
Paternal age in rhesus macaques is positively associated with germline mutation accumulation but not with measures of offspring sociability
- Richard J. Wang1,
- Gregg W.C. Thomas1,2,
- Muthuswamy Raveendran3,4,
- R. Alan Harris3,4,
- Harshavardhan Doddapaneni3,4,
- Donna M. Muzny3,4,
- John P. Capitanio5,
- Predrag Radivojac2,6,
- Jeffrey Rogers3,4 and
- Matthew W. Hahn1,2
- 1Department of Biology, Indiana University, Bloomington, Indiana 47405, USA;
- 2Department of Computer Science, Indiana University, Bloomington, Indiana 47405, USA;
- 3Human Genome Sequencing Center, Baylor College of Medicine, Houston, Texas 77030, USA;
- 4Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas 77030, USA;
- 5California National Primate Research Center, University of California–Davis, Davis, California 95616, USA;
- 6Khoury College of Computer Sciences, Northeastern University, Boston, Massachusetts 02115, USA
Abstract
Mutation is the ultimate source of all genetic novelty and the cause of heritable genetic disorders. Mutational burden has been linked to complex disease, including neurodevelopmental disorders such as schizophrenia and autism. The rate of mutation is a fundamental genomic parameter and direct estimates of this parameter have been enabled by accurate comparisons of whole-genome sequences between parents and offspring. Studies in humans have revealed that the paternal age at conception explains most of the variation in mutation rate: Each additional year of paternal age in humans leads to approximately 1.5 additional inherited mutations. Here, we present an estimate of the de novo mutation rate in the rhesus macaque (Macaca mulatta) using whole-genome sequence data from 32 individuals in four large pedigrees. We estimated an average mutation rate of 0.58 × 10−8 per base pair per generation (at an average parental age of 7.5 yr), much lower than found in direct estimates from great apes. As in humans, older macaque fathers transmit more mutations to their offspring, increasing the per generation mutation rate by 4.27 × 10−10 per base pair per year. We found that the rate of mutation accumulation after puberty is similar between macaques and humans, but that a smaller number of mutations accumulate before puberty in macaques. We additionally investigated the role of paternal age on offspring sociability, a proxy for normal neurodevelopment, by studying 203 male macaques in large social groups.
Paternal age at conception is the single strongest predictor of the number of de novo mutations that a human will inherit. Studies show that children will inherit approximately 1.5 additional mutations per year of paternal age and that the average father contributes new mutations at a rate that is three to four times greater than the mother per year of parental age (Kong et al. 2012; Besenbacher et al. 2015; Francioli et al. 2015; Jónsson et al. 2017). This male bias in the number of de novo mutations has been attributed to the continuous nature of spermatogenesis, which results in the accumulation of errors in the male germline (Crow 2000). As new mutations are responsible for the incidence of spontaneous genetic disorders, this pattern has considerable implications for human health. The male bias in mutation rate has also been observed in other primates (Venn et al. 2014; Thomas et al. 2018), making it an important feature in models of mutation rate evolution (Thomas and Hahn 2014; Amster and Sella 2016; Moorjani et al. 2016; Besenbacher et al. 2019).
Children conceived by older fathers are at greater risk for numerous adverse developmental outcomes (Crow 2000). These include a greater risk of certain genetic disorders due to new mutations inherited at single genes (e.g., Glaser et al. 2003; Green et al. 2010; Goriely et al. 2013). Evidence from epidemiological studies also suggests an association between advanced paternal age and complex neurodevelopmental disorders, including an increased risk of schizophrenia, autism, and bipolar disorder (Sipos et al. 2004; Reichenberg et al. 2006; Durkin et al. 2008; Frans et al. 2008; Menezes et al. 2010; Lee et al. 2011). The mechanisms underlying these epidemiological associations with paternal age have not been conclusively determined. A central question lies in whether the risk inherited from older fathers comes from genetic predisposition or de novo mutations. In the de novo model, neurological disorders are highly polygenic and result from the additive effects of a rising mutational burden (Malaspina et al. 2002; Hultman et al. 2011; Kong et al. 2012; Ronemus et al. 2014). The competing predisposition hypothesis attributes the epidemiological association to underlying pre-existing genetic factors that may actually contribute to delayed reproduction in males (Gratten et al. 2016; Janecka et al. 2017b). For example, a genetic correlation between psychiatric disorders and delayed fatherhood could explain the association seen with advanced paternal age.
