell types based on the presence or absence of a peak call. Then, for a given one of the 127 Roadmap Epigenomics cell types, we determined the maximum posterior probability of the enhancer states in the Roadmap Epigenomics ChromHMM model within each peak. We then used logistic regression to model the relationship between the peak accessibility and the enhancer posteriors (running one model per muscle cell type per Roadmap Epigenomics cell type). Then, for each muscle cell type, the model coefficient was normalized to 1 by dividing by the maximum coefficient across all 127 Roadmap Epigenomics cell types, and this value was used as the enhancer similarity score for that muscle cell type and Roadmap Epigenomics cell type. (G) Similarity of snATAC-seq peak calls for each cell type and species to Roadmap Epigenomics ChromHMM enhancer states based on the logistic regression procedure outlined in F. The Roadmap Epigenomics cell type names have been adjusted for clarity and the sake of space. The full names and the identifiers from the Roadmap Epigenomics paper are: psoas muscle (E100), mesenchymal stem cell–derived adipocyte cultured cells (E023), HUVEC umbilical vein endothelial primary cells (E122), stomach smooth muscle (E111), primary monocytes from peripheral blood (E029), and fetal muscle trunk (E089). (H) Nucleus counts per species for snATAC-seq data. Copyright disclosure for rat cartoon: Rat by Francisca Arévalo from the Noun Project (https://thenounproject.com/search/?q=rat&i=15130).
We used marker genes to assign cell types to each cluster (Supplemental Table S4) and found clear concordance between human snRNA-seq and snATAC-seq (Fig. 2C,D). We found marker gene expression and accessibility in the rat data to be largely consistent with the human data, though examination of the myosin heavy chain genes, used to distinguish different muscle fiber types, indicated that a considerable number of rat type II muscle fiber nuclei were likely present in the type I muscle fiber cluster (the opposite did not seem to occur; i.e., the type II muscle fiber cluster appeared to be relatively free of rat type I muscle fiber nuclei) (Supplemental Fig. S13). This mixing of some rat muscle fiber nuclei is a limitation of our data; because only 1365 of 34,244 (4.0%) of all nuclei used for clustering are rat nuclei, the human data drive the clustering. As expected, the vast majority of the profiled nuclei (88.8%) came from muscle fibers (Fig. 2E).
We sought to independently assess cluster identity without relying on marker gene patterns and therefore focused on cluster-level TSS-distal ATAC-seq peaks, many of which would not be taken into account when assigning cell types using marker genes. We developed a logistic regression approach to score the similarity between these peaks and enhancer chromatin states from 127 Roadmap Epigenomics cell types (Fig. 2F; Roadmap Epigenomics Consortium et al. 2015). We found concordance with the marker gene-based cell type assignment approach (Fig. 2G), and this approach also worked relatively well in assigning rat nuclei, despite the fact that the number of rat nuclei per cluster with chromatin accessibility data ranged between 10 and 35 for the rarest three cell types (Supplemental Table S5; Fig. 2H).
The majority of the nuclei were assigned as type I or type II muscle fibers. Genes previously discovered to be preferentially expressed in type I versus type II muscle fibers (Rubenstein et al. 2020) were usually similarly preferentially expressed in our snRNA-seq data (Supplemental Fig. S14), validating the quality of the data and accuracy of muscle fiber type assignments.
To determine transcription factors (TFs) that may play an important role in each cell type, we used chromVAR (Schep et al. 2017) to score the relative accessibility of TF motifs in nuclei of different cell types (Supplemental Fig. S15). This analysis uncovered many cell type–TF relationships consistent with known biology; for example, PAX7 and MYOG motifs were especially accessible in satellite cells (Seale et al. 2000; Ganassi et al. 2020); GATA and SOX motifs in endothelial cells (Kanki et al. 2017); ATF4 and CEBPB motifs in mesenchymal stem cells (Cohen et al. 2015); SRF motifs in smooth muscle cells (Mack 2011); and STAT5A and SPI1 motifs in immune cells (Friedman 2007; Hennighausen and Robinson 2008; Owen and Farrar 2017).
Integration of cell type–specific ATAC-seq peaks with UK Biobank GWAS reveals cell type roles in complex phenotypes
Genetic variants associated with complex traits and disease are frequently located in noncoding regions of the genome (Maurano et al. 2012; Schaub et al. 2012; Parker et al. 2013). Variants associated with a given complex trait are expected to be enriched specifically in noncoding regulatory elements of the trait-relevant cell types; for example, T2D-associated genetic variants are enriched in regulatory elements specific to pancreatic islets and beta cells (Parker et al. 2013; Pasquali et al. 2014; Finucane et al. 2015; Gaulton et al. 2015; Quang et al. 2015; Varshney et al. 2017, 2021; Mahajan et al. 2018; Thurner et al. 2018; Rai et al. 2020), and variants associated with autoimmune disorders are enriched in immune cell–specific regulatory elements (Finucane et al. 2015). Variant enrichment in cell-specific regulatory elements can therefore be used to determine which cell types are relevant to a given trait or disease. Variants in high linkage disequilibrium (LD) with trait-influencing SNPs are often statistically associated with the trait as well, making it difficult to infer the causal SNP through statistical association alone. Epigenomic data, such as chromatin accessibility in trait-relevant cell types, can be used to nominate causal genetic variants under the assumption that noncoding SNPs in accessible regions of the genome are more likely to be causally related to a trait than noncoding SNPs in inaccessible regions.
To explore the relationship between complex traits and the cell types present in our data, as well as demonstrate the value of our muscle cell type chromatin data in narrowing the post-GWAS search space, we used LD score regression (LDSC) (Finucane et al. 2015; Gazal et al. 2017) to perform a partitioned heritability analysis using GWAS of 404 heritable traits from the UK Biobank (Sudlow et al. 2015) (http://www.nealelab.is/uk-biobank/) and our muscle cell type open chromatin regions (Supplemental Table S6; see Methods; Finucane et al. 2015; Gazal et al. 2017). Results for all traits in which at least one of our cell types showed significant (P < 0.05) enrichment after Benjamini–Yekutieli correction are displayed in Figure 3. Due to the heavy multiple testing correction burden, relatively few traits meet this threshold. However, we observed that immune cell abundance traits show enrichment for the immune cell cluster, blood pressure GWAS SNPs are enriched in smooth muscle ATAC-seq peaks, and SNPs associated with “diseases of veins, lymphatic vessels, and lymph nodes, not elsewhere classified” showed enrichment in endothelial cell ATAC-seq peaks. In addition, we see that several skeletal trait GWAS SNPs are enriched in mesenchymal stem cell peaks. Previous work has shown a central role of bone mesenchymal stem cells in osteoblast development (Xian et al. 2012; Pittenger et al. 2019). In addition, SNPs for several corneal traits are also enriched in mesenchymal stem cell peaks, consistent with previously observed enrichment of corneal thickness GWAS SNPs in mesenchymal stem cell/connective tissue cell annotations (Iglesias et al. 2018). Results using rat peaks projected into human coordinates largely mirror the mesenchymal stem cell enrichment findings (Supplemental Fig. S16), suggesting that cross-species clustering identified comparable cell types between the species. These enrichment results must be interpreted with caution due to the small number of rat nuclei with chromatin accessibility data for the more minor cell types, which limits peak calling power.
