
The Nubeam reference-free approach to analyze metagenomic sequencing reads
- Hang Dai and
- Yongtao Guan
Abstract
We present Nubeam (nucleotide be a matrix) as a novel reference-free approach to analyze short sequencing reads. Nubeam represents nucleotides by matrices, transforms a read into a product of matrices, and assigns numbers to reads based on the product matrix. Nubeam capitalizes on the noncommutative property of matrix multiplication, such that different reads are assigned different numbers and similar reads similar numbers. A sample, which is a collection of reads, becomes a collection of numbers that form an empirical distribution. We demonstrate that the genetic difference between samples can be quantified by the distance between empirical distributions. Nubeam includes the k-mer method as a special case, but unlike the k-mer method, it is convenient for Nubeam to account for GC bias and nucleotide quality. As a reference-free approach, Nubeam avoids reference bias and mapping bias, and can work with organisms without reference genomes. Thus, Nubeam is ideal to analyze data sets from metagenomics whole genome shotgun (WGS) sequencing, where the amount of unmapped reads is substantial. When applied to a WGS sequencing data set to quantify distances between metagenomics samples from various human body habitats, Nubeam recapitulates findings made by mapping-based methods and sheds light on contributions of unmapped reads. Nubeam is also useful in analyzing 16S rRNA sequencing data, which is a more prevalent type of data set in metagenomics studies. In our analysis, Nubeam recapitulated the findings that natural microbiota in mouse gut are resilient under challenges, and Nubeam detected differences in vaginal microbiota between cases of polycystic ovary syndrome and healthy controls.
When identifying variants is not a must and the primary interest is to quantify genetic differences between samples (Ravel et al. 2011; Nayfach and Pollard 2016), it can be beneficial to analyze short sequencing reads without reference genomes. First, it avoids reference bias and mapping bias. Both biases can be mitigated but never overcome because they are intrinsic to the mapping-based approach. Second, it avoids uncertainty related to variant calling, particularly when the sequencing coverage is low. Third, it becomes possible to analyze organisms that have no reference genomes or the reference genomes are incomplete or of low quality.
The prominent reference-free approach is the k-mer method (Jiang et al. 2012; Subramanian and Schwartz 2015; Lu et al. 2017b). Simply put, the k-mer method calculates the frequency of each k-mer (k consecutive nucleotides) in all reads from a sample and computes differences between samples by comparing k-mer frequencies. In practice, however, the k-mer method has several limitations. For example, choosing k can be a challenge—a too small or too large k will make the k-mer frequencies less informative. More importantly, some pairs of k-mers only differ by one nucleotide and other pairs of k-mers differ by k nucleotides, but it is difficult to account for such differences in the k-mer method. Last but not least, how to account for GC bias is an unmet challenge for the k-mer method.
We present a novel method, Nubeam (nucleotide be a matrix), that includes the k-mer method as a special case but with several key advantages. Nubeam can account for nucleotide quality and GC bias, and its computation is efficient. As a reference-free method, Nubeam is particularly suitable to analyze whole genome shotgun (WGS) sequencing data in metagenomics, because it is the norm that a large percent of the shotgun sequencing reads cannot be mapped to reference genomes (Supplemental Fig. S1).
Though recent studies improved mapping rates for gut (Almeida et al. 2019; Nayfach et al. 2019; Pasolli et al. 2019) and other well-studied human body habitats (Pasolli et al. 2019), the mapping rate remains low for other environments like soil and seawater (Nayfach et al. 2016).
We applied Nubeam to analyze WGS reads from the Human Microbiome Project (HMP) (The Human Microbiome Project Consortium 2012), and our main objective was to understand how the unmapped reads affect the genetic distance estimates between samples. The majority of the available microbiome data are 16S rRNA sequencing (usually V3/V4 hypervariable regions) instead of WGS. To demonstrate Nubeam's utility analyzing 16S rRNA sequencing data, we applied Nubeam to analyze a mouse gut microbiota 16S data set (Rosshart et al. 2019) and a human vaginal microbiota 16S data set in a recent case control study (Hong et al. 2020).
Results
Nubeam
We assume each sample is a collection of reads of the same length l, and each read consists of 4 nt, A, T, C, and G, and possibly an unknown nucleotide denoted by N. We first construct four
binary sequences from a read by using each of the 4 nt as a reference, with the reference nucleotide being 1 and the others
0. (Note that our representation allows us to mask a low-quality nucleotide to N to account for nucleotide quality.) We use
a matrix 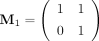 to represent 1, and its transpose
to represent 1, and its transpose 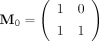 to represent 0. For each binary sequence B = b1b2…bl, we obtain the product matrix
to represent 0. For each binary sequence B = b1b2…bl, we obtain the product matrix 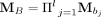 . (When B is an alternating sequence 101010… , the entries in MB are entries in a Fibonacci sequence.) Let W be a weight matrix, we designate log (tr(WMB)) as the Nubeam number to the binary sequence B. (Here, we use
. (When B is an alternating sequence 101010… , the entries in MB are entries in a Fibonacci sequence.) Let W be a weight matrix, we designate log (tr(WMB)) as the Nubeam number to the binary sequence B. (Here, we use 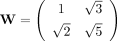 , and this choice is explained further below.) Thus, for each read, we obtain four Nubeam numbers (Nubeam quadruplet) that
jointly represent the read (Fig. 1A).