Primate models provide a powerful experimental system for investigating the relationship between the increasing number of de novo mutations and the increased risk of developmental outcomes with paternal age. Along with estimates of de novo mutations, testing this relationship requires information on neurodevelopmental maturation and behavior. Although such data are much harder to collect, several long-term studies of captive primate populations have tracked multiple aspects of neurological and social abilities. In the important model system, rhesus macaque (Macaca mulatta), researchers have found high levels of social intelligence (Capitanio 1999; Sclafani et al. 2016). As a consequence, the rhesus monkey has become a model for studying schizophrenia (Gil-da-Costa et al. 2013) and autism spectrum disorder (Bauman and Schumann 2018; Parker et al. 2018).
Studying the mutation rate in nonhuman primates also allows us to address fundamental questions concerning its evolution. Because the average parental age at conception in the rhesus macaque is much lower than in human, any process that leads to a relationship between parental age and the number of germline mutations will also lead to a lower per-generation mutation rate in macaques. A smaller number of germline cell divisions from spermatogenesis in male macaque parents predicts a smaller number of replication-dependent errors (Thomas and Hahn 2014). The observation of a significant, though smaller, maternal age effect on the human mutation rate (Goldmann et al. 2016; Wong et al. 2016; Jónsson et al. 2017) raises the possibility that a substantial portion of germline mutations may not originate from replicative errors during spermatogenesis (Gao et al. 2019; Sasani et al. 2019). Nonetheless, any relationship between nonreplicative mutations and parental age would contribute to the evolution of mutation rate between species due to differences in the age of reproduction. Understanding the contribution of each of these processes to possible differences in the per-generation mutation rate between species—as well as their effects on variation in the substitution rate—is a goal of recent mutation studies of nonhuman primates (Thomas et al. 2018; Besenbacher et al. 2019).
Here, we performed whole-genome sequencing on 32 rhesus macaques from four multiple-generation pedigrees maintained at the California National Primate Research Center to identify de novo mutations. We also analyzed behavioral data in 203 male macaques from the same population to examine links between paternal age at conception and behavioral metrics of sociability, including those linked to autism.
Results
The number of inherited mutations increases with paternal age in rhesus monkeys
We sequenced 32 individuals from four three-generation families of rhesus monkeys (Supplemental Fig. S1) to 40× average coverage using Illumina short-read sequencing. These families contained 14 trios with different offspring, from which we identified 307 de novo single nucleotide mutations (Table 1; Supplemental Data S1). We found mutations that could be tracked to a third generation were transmitted 42% of the time, implying a false positive rate of up to 16%. After applying stringent quality filters, comparing two genotype-calling pipelines, and controlling for the callability of sites across the genome (Methods), we estimated an average mutation rate of 0.58 × 10−8 per base pair (bp) per generation for parents with an average age of 7.5 yr (7.8 paternal, 7.1 maternal).
Mutation count and rate by trio
We found a strong association between paternal age and the number of de novo mutations inherited by offspring. For each additional year of paternal age at conception, offspring inherited an additional 1.5 de novo mutations (R2 = 0.79; Poisson regression, P = 2.9 × 10−11). Due to the structure of the pedigree among sampled individuals, we were able to unambiguously phase 139 of the 307 mutations (see Methods). Of these phased mutations, 76.3% (95% CI: [68.5, 82.6]) were determined to be of paternal origin. Figure 1 shows the count of phased mutations as a function of parental age, illustrating the large effect of paternal age on mutation rate (R2 = 0.78; Poisson regression, P = 7.5 × 10−4). In contrast to this relationship, we found no significant association between maternal age and the mutation rate (Fig. 1; Supplemental Fig. S2). Early studies of human trios also found no significant effect of maternal age (Kong et al. 2012; Francioli et al. 2015), though studies with much larger sample sizes have detected a small effect, potentially due to the accumulation of DNA damage in the maternal germline (Goldmann et al. 2016; Jónsson et al. 2017).