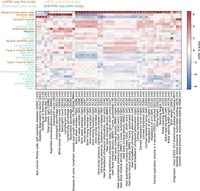
Integration of cell type–specific open chromatin regions with UK Biobank GWAS reveals cell type roles in complex phenotypes. UK Biobank LDSC-partitioned heritability results for traits for which at least one cell type showed significant heritability enrichment after Benjamini–Yekutieli correction (LDSC coefficient P < 0.05) across all cell types and traits. Asterisks denote the significant cell type–trait combinations. One trait name has been shortened to preserve space: “Diseases of veins, lymphatic vessels, and lymph nodes, not elsewhere classified” has been shortened to “Diseases of veins, lymphatic vessels, and lymph nodes.”
Integration of cell type–specific ATAC-seq peaks with T2D GWAS credible sets nominates causal cell types, regulatory elements, and SNPs
It is well-established that T2D GWAS SNPs overlap pancreatic islet/beta cell enhancers (Parker et al. 2013; Gaulton et al. 2015; Varshney et al. 2017; Mahajan et al. 2018; Rai et al. 2020); however, some SNPs may act through other T2D-relevant tissues, such as muscle, adipose, or liver. We therefore used LDSC to perform a partitioned heritability analysis for T2D-associated SNPs (Mahajan et al. 2018) in each of the muscle cell types as well as in beta cell ATAC-seq peaks, adipose ATAC-seq peaks, and liver DNase I hypersensitive sites (see Methods; Fig. 4A). When modeling each cell type separately (adjusting for the cell type–agnostic LDSC baseline annotations and common open chromatin regions), we found significant enrichment (after Bonferroni correction for 40 tests) in type I muscle fibers and beta cells, though when modeling all cell types in a single joint model, only beta cell open chromatin regions showed significant enrichment. We performed a similar analysis on GWAS SNPs for a T2D-related trait, fasting insulin (Fig. 4A; Manning et al. 2012). For fasting insulin, we found significant enrichment in mesenchymal stem cells, smooth muscle, and bulk adipose when modeling each cell type individually, but only adipose showed significant enrichment when modeling all cell types jointly. For fasting insulin, we note that the small sample size of that GWAS means the analysis was likely underpowered, leaving open the possibility that other cell types will show significant enrichment when GWAS with larger sample sizes are available. We also note that the adipose open chromatin regions are derived from bulk tissue open chromatin profiling; it is therefore possible that at least some of the signal from adipose is being driven by cell types shared between our muscle samples and adipose tissue, such as mesenchymal stem cells. This is an area for further exploration when single-nucleus data from adipose is available.
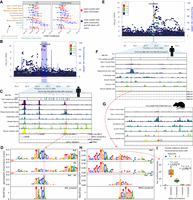
Integration of cell type–specific ATAC-seq peaks with T2D GWAS nominates causal cell types, regulatory elements, and SNPs. (A) LDSC-partitioned heritability results for T2D (BMI-unadjusted) and fasting insulin GWAS (BMI-adjusted), using human peak calls. For each cell type, one model was run adjusting for cell type–agnostic annotations from the LDSC baseline model and common open chromatin regions. Asterisks represent Bonferroni significance (P < 0.05 after adjusting for 40 tests). (B) LocusZoom plot for ITPR2 (T2D). (C) T2D credible set near the ITPR2 gene, consisting of 22 SNPs. Only SNP rs7132434 (highlighted in red) overlaps a peak call in any of the muscle cell types. (D) gkmexplain importance scores for the ref and alt allele (top two rows) and the difference between the ref and alt importance scores (third row); the G allele disrupts an AP-1 motif (bottom row). (E) LocusZoom plot for ARL15 locus (T2D). (F) T2D credible set SNPs near the ARL15 gene. The three SNPs represent the three-SNP credible set discussed in the text. One of these SNPs (rs702634; highlighted in red) overlaps a mesenchymal stem cell–specific peak. (G) Projecting the SNP highlighted in F, rs702634, into the rat genome (projected SNP position indicated by the red vertical line) shows the corresponding region has open chromatin in rat mesenchymal stem cells. (H) gkmexplain importance scores for the ref and alt alleles (top two rows), the difference between them (third row), and a MEF2 motif disrupted by the SNP. (I) Luciferase assay using a construct containing either allele of SNP rs702634 in human adipose-derived mesenchymal stem cells. Each point represents one clone; the experiment was performed twice, once on two different days (“Replicate”). P-values computed using a two-sided unpaired t-test. Copyright disclosure for rat cartoon: Rat by Francisca Arévalo from the Noun Project (https://thenounproject.com/search/?q=rat&i=15130).
We performed similar GWAS enrichments using the rat muscle cell type peaks projected into human coordinates (Supplemental Fig. S17). For T2D, we found muscle fiber types, mesenchymal stem cells, and beta cells were significantly enriched after Bonferroni correction, but as with human muscle cell types, only the beta cell enrichment persisted in a joint model with all cell types. For fasting insulin, no rat muscle cell types showed enrichment after Bonferroni correction, and significant enrichment was only observed for bulk adipose ATAC-seq peaks (Supplemental Fig. S17), again mirroring results using human peak calls.
Although none of our cell types showed significant enrichment in 10-cell-type models after Bonferroni correction, it is still possible that some T2D GWAS loci act through muscle cell types or cell types shared between muscle and other tissues such as adipose. There are a substantial number of T2D GWAS credible sets that show no overlap with pancreatic islet functional annotations (Mahajan et al. 2018). We therefore overlapped 380 previously published T2D GWAS signals with 99% genetic credible set SNPs (Mahajan et al. 2018) with our snATAC-seq peaks to nominate SNPs that may be acting through the muscle cell types, including those that are expected to be shared with other T2D-relevant tissues (Supplemental Table S7).
One locus highlighted by our data is the ITPR2 locus on Chromosome 12 (Fig. 4B). This locus contains 22 credible set SNPs, none with a particularly high posterior probability of association (PPA) in the DIAMANTE genetic fine-mapping (maximum across all credible set SNPs = 0.06). Only one SNP (rs7132434; PPA = 0.042) overlaps any of our muscle cell type peak calls (Fig. 4C). This SNP is in a large mesenchymal stem cell ATAC-seq peak and also overlaps peak calls in smooth muscle and immune cells, though the chromatin accessibility signal in those cell types is lower in our data. The SNP also overlaps a peak call in a subset of adipose and islet samples (Supplemental Fig. S18).
To predict allelic effects on chromatin accessibility at this SNP in our cell types, we trained a gapped k-mer support vector machine model (gkm-SVM) (Ghandi et al. 2014; Lee 2016) to detect k-mers associated with increased or decreased chromatin accessibility using the top ATAC-seq peaks for each of our cell types and then ran deltaSVM (Lee et al. 2015). DeltaSVM predicts a SNP's effect by comparing the gkm-SVM inferred k-mer weights for k-mers created by the reference versus the alternative allele; we transformed the deltaSVM score to a z-score based on the distribution of the predicted impacts of all autosomal 1000 Genomes SNPs (The 1000 Genomes Project Consortium 2015). We validated our models using held-out test data (Supplemental Table S8) and by comparing deltaSVM scores to ATAC-seq allelic bias observed in our data set, where we found a high level of consistency (92% of SNPs had concordant allelic directional effects; binomial P = 5.12 × 10−44) (Supplemental Fig. S19). We found that this SNP had a large deltaSVM z-score in several of the muscle cell types (absolute z-score = 4.06 in mesenchymal stem cells; the T2D risk allele, A, is predicted to result in greater chromatin accessibility) (Supplemental Fig. S20). No other SNP in the credible set had as great a deltaSVM score in any of the cell types (Supplemental Fig. S20). We also attempted to interpret how each allele of the SNP affects the gkm-SVM model's score for the sequence using the gkmexplain software package, which scores the importance of each base in a sequence to the gkm-SVM model score for the sequence (Shrikumar et al. 2019). We ran gkmexplain on the sequence surrounding the SNP in the presence of either the reference or the alternative allele and found the gkmexplain importance scores for the sequence in the presence of the risk allele resembled an AP-1 motif (Fig. 4D; Kheradpour and Kellis 2014). A literature search revealed that the element underlying this SNP has been validated for enhancer activity using a luciferase assay (in the 786-O cell line), and the risk allele showed preferential binding of the AP-1 transcription factor in an EMSA assay in the same study and cell line (Bigot et al. 2016), consistent with our findings. AP-1 subunits are expressed in mesenchymal stem cells in our data (Supplemental Fig. S21). We note that this SNP is also a 95% credible set SNP for waist-hip ratio (one of eight SNPs in the credible set) (Liu et al. 2014). We therefore hypothesize that rs7132434 is the causal SNP at this locus and that it may be acting through mesenchymal stem cells.