, and this choice is explained further below.) Thus, for each read, we obtain four Nubeam numbers (Nubeam quadruplet) that
jointly represent the read (Fig. 1A).
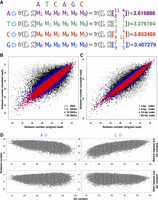
Nubeam assigns numbers to reads. (A) Illustration of how to obtain Nubeam quadruplet for a read. Convert a read to four binary sequences (indicated by the on/off power symbol), turn each binary sequence into a product matrix, and obtain a number from each product matrix. (B,C) Similar binary sequences produce similar numbers. For each simulated binary sequence of length 100, we obtained sequences with 1 or 3 or 10 or 50 random SNVs, or sequences with 1- or 3- or 10- or 50-bp indels, and compared the Nubeam numbers of original sequences with those of mutant sequences. (D) Regressing out GC content from Nubeam numbers of binary sequences; data comes from mapped reads of HMP sample SRS019215. Left: With A as reference (T as reference is similar). Right: With C as reference (G as reference is similar).
For each sample, a collection of reads becomes a collection of Nubeam quadruplets. These quadruplets define a four-dimensional empirical distribution, and the genetic distance between two samples can be quantified by the distance between the two empirical distributions. We obtain a histogram estimate for each collection of quadruplets (Supplemental Material and Methods) and calculate the Hellinger distance between probability mass functions (Methods). We will show that the Nubeam Hellinger distance highly correlates with the genetic distance between two samples.
Nubeam capitalizes on the noncommutative property of matrix multiplication, so that if two reads differ, their Nubeam quadruplets differ. More importantly, if two reads are similar (e.g., with small Hamming distance), their Nubeam quadruplets are close to each other. To demonstrate this through simulations, we first simulated binary sequences of length 100. For each sequence, we introduced SNV by flipping the digit at 1 or 3 or 10 or 50 random positions to obtain mutant sequences. We also introduced indels to the original sequences by inserting or deleting a segment of lengths of 1 or 3 or 10 or 50. Figure 1, B and C, plots Nubeam numbers of original binary sequences (x-axis) versus mutant sequences (y-axis) with different numbers of SNVs or different lengths of indels. Indeed, similar sequences tend to have similar Nubeam numbers. This “continuum” property is important as it is the foundation for the usefulness of the Nubeam representation. The effectiveness of quantitative treatments, including controlling for GC bias and histogram estimates for empirical distributions, implicitly depends on the continuum property.
The usefulness of a method depends on how effectively it handles the known artifacts (Benjamini and Speed 2012; Guo et al. 2012) due to shotgun sequencing technology in real data. One major artifact in sequencing is strand bias (Guo et al. 2012), where one strand may be sequenced in a much higher proportion than the other. Here, we provide a simple solution to account for strand bias. For a read R, we obtain its reverse complement U, and we compute quadruplets for both R and U for building an empirical distribution. The other major artifact in sequencing is the GC content bias, mainly due to the intrinsic property of the polymerase chain reaction (PCR), leading to the phenomenon that some stretches of nucleotides are more likely to be sequenced than others, depending on the GC content (Benjamini and Speed 2012). We control for GC bias by regressing out GC counts or proportions from each quadruplet using a simple linear regression, treating read as the unit of observation (Fig. 1D). We found it more effective to perform regression jointly over all samples, instead of one sample at a time. After GC is regressed out, the residuals are used to define empirical distributions to quantify genetic distances between samples.
Nubeam distance reflects genetic distance
We first demonstrate that Nubeam distance correlates well with the genetic distance in a three-taxa setting (Fig. 2A,B). Let a star tree have three leaf nodes and the inner node is seeded with a substrain of Escherichia. coli. Taxa S1 and S2 have equal branch lengths that correspond to 1 × 10−5 mutations per site per cell division. We varied the branch length of taxa S3, with the corresponding mutation rate ranging from 1 × 10−5 to 1 × 10−3 per site per cell division. In perspective, two phenotypically similar E. coli strains from different environments differ by about 1000 nt (Swick et al. 2013), which corresponds to a mutation rate of 1 × 10−4. This simulation setup produces varying genetic distances between S3 and S1/S2 with fixed distance between S1 and S2 as a baseline. We then simulated genomes of S1, S2, and S3 using iSG (Strope et al. 2006) under the general time reversible (GTR) substitution model (Tavaré 1986; Setti et al. 2012). For each simulated genome, we produced 75-bp error-free reads using a sliding window. The simulations were replicated 100 times. Figure 2A plots the true genetic distance (measured by the Hamming distance normalized by the genome size) versus inferred Nubeam distance, and the Spearman's rank correlation between the Nubeam distance and genetic distance is 0.999.
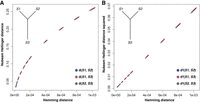
Nubeam Hellinger distance correlates well with genetic distance. (A) Hamming distance (x-axis, normalized by genome size) versus Nubeam Hellinger distance (y-axis). (B) The squared Hellinger distance (y-axis) appears to be linear with the Hamming distance (x-axis).