To determine which gene or genes might be regulated by the regulatory element containing the SNP, we used three methods to determine potential target genes. First, we examined public Hi-C data from 21 cell types (Schmitt et al. 2016). Second, we used CICERO (Pliner et al. 2018) to determine if any ATAC-seq peaks near the TSS of nearby genes were co-accessible with the regulatory element ATAC-seq peak. Third, we used human nuclei from the multiome library to test for significant correlation between the accessibility of the regulatory element and the expression of nearby genes (using the Signac software package [Stuart et al. 2019, 2021]). In mesenchymal stem cell Hi-C data, the genomic region containing the regulatory element interacted with genomic regions containing the TSS of six genes within 1 Mb (RNA5SP354, ITPR2, SSPN, BHLHE41, ASUN, and LMNTD1) (Supplemental Fig. S22). All but one of these genes (RNA5SP354) have nonzero RNA counts in several muscle cell types, including mesenchymal stem cells (Supplemental Fig. S23). The interactions with most of these TSSs was also observed in several other cell types (Supplemental Fig. S22). Using CICERO, we found that three genes had promoter peaks that showed co-accessibility score > 0.05 (Rai et al. 2020) with the regulatory element peak in mesenchymal stem cells (ITPR2, RASSF8, STK38L). ITPR2 and RASSF8 promoter peaks showed co-accessibility > 0.05 in smooth muscle and immune cells as well (Supplemental Fig. S24). In the multiome library, we found little evidence of correlation between the peak and the expression of any nearby gene (Supplemental Fig. S25). The discordance between the three methods at this locus is not necessarily surprising—each method is measuring different outcomes (frequency of contact in 3D space; peak-peak ATAC signal correlation; peak-gene correlation)—but makes it difficult to confidently nominate a target gene at this locus.
A second region highlighted by our data is an intronic locus in the ARL15 gene (Fig. 4E). The DIAMANTE genetic fine-mapping narrowed the list of potentially causal SNPs at this locus to three (two other, larger DIAMANTE genetic fine-mapping credible sets are also annotated to ARL15). Variants in this three-SNP credible set are statistically associated with fasting insulin (Mahajan et al. 2014), and more broadly variants in or near ARL15 associate with metabolic traits including adiponectin, HDL cholesterol levels, and BMI (Richards et al. 2009; Teslovich et al. 2010; Mahajan et al. 2014; Udler et al. 2018), suggesting that the locus may affect T2D risk not through islets but through adipose or a related cell type. None of the SNPs overlap with any of ENCODE's 1.3 million candidate cis-regulatory elements (Moore et al. 2020) or any of the ∼3.6 million DNase I hypersensitive sites (DHSs) annotated in (Meuleman et al. 2020); however, in our data we find that one of the SNPs (rs702634) is in the center of a mesenchymal stem cell–specific ATAC-seq peak (Fig. 4F), and a mesenchymal stem cell peak is likewise present in the corresponding position in the rat genome (Fig. 4G), indicating that this is a highly cell type–specific regulatory element that has been conserved across species. The DIAMANTE genetic fine-mapping assigned this SNP a probability of 0.48 of being the causal SNP at this locus, higher than either of the other two SNPs (0.33 and 0.19, respectively). We examined publicly available beta cell (n = 1), islet (n = 10) (Rai et al. 2020), and adipose (n = 3) (Cannon et al. 2019) ATAC-seq data to see if hints of this peak are present in these T2D-relevant cell types. No convincing signal appears to be present in beta cell or islet data; a weak increase in signal at that SNP is evident in the adipose samples and a peak is called (Supplemental Fig. S26). As mesenchymal stem cells are one component of adipose tissue, it is possible that the weak signal in adipose is due to mesenchymal stem cell populations within adipose; this is one area for follow-up when adipose single-nucleus ATAC-seq data is available. The absolute deltaSVM z-score in mesenchymal stem cells for this SNP was 1.37, indicating that it does not have a particularly large impact on predicted chromatin accessibility (Supplemental Fig. S27); however, the risk allele is predicted to disrupt a MEF2 motif (Grant et al. 2011; Kheradpour and Kellis 2014), and we found the change in gkmexplain importance scores between the reference and alternative allele showed similarity to this motif (Fig. 4H). MEF2 TF family members are expressed in mesenchymal stem cells in our data (Supplemental Fig. S28). To determine if the SNP might alter regulatory element activity, we tested the regulatory element with each allele in a luciferase assay in human adipose-derived mesenchymal stem cells (hAMSCs) and found that the G (nonrisk) allele resulted in significantly greater luciferase expression than the A (risk) allele (fold change = 2.6 and P = 4.57 × 10−6 in the forward orientation; fold change = 1.4 and P = 3.48 × 10−4 in the reverse direction), consistent with the expected allelic directional effect based on the MEF2 PWM (Fig. 4I; Supplemental Fig. S29) and assuming that MEF would act as an activator. We found that the allele-specific effects remained throughout differentiation to adipocytes (Supplemental Fig. S29). This data is consistent with a model in which rs702634 is the causal SNP and acts through mesenchymal stem cells or a closely related cell type.
To determine which gene or genes might be regulated by the regulatory element, we again applied three methods (Hi-C data, peak-peak co-accessibility via CICERO, and peak-gene expression correlation) to probe the relationship between the ATAC-seq peak and nearby genes. Hi-C data showed no SNP-gene TSS connections for any genes within 1 Mb in mesenchymal stem cells; however, connections between the SNP-containing genomic bin and the FST gene TSS were observed in two other cell types (Supplemental Fig. S30). An ATAC-seq peak near the FST promoter showed co-accessibility > 0.05 with the regulatory element ATAC-seq peak (an ATAC-seq peak at the NDUFS4 promoter showed co-accessibility > 0.05 as well) (Supplemental Fig. S31). FST is expressed in mesenchymal stem cells (Supplemental Fig. S32), and FST gene expression showed relatively high correlation with the element accessibility in the multiome single-nucleus library (z-score = 2.65 relative to control ATAC-seq peak accessibility) (Supplemental Fig. S33). These results suggest that FST may be a promising target gene candidate for the regulatory element at this locus. The FST gene encodes follistatin, which binds and antagonizes members of the transforming growth factor beta (TGFB) family (Michel et al. 1993; Fainsod et al. 1997; Amthor et al. 2004). FST has a well-established role in driving muscle growth (Amthor et al. 2004; Kota et al. 2009) but is involved in other biological processes as well (Patel 1998; Bilezikjian et al. 2004; Tang et al. 2020). Knockout of FST is associated with decreased adipogenesis (Braga et al. 2014; Chen et al. 2020) and overexpression of FST promotes white adipose tissue browning (Braga et al. 2014; Tang et al. 2020). Therefore, in addition to the genomic evidence suggesting FST is the target gene at this locus, FST’s established roles in T2D-relevant cell types and mesenchymal stem cell–related processes (adipogenesis) render it a biologically compelling candidate.
Discussion
Here, we present snATAC-seq and snRNA-seq for human skeletal muscle and snATAC-seq for rat skeletal muscle, which we use to map the transcriptomes and chromatin accessibility of cell types present in skeletal muscle samples. The cell types identified are consistent with known biology and with previous studies of human (Rubenstein et al. 2020) and mouse (The Tabula Muris Consortium 2018; Dell'Orso et al. 2019; Giordani et al. 2019) skeletal muscle tissue. However, our use of single-nucleus rather than single-cell techniques allows us to capture muscle fiber nuclei, cell types missing from previously published snRNA-seq data sets. To our knowledge, this is the first published snATAC-seq data set for human and rat skeletal muscle tissue. We therefore anticipate that this data set will be useful in nominating causal GWAS SNPs and affected genes and demonstrate this by integrating the data with previously published T2D GWAS credible sets, highlighting potentially causal SNPs at the ARL15 and ITPR2 loci. At the ARL15 locus, we also demonstrate that rat mesenchymal stem cells have an orthologous ATAC-seq peak. This (1) provides additional evidence that this peak is real (not a false peak call), (2) reinforces the cell type–specificity of this peak, and (3) suggests that rats may be a suitable model in which to study this locus. Rats are a common model organism for skeletal muscle biology (Chaillou 2018; Christian and Benian 2020), and we expect that the rat single-nucleus data will be a valuable resource for those utilizing rat muscle in their research. Just as the ATAC-seq signal at the ARL15 locus suggests rats would be an ideal model organism for studying the regulatory mechanisms at this locus, researchers investigating other loci can use the data generated here to help determine which human loci might be reasonably studied in rat.
Additionally, we explore the effect of two technical parameters on snRNA-seq and snATAC-seq results. First, we find that FANS (using DRAQ7 DNA staining) prior to Tn5 tagmentation substantially alters snATAC-seq results. Though the stereotypical ATAC-seq fragment length distribution is observed, signal-to-noise (as measured by TSS enrichment and fraction of reads in peaks, as well as by visual inspection) appears to decrease substantially relative to non-FANS libraries. We note that the effect of FANS (nucleus sorting) may differ from that of FACS (cell sorting) carried out prior to nuclear isolation. 10x Genomics, the producer of the single-nucleus platforms utilized here, states that FANS is compatible with successful snATAC-seq, provided certain guidelines and particular dyes are used (others may disrupt chromatin structure) (10x Genomics 2020). To our knowledge the data presented here represent the first published direct comparison between FANS and non-FANS snATAC-seq libraries and suggest that FANS using a DRAQ7 DNA stain prior to tagmentation, at least with the protocol tested here, compromises snATAC-seq data quality. It is possible that this could be overcome by using a different dye.
In contrast to snATAC-seq, snRNA-seq results appear to be substantially less sensitive to FANS—the pseudobulk gene expression from FANS libraries correlates strongly with that from non-FANS libraries—suggesting that chromatin is more sensitive to FANS than is RNA. This finding is in line with previous work successfully utilizing FANS prior to snRNA-seq (Denisenko et al. 2020). We also observed higher nucleus yield in our FANS snRNA-seq libraries than our non-FANS libraries. There are several potential explanations for this. One is that the nuclei counting step that necessarily precedes loading of the 10x platform may be sensitive to debris. If greater amounts of debris are observed in non-FANS libraries, nucleus concentration may be systematically overestimated in non-FANS libraries, resulting in more nuclei actually being loaded onto the 10x platform from FANS libraries. Although not mutually exclusive, FANS may also decrease the amount of debris being loaded into the 10x platform and thereby improve nucleus capture.
We found that snATAC-seq and snRNA-seq results were highly consistent at different loading concentrations. Previous work utilizing customized protocols and indexing strategies has demonstrated quality scRNA-seq/snRNA-seq results can be obtained at loading concentrations exceeding standard 10x recommendations (Stoeckius et al. 2018; Gaublomme et al. 2019; Datlinger et al. 2021); our data suggest that snATAC-seq results are relatively robust to loading concentration as well. One clear caveat is that this may change as the loading concentration is further increased. It is also important to note that the actual number of nuclei loaded may differ from the estimated 20k or 40k nuclei. The number of nuclei passing QC for these libraries was approximately an order of magnitude lower than the number of nuclei ostensibly loaded (e.g., 3839 and 2118 nuclei for snRNA-seq). There are several steps between nuclei isolation and computational quality control that might explain this. First, the initial nucleus count may have been incorrect. As discussed above, it is possible that debris in the input preparation makes nucleus counting less accurate, in which case our cited values may not reflect the true values. Second, some nuclei loaded onto the Chromium platform may not be captured in gel beads. Third, some of the captured nuclei may be unhealthy or may receive an insufficient dose of the enzymes necessary for successful library construction (e.g., in the case of snATAC-seq, some captured nuclei may receive an inadequate dose of Tn5). Previous work on the 10x Genomics scRNA-seq platform suggests that ∼50% of the loaded nuclei may be captured and undergo successful library construction (Zheng et al. 2017). Last, some quality nuclei may simply be filtered out computationally, for example, as likely doublets; however, the barcode rank plots (Supplemental Fig. S34) suggest that the number of gel beads with above-background numbers of UMIs/reads was simply substantially less than the number of nuclei loaded.
The GWAS enrichments presented here will be one interesting area to follow up on as more snATAC-seq data are published. Interpretation of the results is complicated by the fact that many tissues share cell types. For example, mesenchymal stem cell–like populations exist in many tissues besides muscle, such as adipose tissue and bone marrow. Taking the fasting insulin enrichments as an example, we found that the enrichment of GWAS SNPs in muscle cell type ATAC-seq peaks disappeared when adipose tissue was included in the enrichment model. However, it is possible that the adipose enrichment is being driven in part by mesenchymal stem cell populations within adipose itself. Direct comparison of snATAC-seq and snRNA-seq profiles from mesenchymal stem cells from a wider array of tissues will help tease apart commonalities and tissue-specific differences in this interesting population.
Methods
Ethics approval and consent to participate
Human samples were approved by the University of Michigan IRB protocol #HUM 000060733. Collection of the rat muscle sample was approved by the University of Michigan Institutional Animal Care and Use Committee.
snATAC-seq and snRNA-seq, FANS versus no FANS experiment
Three separate pieces of tissue were cut from a single human skeletal muscle sample (weighing 60 mg, 50 mg, and 50 mg; sample HSM1, quadriceps femoris muscle group). Nuclei were isolated using a modified version of the ENCODE protocol (Supplemental Protocol S1) (The ENCODE Project Consortium 2012), customized from step 5 onwards to accommodate FANS. In step 5, the nuclei were resuspended in 700 µL of sort buffer (1% BSA, 1 mM EDTA in PBS) and filtered through a 30-µm filter. Three different nuclei isolations were performed and the nuclei suspended in sort buffer were mixed, pooled together, and divided into two groups, one with FANS and one without FANS. FANS nuclei were sorted according to the previously published FANS protocol using DRAQ7 (Preissl et al. 2018). DRAQ7 (0.3 mM from Cell Signaling Technology) was added to the FANS nuclei suspension, at 100-fold dilution to get a final concentration of 3 μM. Nuclei were gently mixed and incubated for 10 min on ice. Nuclei were analyzed in the presence of DRAQ7 and sorted for high DRAQ7 positive signal using Beckman Coulter's Astrios MoFlo. We followed the gating strategy outlined in the FANS protocol (Preissl et al. 2018). The sorted nuclei were collected in a recovery buffer (5% BSA in PBS). The nuclei with and without FANS were spun at 1000g for 15 min at 4°C. The nuclei were resuspended in 100 µL of 1× diluted nuclei buffer and counted in the Countess II FL Automated Cell Counter. The appropriate amount of nuclei were split for snRNA-seq and spun down at 500g for 10 min at 4°C and resuspended in RNA nuclei buffer (1% BSA in PBS containing 0.2 U/μL of RNase inhibitor). The nuclei at appropriate concentration for snATAC-seq and snRNA-seq were submitted to the Advanced Genomics core for all the snATAC-seq and snRNA-seq processing on the 10x Genomics Chromium platform (v. 3.1 chemistry for snRNA-seq). For each modality nuclei were loaded at 15.4k nuclei/well.