Next, we demonstrate that true phylogenies could be reconstructed using Nubeam distance. We simulated sets of sequencing reads using eight-taxonomic-unit trees (Supplemental Material and Methods), computed Nubeam distance between each pair of taxa to reconstruct the phylogenetic tree, and assessed the accuracy of phylogeny reconstruction by comparing the inferred tree with the true tree (Chan et al. 2014). We used six combinations of internal and terminal branch lengths of 1 × 10−5 and 5 × 10−5 to represent different degrees of genomic difference and different levels of difficulties for phylogeny inference (Supplemental Fig. S2). For each of the 100 replicates of a tree, hierarchical clustering was applied on the Nubeam distance matrix to reconstruct the phylogeny. The reconstructed phylogenies were compared with the true trees using Compare2Trees (Nye et al. 2005), and Nubeam perfectly reconstructed phylogeny for each of 100 replicates for all six generating trees. When applied to complete genomes of bat betacoronavirus and the newly identified pandemic coronavirus strain SARS-CoV-2, Nubeam clustered the virus according to subgenera, correctly grouped human SARS and MERS coronavirus with the closest bat strains at the genome level (Hu et al. 2015), and recapitulated the findings that the genome of bat coronavirus RaTG13 is highly similar to that of SARS-CoV-2 (Supplemental Fig. S2; Zhou et al. 2020).
We further demonstrate that the Nubeam distance correlates well with composition-based dissimilarities using synthetic communities. We generated synthetic communities composed of 10 equidistant or unequal-distant E. coli strains at different complexity levels (Fig. 3A,B); the pair-wise Nubeam distance correlates well with Bray-Curtis dissimilarity or weighted UniFrac distance (Lozupone et al. 2007) even at a high level of complexity (Fig. 3B). The k-mer frequency-based method (Lu et al. 2017b) and reference-based method (Lu et al. 2017a) are not as effective as Nubeam (Supplemental Fig. S3), presumably because Nubeam accounts for similarities between k-mers. Further, Nubeam consumes <20% of CPU time compared to the k-mer method (Supplemental Table S1).
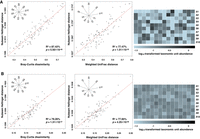
Nubeam Hellinger distances correlate well with composition-based dissimilarities among synthetic communities. We generated four sets of synthetic communities; each set contains 15 samples composed of 10 taxonomic units. The relative abundances of taxonomic units in each sample follow Dir(α1×10), with α being 1 and 10 in A and B, respectively, controlling for the complexity of the community. The relationship among the taxonomic units is shown in the star tree, with branch lengths of either 1 × 10−4 or 2 × 10−4. Each sample has 50 million 75-bp reads. The 105 pair-wise Nubeam distances were calculated among the 15 samples. The significance of linear relationship is measured by R2 and P-value for regression coefficient.
Nubeam controls for GC bias
Based on the E. coli genome, we simulated 15 samples without GC content bias and 30 samples with GC content bias, 10 for each bias scheme. The
bias schemes are detailed in the Methods section, and the severity of the GC bias in each schedule can be observed in Figure 4A. These three schemes reflect typical GC bias patterns observed in libraries of human whole genome sequencing data (Benjamini and Speed 2012; Xu et al. 2018). These bias schemes are intrinsic to the shotgun sequencing technology so that they also apply to libraries of mouse and
bacteria. Let d0 represent the Nubeam distance between two samples without GC bias; let db represent Nubeam distance between a sample without GC bias and a sample with GC bias without controlling for GC bias; and
let dc represent Nubeam distance between a sample without GC bias and a sample with GC bias but controlling for GC bias. To see
the effectiveness of Nubeam controlling for GC bias, we compared 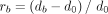 with
with 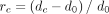 both visually and numerically (Fig. 4B–D). The GC biases were reduced effectively in all three simulated GC bias schemes.
both visually and numerically (Fig. 4B–D). The GC biases were reduced effectively in all three simulated GC bias schemes.
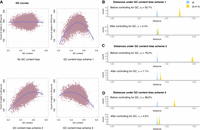
The effect of controlling for GC content bias in Nubeam. (A) GC content (x-axis) versus bin coverage (y-axis) to demonstrate patterns of GC bias for three simulation schemes, with LOESS curve marked in blue. Recall that 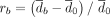 and
and 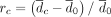 measure relative differences before and after controlling for GC bias, respectively. (B) rc = 2.4% after controlling for GC bias versus rb = 50.7% before controlling for GC bias for simulation scheme 1. (C) rc = 7.1% after controlling for GC bias versus rb = 78.2% before controlling for GC bias for simulation scheme 2. (D) rc = 4.9% after controlling for GC bias versus rb = 38.6% before controlling for GC bias for simulation scheme 3.
measure relative differences before and after controlling for GC bias, respectively. (B) rc = 2.4% after controlling for GC bias versus rb = 50.7% before controlling for GC bias for simulation scheme 1. (C) rc = 7.1% after controlling for GC bias versus rb = 78.2% before controlling for GC bias for simulation scheme 2. (D) rc = 4.9% after controlling for GC bias versus rb = 38.6% before controlling for GC bias for simulation scheme 3.
Nubeam includes the k-mer method as a special case
We first show that if two binary sequences are different, then their corresponding product matrices differ (Proposition 1).