snATAC-seq and snRNA-seq, loading 20k or 40k nuclei
Two pieces of tissue (weighing 85.3 mg and 85.8 mg) were cut from one human skeletal muscle sample (HSM1) and two tissue pieces (weighing 95.9 mg and 92.6 mg) were cut from a second human skeletal muscle sample (HSM2; quadriceps femoris muscle group). Each of the samples was cut on dry ice using a frozen scalpel to prevent thawing. The samples were pulverized using a CP02 cryoPREP automated dry pulverizer (Covaris 500001). We developed a customized nuclei isolation protocol (Supplemental Protocol S2) derived from the previously published ENCODE protocol (The ENCODE Project Consortium 2012). All four pulverized tissue pieces were mixed and redistributed to perform four different nuclei isolations. The desired concentration of nuclei was achieved by resuspending the appropriate number of nuclei in 1× diluted nuclei buffer for snATAC-seq and RNA nuclei buffer (1% BSA in PBS containing 0.2 U/µL of RNase inhibitor) for snRNA-seq. The nuclei at appropriate concentration for snATAC-seq and snRNA-seq were submitted to the Advanced Genomics core for all the snATAC-seq and snRNA-seq processing on the 10x Genomics Chromium platform (v. 3.1 chemistry for snRNA-seq). For each modality, nuclei were loaded at two different concentrations, 20k nuclei/well and 40k nuclei/well.
snATAC-seq, human and rat mixed library
Tissue from human (49 mg of pulverized human skeletal muscle; sample HSM1) and rat (45 mg of pulverized gastrocnemius samples) were used in this single nuclei ATAC experiment. We used the previously published ENCODE protocol (Supplemental Protocol S1) (The ENCODE Project Consortium 2012) to isolate nuclei. After isolating nuclei from each sample (species) individually, the nuclei were mixed in equal proportions. The desired concentration of nuclei was achieved by resuspending the appropriate number of nuclei in 1× diluted nuclei buffer for snATAC-seq. The nuclei at the appropriate concentration for snATAC were submitted to the University of Michigan Advanced Genomics core for all the snATAC-seq processing on the 10x Genomics Chromium platform; 15.4k nuclei were loaded into a single well.
Bulk ATAC-seq
Two tissue pieces weighing 99.4 mg and 80.7 mg were cut from one human skeletal muscle sample (HSM1) and two pieces weighing 67.6 mg and 103.5 mg were cut from a second human skeletal muscle sample (HSM2). Each of the samples was cut on dry ice using a frozen scalpel to prevent thawing. The samples were pulverized using a CP02 cryoPREP automated dry pulverizer (Covaris 500001). For bulk ATAC-seq, we followed the nuclei isolation protocol outlined in Supplemental Protocol S2, except in the final step the nuclei were resuspended in 250 μL of 1% BSA in PBS. The nuclei were counted in a Countess II FL Automated Cell Counter, and the appropriate volume of the suspension for 50k nuclei was spun down and used for the downstream transposition reaction (a modified version of the ENCODE protocol; Supplemental Protocol S3) (The ENCODE Project Consortium 2012).
Multiome library
Tissue from human (74 mg of pulverized human skeletal muscle; sample HSM2) and rat (79 mg of pulverized gastrocnemius samples) were used in the multiome experiment. The pulverized tissues were mixed together prior to isolation. For this experiment, we developed a customized protocol based on those recommended by 10x Genomics (https://www.10xgenomics.com/resources/demonstrated-protocols/) and on the previously developed Supplemental Protocol S2 to isolate nuclei (Supplemental Protocol S4). The desired concentration of nuclei was achieved by resuspending the appropriate number of nuclei in 1× diluted nuclei buffer for joint (on the same nucleus) snATAC-seq and snRNA-seq. The nuclei at appropriate concentration for joint snATAC-seq and snRNA-seq were submitted to the Advanced Genomics core for processing on the 10x Genomics Chromium platform; 16.1k nuclei were loaded into a single well.
Processing of muscle bulk ATAC-seq data
Adapters were trimmed using cta (v. 0.1.2; https://github.com/ParkerLab/cta). Reads were mapped to hg19 using BWA-MEM (-I 200,200,5000 -M; v. 0.7.15-r1140) (Li and Durbin 2009). Duplicates were marked using Picard MarkDuplicates (v. 2.21.3; https://broadinstitute.github.io/picard/). We used SAMtools to filter to high-quality, properly-paired autosomal read pairs (-f 3 -F 4 -F 8 -F 256 -F 1024 -F 2048 -q 30; v. 1.9 using htslib v. 1.9) (Li et al. 2009). To call peaks, we used BEDTools bamtobed to convert to a BED file (v. 2.27.1) and then used that file as input to MACS2 callpeak (‐‐nomodel ‐‐shift -100 ‐‐seed 762873 ‐‐extsize 200 ‐‐broad ‐‐keep-dup all ‐‐SPMR; v. 2.1.1.20160309) (Zhang et al. 2008; Quinlan 2014). To visualize the signal, we converted the bedGraph files output by MACS2 to bigWig files using bedGraphToBigWig (v. 4) (Kent et al. 2010).
Processing of snATAC-seq data
Adapters were trimmed using cta. We used a custom Python script, available in the GitHub repository, for barcode correction (see Supplemental Methods). Reads were mapped using BWA-MEM with flags “-I 200,200,5000 –M”. We used Picard MarkDuplicates to mark duplicates and filtered to high-quality, nonduplicate autosomal read pairs using SAMtools view with flags “-f 3 -F 4 -F 8 -F 256 -F 1024 -F 2048 -q 30”. Quality control metrics were gathered on a per-nucleus basis using ataqv (v. 1.1.1) on the BAM file with duplicates marked. In the case of mixed rat and human libraries, all reads were mapped to the hg19 and rn6 genomes separately, and then a nucleus was assigned as either rat or human by counting the number of high-quality, nonduplicate autosomal reads after mapping to either genome. If (# reads mapping to genome of species A)/(# reads mapping to genome of species A + # reads mapping to genome of species B) was greater than 0.87, then the nucleus was assigned to species A. If a nucleus did not meet this threshold for either species, it was dropped.
For the two snATAC-seq libraries that contained a mix of nuclei from the two human individuals, we assigned nuclei to biological samples and determined doublets using demuxlet (Kang et al. 2018) (see Supplemental Methods).
When comparing aggregate snATAC-seq signal to bulk ATAC-seq signal (Fig. 1), we eliminated sequencing reads corresponding to nucleus barcodes that couldn't be matched to the 10x barcode whitelist but otherwise processed it as bulk ATAC-seq data (i.e., marking duplicates ignoring cell-level information, and not filtering to quality nuclei).
To select quality nuclei from each library, we selected nuclei (barcodes) meeting the thresholds in Supplemental Table S1. In addition to setting a threshold for minimum fragments (to filter out barcodes that only capture ambient DNA fragments), we set a threshold for maximum fragments, because barcodes with very high fragment counts may be enriched for doublets (Rai et al. 2020). We also set a threshold for minimum TSS enrichment (because ATAC-seq signal for healthy nuclei is expected to be enriched near TSSs) (Buenrostro et al. 2013, 2015; Rai et al. 2020), and we filtered out barcodes that showed an unexpectedly large fraction of reads coming from a single autosome (see Orchard et al. 2020).
Processing of snRNA-seq data
snRNA-seq data was processed using starSOLO (STAR v. 2.7.3a, with GENCODE v. 19 annotation; options ‐‐soloUMIfiltering MultiGeneUMI ‐‐soloCBmatchWLtype 1 MM_multi_pseudocounts ‐‐soloCellFilter None), which outputs the count matrices needed for most of the analyses (Dobin et al. 2013). To select quality nuclei from each library, we selected nuclei meeting the thresholds in Supplemental Table S2 (we set a threshold for minimum UMIs to filter out barcodes that only capture ambient RNA; a threshold for maximum fragments, because barcodes with very high UMI counts may be enriched for doublets; and a threshold for maximum mitochondrial contamination, because barcodes with quality nuclei and low ambient RNA should show reduced mitochondrial contamination) (Alvarez et al. 2020). For the snRNA-seq libraries containing nuclei from two human individuals, nuclei were assigned to samples (and doublets called) using demuxlet (see Supplemental Methods). Prior to clustering and downstream analysis, we used DecontX (Yang et al. 2020) to adjust the nucleus × gene expression count matrices for ambient RNA (see Supplemental Methods).
Processing of single-nucleus multiome data
The reads from the chromatin accessibility component of the multiome data were trimmed, mapped (to both the hg19 and rn6 reference genomes separately), and filtered (including duplicate marking) as described above for snATAC-seq libraries.
The reads from the gene expression component of the multiome data were processed using starSOLO (STAR v. 2.7.3a). All reads were mapped to the hg19 and rn6 genomes separately. For rn6, we used NCBI Rattus norvegicus Annotation Release 106 (for hg19, we used the same reference as for snRNA-seq).
To select quality nuclei from the library, we first assigned nuclei to the human or rat sample by counting the number of high-quality, nonduplicate autosomal ATAC reads after mapping to either genome (“HQAA” statistic from ataqv) (Orchard et al. 2020). If (# reads mapping to genome of species A)/(# reads mapping to genome of species A + # reads mapping to genome of species B) was greater than 0.85, then the nucleus was assigned to species A. If a nucleus did not meet this threshold for either species, it was dropped. Next, we filtered out nuclei that did not meet any of the following thresholds: at least 20,000 ATAC reads pass filtering; at least 500 RNA UMIs (based on starSOLO counts matrix); minimum TSS enrichment of two; no more than 15% of ATAC reads derived from any single autosome; and no more than 2% of RNA UMIs derived from mitochondrial genes (Supplemental Fig. S11).
The fraction of species-unique ATAC fragments or RNA UMIs derived from human (i.e., the number of fragments mapping to human but not rat, divided by the total number of fragments mapping to rat or human but not both) in each pass-QC nucleus is shown in Supplemental Figure S10 (bottom right panel). We note that many points are closer to 0.5 along the RNA axis (y-axis) than along the ATAC axis (x-axis). A similar pattern is observable in previously published joint chromatin accessibility and gene expression data from another platform (Ma et al. 2020). It is possible that this could be explained by the sources of ambient contamination being different for RNA than for DNA.
Specifically for the DropletUtils panel in Supplemental Figure S10 (top right panel), we reprocessed the raw RNA data using starSOLO to map to a chimeric reference (concatenating the human and rat references used when mapping to each species individually), resulting in a single barcode × gene UMI matrix. We then used DropletUtils’ emptyDrops function (Lun et al. 2019) (default parameters except niters = 1 × 105) to determine whether each barcode represented a nucleus by comparing the UMI counts for each barcode to the ambient contamination estimated from barcodes with less than 100 UMIs. We used a strict FDR threshold of 1 × 10−5 for calling nuclei.
Prior to clustering and downstream analysis, we used DecontX (Yang et al. 2020) to adjust the nucleus × gene expression count matrices for ambient RNA (see Supplemental Methods).
Clustering with Seurat
All pass-qc nuclei were clustered using Seurat (Butler et al. 2018; Stuart et al. 2019) (v. 3.9.9.9010, in R v. 3.6.3) (R Core Team 2021). Multiome library nuclei were clustered using RNA UMI counts. First, we integrated RNA from each individual (three matrices; two human samples and one rat sample), identifying the top 2000 variable features for each individual and then finding integration anchors and integrating (dims = 1:20). After scaling and PCA, we found neighbors (k = 20, using PCs 1–10, except PC9 which otherwise drove a modality-specific cluster) and clustered using the Louvain algorithm (resolution = 0.05). Next, we integrated ATAC from each individual (three matrices as for the RNA data). As ATAC peak calls on the aggregate snATAC-seq libraries would be heavily biased toward muscle fiber type peaks and against peaks specific to minor cell populations, we worked only with per-gene scores calculated for each nucleus as the number of fragments overlapping with each gene's promoter/gene body using BEDTools intersect. Gene promoter/body was calculated based on NCBI annotation GTF files (NCBI Rattus norvegicus Annotation Release 106 and Homo sapiens Updated Annotation Release 105.20190906), filtered to include only protein-coding/lncRNA genes with source “BestRefSeq”/BestRefSeq%2CGnomon’/’Curated Genomic’. Genes assigned to multiple chromosomes/strands were excluded, and then the regions for each gene were merged to get the gene body. Promoters were taken as the 3 kb upstream of the TSS; after this, genes represented by multiple noncontiguous genomic stretches were excluded. Using these per-nucleus, per-gene scores as input to Seurat, we normalized the matrices, integrated them using the top 2000 variable genes from the integrated RNA data (setting dims = 2:20 for FindIntegrationAnchors and IntegrateData), and scaled the resulting integrated ATAC data. Next, we transferred cluster labels from the RNA to the ATAC using the FindTransferAnchors and TransferData functions (FindTransferAnchors reduction = CCA, using the top 2000 RNA variable features; TransferData anchor weighting using the top 20 PCs from the integrated ATAC data). In order to co-embed the RNA and ATAC in the same UMAP, we used the TransferData function to impute RNA values (for the top 2000 RNA variable genes) in the ATAC nuclei (using same TransferData parameters as used to transfer cluster labels), merged the ATAC and RNA nuclei, and created the UMAP using the top 20 PCs on the centered data. Lastly, as recommended in the Seurat RNA-ATAC integration vignettes, we removed ATAC nuclei with low-confidence cluster assignments (prediction.score.max < 0.7).
Per-cluster ATAC-seq peak calling
The filtered reads from all snATAC-seq nuclei in each cluster were merged using SAMtools merge. Peaks were called and bigWig files produced as described for the bulk ATAC-seq data. Peak files were filtered against blacklist files available from http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeMapability/wgEncodeDacMapabilityConsensusExcludable.bed.gz and http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeMapability/wgEncodeDukeMapabilityRegionsExcludable.bed.gz (hg19) (The ENCODE Project Consortium 2012) and GitHub (https://github.com/shwetaramdas/maskfiles/blob/master/rataccessibleregionsmaskfiles/strains_intersect.bed) for rn6 (Ramdas et al. 2019).
For analysis of rat peak overlap with human GWAS data, rat peaks were projected into the human genome using bnMapper (v. 0.8.6) and the chain file at http://hgdownload.cse.ucsc.edu/goldenpath/rn6/liftOver/rn6ToHg19.over.chain.gz.