By definition of 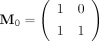 and
and 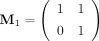 , a matrix right multiplying an M0 is to add the second column of the matrix to the first, and right multiplying an M1 is to add the first column to the second. Thus, for a given product matrix, one can solve for the binary string using the
following algorithm. Let the binary string be b1b2…bl. Compare the columns of the product matrix, pick the large column (whose entries are numerically not less than the corresponding
ones of the other); if it is the first column, then bl = 0 and if it is the second, then bl = 1. Subtract the small column from the large column to obtain a new matrix, let l decrease by 1 and repeat the procedure. Because the algorithm is deterministic, the solution must be unique. Thus, two different
binary sequences must correspond to two different product matrices.
, a matrix right multiplying an M0 is to add the second column of the matrix to the first, and right multiplying an M1 is to add the first column to the second. Thus, for a given product matrix, one can solve for the binary string using the
following algorithm. Let the binary string be b1b2…bl. Compare the columns of the product matrix, pick the large column (whose entries are numerically not less than the corresponding
ones of the other); if it is the first column, then bl = 0 and if it is the second, then bl = 1. Subtract the small column from the large column to obtain a new matrix, let l decrease by 1 and repeat the procedure. Because the algorithm is deterministic, the solution must be unique. Thus, two different
binary sequences must correspond to two different product matrices.
Let F and G be two matrices whose entries are integers, and recall 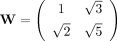 , then tr(WF) = tr(WG) if and only if F = G (Proposition 2). This is true because any entry in
, then tr(WF) = tr(WG) if and only if F = G (Proposition 2). This is true because any entry in 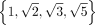 is not a linear combination of the other three entries with rational coefficients (Boreico 2008).
is not a linear combination of the other three entries with rational coefficients (Boreico 2008).
If two reads are different, then at least one pair of binary strings (among four such pairs) is different, and consequently (by Proposition 2), these two binary strings get assigned different numbers. Thus, if two reads are different, then the two quadruplets are different, and vice versa. In other words, k-mer has a one-to-one correspondence with the Nubeam quadruplet. When we use each unique quadruplet as a bin to compute distance between empirical distributions, then the practice is equivalent to the k-mer method (see Supplemental Material and Methods for more details).
Compared with Nubeam, the k-mer method is rather primitive; it has difficulty in controlling for GC bias, it disregards the different degree of similarities among k-mers, and its computation becomes difficult rather quickly as k increases.
Nubeam analyzing WGS sequencing data
Nubeam is ideal in analyzing metagenomics WGS sequencing data, simply because about half of reads cannot be mapped anywhere in more than 3000 microbial reference genomes. We analyzed 690 HMP (The Human Microbiome Project Consortium 2012) phase I samples collected from seven body habitats (excluding a habitat called “other oral”). HMP performed quality control and removed human sequence contamination; HMP then aligned reads passing a low-complexity filter to reference genomes (https://www.hmpdacc.org/HMREFG/), with an average of 57% of reads mapped per sample (SEM 13%), but mapping rates vary for samples from different body habitats (Supplemental Fig. S1). The inadequacy of the reference genomes and mapping bias both contribute to the high percentage of unmapped reads (Nayfach and Pollard 2016). We would like to mention here that the most recent de novo assembly resulted in 29.14% and 26.40% increases in mappability on average for gut and oral habitats, respectively (Pasolli et al. 2019). We split each sample (whole sample) into mapped reads (mapped pseudosample) and unmapped reads (unmapped pseudosample). Our analysis has two aims: whether we can recapitulate the findings made by the mapping-based method through analyzing mapped reads using Nubeam; and whether Nubeam enables the unmapped reads to provide additional information.
Nubeam within-sample diversity
We used the second moment of Nubeam numbers of a sample to quantify within-sample diversity (see Methods). To validate our definition of Nubeam within-sample diversity, we simulated a mixture of two bacteria species (E. coli and Proteus mirabilis) by mixing reads at various mixing proportions. The inferred within-sample diversities reflected our intuition that the intermediate mixing proportions led to higher within-sample diversity than extreme mixing proportions (Supplemental Fig. S4).
We further simulated 10-species communities (Supplemental Table S2) and compared our definition of Nubeam within-sample diversity with reference-based alpha diversity measures such as an inverse Simpson's index and Shannon index. Communities with a smaller inverse Simpson's index and Shannon index generally have smaller Nubeam within-sample diversity; however, communities with the same inverse Simpson's (or Shannon) index can have divergent Nubeam within-sample diversities (Supplemental Table S3). This is because the Nubeam within-sample diversity of a community takes into account within-sample diversities of each component (Supplemental Fig. S5) and the relatedness (or similarity) between components, whereas the reference-based alpha diversity measures are oblivious to these factors.
We quantified Nubeam within-sample diversities of mapped and unmapped pseudosamples. Being able to quantify within-sample diversity of unmapped pseudosamples is a strength of Nubeam. For mapped pseudosamples, oral and gastrointestinal habitats have high within-sample diversity, whereas skin and vagina have low within-sample diversity (Fig. 5)—the ranking of within-sample diversity by body habitats is largely consistent with that in a published study of HMP (The Human Microbiome Project Consortium 2012). For unmapped pseudosamples, the within-sample diversity is generally higher than mapped ones, particularly for nasal and urogenital samples (Fig. 5); the ranking by body habitats is also different from mapped ones.
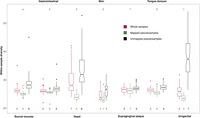
The Nubeam within-sample diversity of the whole sample, mapped, and unmapped pseudosamples for each body habitat. The rankings for the Nubeam within-sample diversity of different body habitats are shown at the bottom; the rankings in the published study (The Human Microbiome Project Consortium 2012) of the same data set using mapping-based methods are shown at the top.
Nubeam between-sample diversity
In metagenomics literature, the beta-diversity measures the difference of microbial compositions between two samples. Here, we used Nubeam distance to quantify beta-diversity between a pair of samples and examined beta-diversity estimates using hierarchical clustering. In parallel, we applied UMAP (McInnes et al. 2018) for nonlinear dimensionality reduction and visualization to examine beta-diversity estimates (Supplemental Fig. S6). UMAP clustering corroborates hierarchical clustering on big pictures, but for finer details such as outliers, we trust hierarchical clustering over UMAP.
We first examined the beta-diversity of mapped pseudosamples (Fig. 6A). The primary clustering is by body habitats, with gastrointestinal, oral, and urogenital samples well separate. Skin samples intermingle with a subset of nasal samples. Within the oral cavity, samples from supragingival plaque clearly separate from those from buccal mucosa and tongue dorsum, and a portion of buccal mucosa samples intermingle with tongue dorsum samples. These results are comparable with published studies of the same data sets using mapping-based methods (The Human Microbiome Project Consortium 2012).
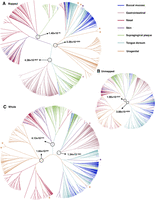
Hierarchical clustering (Ward's minimum variance method) of mapped pseudosamples (A), unmapped pseudosamples (B), and whole samples (C), respectively. If a sample is marked by * in A, then its corresponding whole sample is again marked by * in C. For a selected pair of subclusters, we used a Kruskal–Wallis test to check whether there is a significant difference between within-group distances and between-group distances for the pair of subclusters.
We then examined the beta-diversity of unmapped pseudosamples (Fig. 6B). The overall clustering pattern still follows the body habitats. The most striking difference is that the urogential samples intermingle with the nasal samples, which is also evident in Supplemental Figure S6B. For each body habitat, we used Mantel's test to compare the distance matrices calculated using mapped pseudosamples with those calculated using unmapped pseudosamples. We found nonsignificant correlation for vagina, and significant yet moderate correlations for the rest of the body habitats (Supplemental Table S4), revealing the necessity to study the contribution of unmapped reads in metagenomics studies. Lastly, we examined the beta-diversity of whole samples. Figure 6C shows that the clustering of whole samples is also by body habitats, which largely agrees with those of mapped pseudosamples. Noticeable differences do exist, however, presumably due to unmapped reads. For example, there are only two outliers for skin in mapped pseudosamples, but eight outliers for skin in whole samples. For another example, one gastrointestinal outlier among mapped pseudosamples (marked with an asterisk in Figure 6A), which distinguishes itself by its unusually high contents (7.5%) of pathogen Shigella spp., is no longer an outlier among whole samples. Finally, one skin outlier is clustered with oral mapped pseudosamples, likely due to its 46.1% of Finegoldia magna, an opportunistic pathogen that can be found in skin, oral, gastrointestinal, and urogenital habitats (Rosenthal et al. 2012), and its corresponding whole sample is still an outlier but with a new companion.
It is worth noting that nasal samples have been reported to bridge skin and oral samples in ordination analysis using 16S rRNA sequencing data (The Human Microbiome Project Consortium 2012); our analysis shows that, for both mapped and unmapped pseudosamples and whole samples, it is supragingival plaque samples but not buccal mucosa or tongue dorsum samples that are close to nasal samples (Fig. 6; Supplemental Figs. S6, S7).
Urogenital (vaginal) samples
There are four urogenital outliers in the mapped pseudosamples (Fig. 6A). To make sense of these four samples, we compared the clustering with the taxonomic compositions of all urogenital samples (Methods). Figure 7 demonstrates that the clustering is in concordance with their taxonomic compositions and abundances. In particular, two outlier samples have almost no Lactobacillus bacteria but instead have high proportions of anaerobic Gardnerella and Atopobium bacteria; the other two outlier samples contain large proportions of Bifidobacterium bacteria (10% and 31% for the two samples, respectively), in addition to high proportions of Lactobacillus gasseri.
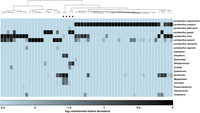
Hierarchical clustering (Ward's minimum variance method) of vaginal samples using a Nubeam distance matrix is consistent with community state types defined by relative abundances of microbial taxa. The heat map was generated using relative abundances of microbial taxa, with only the most abundant ones chosen. The four outlier samples described in the main text are marked by *.
Vaginal microbiomes are known to have simple taxonomic compositions dominated either by a single Lactobacillus species or by strictly anaerobic bacteria (Ravel et al. 2011; Romero et al. 2014). However, our analysis suggests that might be true only for mapped pseudosamples, as the unmapped urogenital pseudosamples have extraordinarily high Nubeam within-sample diversities. We thus performed de novo assembly using state-of-the-art metagenomics assembler metaSPAdes (Nurk et al. 2017) for all unmapped reads to produce contigs (minimum length 228 bp) and remapped reads to contigs. To our surprise, 54% of the reads fail to be remapped, indicating that they are isolated reads; 27% of reads can be mapped to contigs with null BLAST results, indicating that they are from unknown organisms; only 19% of reads can be mapped to contigs from known microorganisms (according to BLAST results). De novo assembly using MEGAHIT (Li et al. 2015) produced qualitatively similar results with metaSPAdes (Supplemental Table S5). These results suggest that the sequencing depths for vaginal samples in HMP are far from sufficient, and the composition of the vaginal microbiome needs to be further studied using WGS with sufficient sequencing depths.