Roadmap enhancer regression
We called peaks on the aggregate of the nuclei in each cluster and then took the union of peaks across all clusters to generate a master peak list. We then used logistic regression to model, for each cluster and each Roadmap Epigenomics cell type in the Roadmap 15-state ChromHMM model, the accessibility of each TSS-distal master peak (>5 kb from a RefSeq TSS) in that cluster as a function of the posterior probability that that master peak is an enhancer in that Roadmap cell type according to the Roadmap ChromHMM model (Roadmap Epigenomics Consortium et al. 2015). Because the posteriors are given in 200-bp windows, and there are also three different enhancer states (“Genic enhancers,” “Enhancers,” and “Bivalent Enhancer”), multiple windows overlap with each master peak—the posterior for the master peak is therefore taken as the maximum of the 200-bp window posteriors, across all three of the enhancer states. The model coefficient was used as the (unnormalized) score for that Roadmap cell type in that cluster, and the normalized score was simply the score for that Roadmap cell type in that cluster divided by the max score across all cell types for that cluster.
For rat peaks, in addition to removing master peaks near TSS in rat coordinates, we additionally removed master peaks that were within 5 kb of a TSS after projecting into human coordinates.
Motif scores
Motif scores were generated using chromVAR (v. 1.8.0) (Schep et al. 2017) in R v. 3.6.0 (see Supplemental Methods).
UK Biobank GWAS enrichment
We downloaded UK Biobank GWAS summary statistics made available by the Benjamin Neale lab (Sudlow et al. 2015) (see Supplemental Methods).
The LDSC software package (v. 1.0.1) includes a “baseline” model with 59 categories derived from 28 genomic annotations (Finucane et al. 2015; Gazal et al. 2017). Many of these annotations are cell type–agnostic; for example, a SNP's minor allele frequency does not change between cell types. However, other annotations in the baseline model are not cell type–agnostic; for example, the FANTOM5 enhancer annotation is derived from experiments performed on a range of different cell types and may change substantially if the cell types used to create the annotation were to change. When performing the UK Biobank GWAS enrichments, we utilized the cell type–agnostic annotations from the LDCS baseline model (Supplemental Table S9). In order to reduce the likelihood of model misspecification, we then added common open chromatin regions and open chromatin regions from a range of cell types. Specifically, we added (1) the ATAC-seq peaks from all seven of our snATAC-seq cell types, (2) beta cell ATAC-seq peaks, (3) adipose ATAC-seq peaks, and (4) DNase-seq peaks derived from 23 additional tissues/organs (see Supplemental Methods). The various annotation files (regression weights, frequencies, etc.) required for running LDSC were downloaded from https://data.broadinstitute.org/alkesgroup/LDSCORE. LD scores were calculated using the Phase 3 1000 Genomes data, keeping only the HapMap3 SNPs as recommended by the LDSC authors and using only SNPs with minimum MAF of 0.01. GWAS summary statistics were prepared for LDSC using the munge_sumstats.py script, with option ‐‐merge-alleles w_hm3.snplist (where w_hm3.snplist is the file in the data download). When running the regression, we required a minimum MAF of 0.05 and utilized the Phase 3 1000 Genomes SNP frequencies/weights.
Hi-C contacts
Hi-C contacts were inferred using the Fit-Hi-C (Ay et al. 2014) results from Schmitt et al. (2016), at a strict FDR threshold of 1 × 10−6 (used throughout that publication). If the genomic bin occupied by the ATAC-seq peak significantly interacted with the bin occupied by any of the TSSs from a given gene, the peak was considered to interact with that gene's promoter. The TSS list was generated from the GENCODE v. 19 annotation, after removal of genes with gene_status = “NOVEL” or with gene_type not amongst (“lncRNA”, “rRNA”, “protein_coding”, “retained_intron”, “processed_transcript”, “non_coding”, “ambiguous_orf”, “lincRNA”, “macro_lncRNA”, “bidirectional_promoter_lncRNA”).
Peak-peak co-accessibility
We ran CICERO (Pliner et al. 2018) (v. 1.4.0; R v. 3.6.1) on the broad peak fragment counts to score peak-peak co-accessibility. CICERO was run once for each cell type. We used UMAP dimensions 1 and 2 (Fig. 2) as the reduced coordinates and set window size to 1.75 Mb. A peak was considered to be a TSS peak for a gene if it overlapped the 5-kb window upstream of that gene's TSS. If multiple TSS peaks were present for a gene, the maximum co-accessibility score was shown in the heat map (Supplemental Figs. S24, S31). The TSS list was generated as described for the Hi-C contact analysis.
Peak accessibility—gene expression correlation in multiome library
We used Signac (Stuart et al. 2021) (v. 1.1.0; R v. 4.0.3) to score the Spearman correlation between fragment counts in ATAC-seq peaks and gene expression in human nuclei in the multiome library. We set EnsDb.Hsapiens.v75 (https://bioconductor.org/packages/EnsDb.Hsapiens.v75) as the annotation and normalized the RNA counts using the SCTransform normalization (Hafemeister and Satija 2019). We required that at least five nuclei be positive for the peak and gene to be included in the test and set the distance threshold to 1.5 Mb.
T2D and fasting insulin GWAS enrichment
We used the T2D (BMI-unadjusted) and fasting insulin (BMI-adjusted) GWAS summary statistics from Mahajan et al. (2018) and Manning et al. (2012), respectively.
Because the cell types relevant to T2D are generally thought to be pancreatic beta cells, adipose, muscle, and liver, we performed enrichments using each of these cell types, common open chromatin, and the cell type–agnostic LDSC baseline annotations. First, for each of these muscle/beta cell/adipose/liver cell types, we ran one model containing the open chromatin from that cell type, the common open chromatin regions, and the cell type–agnostic LDSC baseline annotations. Then, we ran one joint model containing all of those cell types and annotations. LDSC parameters were the same as for the UK Biobank GWAS enrichments.
T2D GWAS locus genome browser screenshots and peak overlaps
All signal tracks in the genome browser were created by converting the normalized bedGraph files output by MACS2 to bigWig files using bedGraphToBigWig (v. 4) (Kent et al. 2010).
Processing and provenance of adipose ATAC-seq and beta cell ATAC-seq is described in the Supplemental Methods. The 10 bulk islet libraries were from Rai et al. (2020). These libraries were processed as described in that manuscript, except we used the 10% FDR peak set from peak calling on the unsubsampled libraries.
Allelic bias analysis
Prior to the allelic bias analysis, we reprocessed snATAC-seq reads using the WASP software package (commit 36c0e5f8b5) (van de Geijn et al. 2015) to reduce reference allele mapping bias (see Supplemental Methods). To determine if a SNP showed allelic bias, we used a two-sided binomial test with expected fraction of reads derived from the ref allele = 0.5. We tested only SNPs that had coverage of at least 15 and at least one count for each allele. Multiple testing correction (Benjamini–Hochberg correction [Benjamini and Hochberg 1995]) was performed within each cell type.