Nubeam analyzing 16S rRNA sequencing data
The 16S rRNA sequencing data is more abundantly available than WGS data in current microbiome studies. By design, 16S rRNA sequencing data is more suitable for mapping-based methods. To demonstrate that Nubeam is also useful in analyzing 16S rRNA sequencing data, we analyzed two sets of 16S rRNA sequencing data.
Mouse gut microbiota samples
In a recent study (Rosshart et al. 2019), mice of the same genetic background but with different gut microbiota states—natural or conventional laboratory—were subjected to antibiotic, dietary, and microbial challenges. We recapitulated the findings in the original study (Rosshart et al. 2019) that natural microbiota are resilient against these challenges, whereas conventional laboratory microbiota are not (Fig. 8A–D). Our analysis also produced some interesting and novel findings. First, for the antibiotic challenge (Fig. 8A), the change of gut microbiota of Taconic laboratory and wildling mice are highly consistent at the end of antibiotic treatment, whereas microbiota of Jackson Laboratory mice change little comparatively. However, during the recovery period, the microbiota of both Taconic and Jackson Laboratory mice differ greatly from that of wildling mice, though there is a tendency of reverting back to normal. Second, for microbial challenge through cohousing, the gut microbiota of wildR mice are indeed resilient (Fig. 8C,D); however, gut microbiota of some laboratory mice appear to be as resilient as that of wildR mice, as can be seen for two Taconic laboratory samples (Fig. 8C) and one Jackson Laboratory sample (Fig. 8D).
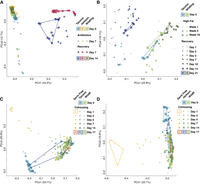
Natural mice microbiota are resilient against interruptions, including (A) antibiotic challenge, (B) high fat dietary challenge, and (C,D) microbial challenge through cohousing, whereas conventional laboratory mice microbiota are generally not. Samples at the beginning and the end of experiments are annotated by convex hulls.
Human vaginal microbiota samples
In a recent study (Hong et al. 2020), vaginal microbiota samples were collected from 39 individuals newly diagnosed with polycystic ovary syndrome (PCOS) and 40 healthy control individuals. First, we performed permutation multivariate analysis of variance (PERMANOVA) (McArdle and Anderson 2001) based on a Nubeam distance matrix and found significant (P-value = 8.59 × 10−3 under 105 permutations) association between vaginal microbiota and case control status. Second, we performed multidimensional scaling (MDS) for the Nubeam distance matrix. A logistic regression shows significant association between the top six principal coordinates, which account for 60.55% of variation, with the case control status (P-value =9.19 × 10−4). There is a significant difference (Kruskal–Wallis test P-value =9.44 × 10−7) between case and control groups in the predicted probability of a sample being a PCOS case (Supplemental Fig. S8A). Third, hierarchical clustering of samples based on the Nubeam distance matrix shows visibly recognizable clustering of samples according to PCOS status (Supplemental Fig. S8B). To summarize, using Nubeam we confirmed the association between vaginal microbiota and PCOS discovered in the original study using mapping-based methods (Hong et al. 2020).
Discussion
We presented a novel reference-free approach, Nubeam, to analyze short sequencing reads. Nubeam can account for strand bias and GC bias when quantifying genetic distance between samples. We showed that the k-mer method is equivalent to a special case of Nubeam, without its ability to account for GC bias. We demonstrated its usefulness by applying Nubeam to analyze both WGS and 16S rRNA sequencing data sets.
Our analysis sheds new light on the HMP WGS sequencing samples in several aspects. First, similar to the clustering of mapped pseudosamples, the clustering of unmapped pseudosamples is also dominated by the body habitats; however, the moderate or even nonsignificant correlations between distance matrices calculated using mapped and unmapped pseudosamples indicate a different within-habitat sample relationship using different sets of reads. Second, the clustering differences between mapped pseudosamples and whole samples do exist, and they are best represented by outlier samples of particular body habitats. Third, analysis of the unmapped reads suggests that the sequencing depths for vaginal samples are far from sufficient, and deeper sequencing might challenge the current belief that vaginal microbiomes have simple taxonomic compositions. A limitation of our analysis, as pointed out by one reviewer, is that we used the same reference database as HMP, which does not reflect the most recent progress in the field (Almeida et al. 2019; Nayfach et al. 2019; Pasolli et al. 2019). We note, however, reference bias and mapping bias are intrinsic to the mapping-based approaches, and both biases can be mitigated but never overcome.
We also demonstrated that Nubeam is useful for analyzing 16S rRNA sequencing data, even though, by design, the 16S data is more suitable for mapping-based method. Our analysis recapitulates findings that mouse natural microbiota are resilient against challenges, whereas conventional laboratory microbiota are not. However, our analysis also revealed that the gut microbiota of some laboratory mice can be as resilient as those of wild mice (natural microbiota) in cohousing experiments. The analysis demonstrates that Nubeam can gain novel insights that are undetectable by other reference-free methods (Callahan et al. 2016; Rosshart et al. 2019). Using the 16S rRNA sequencing data set from a case control study, we were able to detect the significant association between the Nubeam pairwise distance matrix and the case control status.
The unmapped reads from WGS sequencing are more prone to artifacts produced in DNA handling and sequencing. Since Nubeam is a reference-free approach, we paid extraordinary attention to guard against possible artifacts. We performed additional quality controls on sequencing reads and made an effort to remove human contamination by removing reads that can be mapped to the human reference genomes. Yet, it was still possible—although not probable—that the unmapped reads were a mixture of microbial reads and human reads from complex genomic regions, large structural variations, and population-specific sequences, regardless of how small the proportion of human reads. The clustering of unmapped pseudosamples, which largely agrees with that of mapped pseudosamples, reassured us that our additional quality control procedure was effective, and our novel insights based on unmapped reads were not driven by data artifacts.
To remove human contamination, we used human reference genome hg19 (out of convenience) instead of a newer version hg38. We argue that using hg38 will not significantly affect our results of decontamination, for two reasons. First, hg19 is a comprehensive mapping target for human reads: 99.92% of reads from a human sequencing sample could be mapped to the hg19 primary assembly (Schneider et al. 2017). Second, hg38 adds the modeled centromere, 261 scaffolds of alternate loci, gaps closure by WGS contigs, and CHM1 BAC clones (Schneider et al. 2017); many of the new sequence additions were already represented in the human sequences in the NCBI Nucleotide Collection, which was used in addition to hg19 to remove possible human contamination (Methods).
Many aspects of Nubeam invite continuing investigations. One possibility is to design better matrices to represent nucleotides for specific applications. For example, we discovered that the matrix format of quaternion performs better in certain simulations. Another possibility is the computation. For example, computing Nubeam Hellinger distance between two (four dimensional) empirical distributions may be made more effective via shrinkage density estimates (Ma 2017). Other areas of further investigation include accounting for GC bias and sequencing error.
Controlling for GC content bias is the key advantage of Nubeam, and such control is mandatory in real sequencing data analysis. The downside for correcting GC bias is that it may diminish the difference between samples when such a difference is correlated with GC content. We propose the following approach to mitigate the adverse effect of controlling for GC content bias: partition the Nubeam quadruplets into bins according to the GC counts of the corresponding reads; for each GC bin, compute pair-wise distance between samples after controlling for GC bias, and then take a weighted sum of distances from difference bins.
The “continuum” property makes the method more robust to sequencing error. Moreover, we can take advantage of the Nubeam representation to explicitly account for sequencing error. Specifically, if the base quality score is lower than a designated threshold, we mask it with “N”. When the low quality nucleotide happens to be an error, only one Nubeam number in a quadruplet would be changed, whereas a sequencing error in the read affects two Nubeam numbers in a quadruplet. Thus, the masking will mitigate the effect of sequencing errors.
We believe Nubeam can find itself in a broad range of applications. One application we recently developed is to perform reads deduplication before mapping (Dai and Guan 2020). Since a unique read is assigned a unique quadruplet, the deduplication can be done efficiently. Another possible application is to identify sequences that contribute to the difference between sequencing samples of different states, as a distribution of Nubeam numbers of a sample can be viewed as a mixture distribution with component distributions of different weights. Nubeam can be effective in some areas where the k-mer approach is useful, such as characterizing protein binding motif (Newburger and Bulyk 2009), CpG island by the flanking regions (Chae et al. 2013), and sequence feature for haplotype grouping (Navarro-Gomez et al. 2015), analyzing RNA-seq data to gain novel insights on new exons and splicing isoforms (Bray et al. 2016; Audoux et al. 2017), and even detecting genetic associations (Rahman et al. 2018).
A recent study suggested that environment, not genetics, primarily shapes the host microbiome composition (Rothschild et al. 2018). Through quantifying microbiome difference between samples, Nubeam can be used to quantify the environmental factors to facilitate the study of gene × environment interactions for many phenotypes such as diabetes and obesity (Li 2019). Finally, Nubeam enables us to study the contribution of unmapped reads to the genetic distance between samples. Using WGS sequencing for human subjects, Nubeam has the potential to investigate whether, and to what extent, the unmapped reads can contribute to explain the “missing heritabilities” (Maher 2008).
Methods
Controlling for GC bias
The GC content bias is a major sequencing artifact that leads to the dependence between regional coverage and GC content. When the signal of interest is the abundance of reads originating from certain genomic regions, GC content bias is a confounding factor. We correct the GC content bias at the read level by regression. We fit the standard linear regression model yi = Xβ + ε, where yi is an n × 1 matrix of numbers assigned to reads by Nubeam, X is an n × 3 matrix of AT-count and GC-count of reads including a column vector of 1, β is a 3 × 1 vector of corresponding regression coefficients including the intercept, and ε is an n × 1 vector of residuals. The residuals are then assigned to reads.