Predicting SNP regulatory impact
We used the lsgkm package modified by the Kundaje lab with gkmexplain (https://github.com/kundajelab/lsgkm; commit c3758d5bee7) (Ghandi et al. 2014; Lee 2016; Shrikumar et al. 2019). For each cell type, we took the 150 bp on either side of the summits of the top 40,000 narrowPeaks (by P-value) as the positive sequences for gkmSVM. To generate negative sequences, we took windows across the genome (step size = 200), removed those containing Ns, overlapping hg19 blacklists, overlapping any FDR 10% broadPeaks from that cell type, or having repeat content > 60%, and then for each positive sequence selected a negative sequence with matching GC content and repeat content (repeat content was calculated based on the hg19 simpleRepeat table from the UCSC Genome Browser [Kent et al. 2002; Casper et al. 2018], downloaded on March 29, 2020, which contains simple tandem repeats annotated by Tandem Repeats Finder [Benson 1999]; GC content and repeat content for the negative sequence was required to be within 2% of that of the positive sequence; in the case that no such negative sequence could be found, the positive sequence was dropped from the analysis). We held out 15% of sequences as test data and trained the gkmSVM model on the remaining 85% of sequences, setting l = 10 and k = 6 and using the gkm kernel. The model performance on the test data for each cell type is given in Supplemental Table S8. Using this model and deltaSVM (Lee et al. 2015), we predicted the effect of all autosomal 1000 Genomes phase 3 SNPs (downloaded on May 27, 2015 from ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502) (The 1000 Genomes Project Consortium 2015). For each muscle cell type, deltaSVM scores were converted to z-scores based on the distribution of scores across all SNPs for that cell type. We additionally passed the gkmSVM model to gkmexplain to generate importance scores for sequences containing the ref/alt alleles.
Cell culture and differentiation
Human adipose-derived mesenchymal stem cells purchased from ATCC were grown in a culture medium of DMEM supplemented with 10% FBS (Sigma-Aldrich) at 37°C with 5% CO2 in a humidified incubator. To differentiate hAMSCs into adipocytes, three induction periods were performed over 10 days, as previously described (Gojanovich et al. 2018). Briefly, hAMSCs were seeded in 24-well plates (40,000 cells/well), and at 90% confluency, cells were chemically induced with an adipogenic differentiation medium (ADM) comprised of culture medium and supplemented with 250 nM dexamethasone, 0.5 mM 3-isobutyl-1-methylxanthine, 2 µM rosiglitazone, and 10 µg/mL insulin. Cells were incubated in the ADM cocktail for 3 d, then switched to culture medium supplemented with insulin (10 µg/mL) for 2 d. This induction was repeated once more, with 2 d (days six and seven) of the ADM cocktail and 1 d (day eight) of insulin, followed by two additional days (days nine and ten) of incubation in the ADM cocktail.
Lipid droplets were first visible after day five of induction and grew in size and number as adipogenic differentiation progressed. To differentiate hAMSCs to preadipocytes, cells were induced for 24 h with ADM induction medium. Transfections for both preadipocytes and adipocytes were carried out in culture media containing 10 µg/mL insulin.
Transcriptional reporter assays
To test for allele-specific differences in transcriptional activity, we designed PCR primers (5′-AGCTGGGACTTGATTTGGTG and 5′-AGCGGGTAGCTTTCCTTGAT) to amplify a 647-bp region (hg19 Chr 5: 53,271,139–53,271,785) containing rs702634. Genomic DNA of individuals homozygous for the reference allele was used as a template. The PCR products were cloned into the multiple cloning site of the firefly luciferase reporter vector pGL4.23 (Promega) in both orientations, as previously described (Fogarty et al. 2014). To create the alternate allele constructs at rs702634, the resulting construct was altered using a QuikChange site-directed mutagenesis kit (Agilent Technologies). Isolated clones were confirmed by Sanger sequencing.
One day prior to transfection, human adipose-derived mesenchymal cells (40,000/well) were seeded in 24-well plates. These hAMSCs, in addition to preadipocytes (hAMSCs after 24 h of adipogenic differentiation) and adipocytes (hAMSCs after 11 d of adipogenic differentiation), were transfected in triplicate with Lipofectamine 3000 (Thermo Fisher Scientific). For each allele, four to five independent luciferase constructs were cotransfected with a Renilla internal control reporter vector (phRL-TK, Promega). We incubated the transfected cells at 37°C with 5% CO2 for 30 h, then measured the luciferase activity using the Dual-Luciferase Reporter Assay System (Promega). Firefly luciferase readings were normalized to Renilla luciferase readings, then to the average of readings from two empty pGL4.23 vectors.
Overlap of SNPs and peaks with ENCODE candidate cis-regulatory elements
The set of 1,310,152 candidate cis-regulatory elements in ENCODE's “Registry of Candidate Regulatory Elements” (in hg19 coordinates) were fetched from the ENCODE web portal on April 7, 2020 (Moore et al. 2020).
LocusZoom plots
LocusZoom plots were created for the DIAMANTE T2D GWAS summary statistics with the LocusZoom standalone v. 1.4, using the Nov. 2014 EUR 1000 Genomes data included in the download (‐‐pop EUR ‐‐source 1000G_Nov2014) (Pruim et al. 2010).
MEF2 motif disruption
To determine which motifs were disrupted by SNP rs702634, we scanned the surrounding hg19 DNA sequence, and the same sequence with the alternative SNP allele switched in, for motif hits. The motif scan was performed using FIMO (v. 5.0.4) with a background model calculated from the hg19 reference genome (Grant et al. 2011) and otherwise default parameters. We used the motif library from Kheradpour and Kellis (2014), excluding “*_disc” motifs. The MEF2_known10 motif (displayed in Fig. 4H) received a minimum P-value of 4.51 × 10−5 using the reference sequence; when changing the SNP allele to the alternate allele, no motif was called at the default FIMO P-value threshold of 1 × 10−4 (the minimum P-value for the sequence with the alternative allele was 0.00183).
Data access
Raw sequencing data generated in this study have been submitted to the European Genome-phenome Archive (EGA; https://ega-archive.org/) under study accession number EGAS00001005730. Processed sequencing data, including anonymized BAM files (see Supplemental Methods), have been submitted to Zenodo (doi:10.5281/zenodo.5009200) and to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE178735. Codes used for analyses in this manuscript are available at GitHub (https://github.com/ParkerLab/2021-03-sn-muscle) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grant R01 DK117960 and American Diabetes Association Pathway to Stop Diabetes grant 1-14-INI-07 to S.C.J.P., and National Institutes of Health (NIDDK) grant R01 DK099034 to C.F.B. P.O. was funded by a University of Michigan Rackham Predoctoral Fellowship and grant T32 HG00040 from the National Human Genome Research Institute of the National Institutes of Health. K.L.M. was supported by NIDDK grants R01 DK072193 and R01 DK093757. The funding agencies had no role in the study design, sample collection, data analysis/interpretation, and writing of the manuscript. We thank the University of Michigan Advanced Genomics Core for their assistance in generating the snRNA-seq, snATAC-seq, and multiome libraries, and the University of Michigan Flow Cytometry Core for their help performing FANS. We also thank the Benjamin Neale lab for providing their UK Biobank GWAS and LDSC results to the scientific community, and we thank members of the Parker lab for their helpful feedback.
Author contributions: N.M. generated the bulk ATAC-seq data and performed the nuclear isolations for many single-nucleus data sets. C.V. performed nuclear isolation for the multiome single-nucleus data set. A.V. and V.R. processed existing islet and adipose ATAC-seq data. S.V. performed the luciferase assays, and K.L.M. supervised luciferase reporter studies. J.K. helped set up gkm-SVM models. C.L. helped coordinate production of the single-nucleus data. C.F.B. provided the rat muscle sample, and K.G. provided human muscle samples. P.O. performed all computational processing and analyses of the data not attributed to others and contributed to manuscript writing. S.C.J.P. designed and supervised the study and contributed to manuscript writing. All authors read and approved the manuscript.
Footnotes
-
↵9 Co-first authors
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.268482.120.
- Received July 8, 2020.
- Accepted September 16, 2021.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