Simulating GC content bias
We simulated sequencing samples with GC content bias using the following method (Benjamini and Speed 2012). For a position x on the genome, the number of the 75-bp read originating from x to be sampled follows Pois(λ), where λ is the expected count for the read. Denote the GC content (ranging from 0 to 1) of the 200-bp fragment originating from x as gc. When there is no GC content bias, λ = 1 regardless of the value of gc; when there is GC content bias, λ is determined by the Gaussian function λ = a exp( − (gc − b)2/2c2) (Frampton and Houlston 2012). We simulated three schemes of GC content bias: a = 1, b = 0.5, c = 0.2 for scheme 1; a = 1, b = 0.6, c = 0.3 for scheme 2; a = 1, b = 0.4, c = 0.3 for scheme 3. These three schemes reflect typical GC bias patterns we observed in human WGS sequencing data.
Quantifying Nubeam within-sample diversity
Let X, an n × 4 matrix, be a collection of quadruplets. Define Σ = XTX, and perform eigen-decomposition for Σ. The Nubeam within-sample diversity can be quantified as the sum of eigenvalues (each eigenvalue is nonnegative).
Distance between two empirical distributions
Let X and Y be two collections of quadruplets. We divided samples into bins and obtained {xj} and {yj} as probability mass functions for X and Y, respectively. The Hellinger distance is defined as 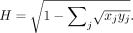 A detailed algorithm to partition bins in a balanced manner can be found in Supplemental Material and Methods.
A detailed algorithm to partition bins in a balanced manner can be found in Supplemental Material and Methods.
Human Microbiome Project WGS samples
We downloaded BAM files of 754 WGS samples from http://hmpdacc.org/HMSCP/ and extracted reads using SAMtools (Li et al. 2009). For a sample to be included in the study, it had to pass the QC procedure described in http://hmpdacc.org/hmp/HMASM/, which leaves us with 690 samples. Five samples had corrupted files and were removed. We further removed 29 samples from body habitat “other oral” and analyzed a total of 656 samples from seven body habitats. We deduplicated reads by an in-house software based on Nubeam numbers of reads. Deduplicated reads were then mapped to human reference genome hg19 and human sequences from the NCBI Nucleotide Collection (https://www.ncbi.nlm.nih.gov/nucleotide/) (downloaded on June 7, 2019) by BWA (Li and Durbin 2009) to remove human sequence contamination. The hierarchical clustering (Ward's minimum variance method) was presented using ggtree (Yu et al. 2017) in R (R Core Team 2018).
Clustering of vaginal samples
For the 56 urogenital (vaginal) samples, we applied hierarchical clustering (Ward's minimum variance method) on the Nubeam distance matrix. The heat map of relative abundances of microbial taxa was generated using data from Kraal et al. (2014), with the abundance tables of each sample downloaded from http://hmpdacc.org/HMSCP/. The abundances of strains in each sample were estimated by the product of breadth and depth of coverage, and relative abundances were obtained by normalization, as described in Kraal et al. (2014). The relative abundances of species were calculated by adding up the relative abundances of strains belonging to the same species, while the relative abundances of genera were calculated similarly. The selected strains/genera each have a cumulative relative abundance of more than 1 × 10−3 across 56 samples; and for each sample, their abundances account for more than 99.9% of overall abundance. Lactobacillus has all the species listed, whereas other genera only have their genera names listed. The hierarchical clustering and heat map were presented using heatmap.2 in R (R Core Team 2018) packagegplots.
Mouse 16S rRNA sequencing samples
We downloaded 16S rRNA hypervariable region V4 sequencing data of mouse gut microbiota samples from the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA540893 and extracted reads using fastq-dump. We merged the PE reads into a single one and then removed the primers from both ends of the merged read using AdapterRemoval (version 2.3.1) (Schubert et al. 2016). We generated the Nubeam quadruplets for sequences defined by a window of 50 bases slid along a merged read with a step of one base. The GC content was regressed out using the logarithm of AT and CG percentages in the merged read. Multidimensional scaling was applied on Nubeam distance matrices; the resulted first two principal coordinates were plotted against each other.
Human vaginal 16S rRNA sequencing samples
We downloaded 16S rRNA hypervariable regions V3–V4 sequencing data of human vaginal microbiota samples from figshare (https://figshare.com/s/e56859e336e71ecc2867). We merged the PE reads into a single one and then removed the primers from both ends of the merged read using AdapterRemoval. We generated the Nubeam quadruplets for sequences defined by the first 400 bases of merged reads. The GC content was regressed out using the logarithm of AT and CG percentages in the merged read. To quantify the association between beta-diversity and PCOS status, PERMANOVA was performed using adonis in R (R Core Team 2018) package vegan (version 2.5-5) (https://rdrr.io/cran/vegan/). Multidimensional scaling was applied on the Nubeam distance matrix; a logistic regression model based on the resulted first six principal coordinates was built.
Software availability
Nubeam source code is available on both GitHub (https://github.com/daihang16/Nubeam) and Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
The work was partially supported by United States Department of Agriculture/Agriculture Research Service under contract number 6250-51000-057 and the start-up fund from Duke University School of Medicine. Most of the work was done at the Children's Nutrition Research Center of Baylor College of Medicine, the authors’ prior institution, where H.D. was a graduate student and Y.G. was an Assistant Professor. The authors thank editors and three referees for their constructive comments that significantly improved the presentation of the manuscript, and Shannon Clarke from Duke University School of Medicine for her editorial assistance.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.261750.120.
-
Freely available online through the Genome Research Open Access option.
- Received August 26, 2019.
- Accepted July 30, 2020.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