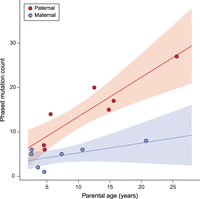
Phased mutation count and parental age. The number of phased mutations identified from seven rhesus macaque trios attributed to paternal (red) and maternal (blue) transmission. There is a strong linear relationship between the number of transmitted paternal mutations and the paternal age at conception (R2 = 0.78; Poisson regression, P = 7.5 × 10−4). The number of maternally transmitted mutations was not significantly associated with the maternal age at conception in our data (R2 = 0.29; Poisson regression, P = 0.11). Shaded areas show respective regression 95% CI. These seven trios represent cases in which mutations can be tracked through the following generation (Supplemental Fig. S1).
The mutation spectrum in rhesus macaques is similar to that found in other primates, except for a slight excess of C > T transitions (Fig. 2; see Supplemental Results). We found that C > T transitions at CpG sites accounted for 24% of all observed point mutations (similar to the 17% and 22%–24% reported in humans and chimpanzees, respectively) (Kong et al. 2012; Venn et al. 2014; Besenbacher et al. 2019). We estimated the mutation rate at CpG sites to be 1.43 × 10−7 per bp per generation, an order of magnitude higher than the rest of the genome (cf. Bird 1980). The number of mutations at CpG sites was also significantly associated with paternal age (R2 = 0.20; Poisson regression, P = 0.015) (Supplemental Fig. S3).
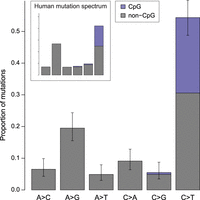
Mutation spectrum in rhesus macaque. The frequency of each type of mutation from among the 307 identified. Error bars show binomial proportion 95% CI (Wilson score interval) for totals at each type. Mutations at CpG sites accounted for 24% of all mutations and represent 43% of all strong-to-weak transitions. Mutation categories represent their reverse complement as well.
A lower per-generation mutation rate in the rhesus macaque
Our overall estimate of the per-generation mutation rate in rhesus monkeys (0.58 × 10−8 per bp per generation) is lower than direct estimates from other primates, but both the average age of parents and the average age at puberty differ among species (Table 2). The average paternal age at conception explains most of the variation among studies in direct estimates of the human mutation rate (Kong et al. 2012; Rahbari et al. 2016; Jónsson et al. 2017). Figure 3 shows the rate of mutation accumulation with paternal age at conception in macaque trios compared to the rate observed in humans. Because parental age explains much of the variation in mutation rate within species, we compared the mutation rate estimate from rhesus macaque to other species using a model of reproductive longevity (Thomas et al. 2018).
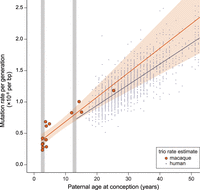
Similar rates of mutation accumulation postpuberty in human and rhesus macaque. Mutation rate accumulation with paternal age estimated from trios in macaques (orange) and humans (black) (data from Jónsson et al. 2017). Approximate ages at male puberty in the macaque (3.5 yr) and human (13.5 yr) are shown in gray. Human trios with paternal age up to 50 are shown here, but the human regression line is from the full data set. The rate at which the mutation rate increases with paternal age is slightly higher in the macaque (4.3 × 10−10 per bp per year; Poisson regression) than in human (3.4 × 10−10 per bp per year). The intercept with puberty is much lower in macaque (3.9 × 10−9 per bp) than in human (7.1 × 10−9 per bp).
Per-generation mutation rate and reproductive age in primate studies
Reproductive longevity is defined as the amount of time a parent is in a reproductive state prior to offspring conception: Here, we use the paternal age at conception minus the age at sexual maturity. In this model, the mutation rate can evolve between species if the rate of error per division changes, if the rate of germline cell-division postpuberty changes, if the period between puberty and conception changes, or if there are different numbers of mutations that accumulate prior to puberty (Thomas and Hahn 2014; Thomas et al. 2018). Though key parameters of germline development surely differ among species, dividing mutation accumulation into two piecewise regimes—linearly increasing with age after puberty and a discrete number of mutations from before puberty—makes a compelling null model for understanding mutational variation between species. If, instead, mutations largely arise in a replication-independent manner, a model of longevity without regard for time at puberty would still be needed to fit the linear rate of mutation accumulation.
We performed a Poisson regression of mutation count on parental age to model the rate of accumulation with reproductive longevity and compared it to a regression from a large human data set (Jónsson et al. 2017). If we assume both that the number of mutations before puberty and the rate of accumulation of mutations after puberty are the same in humans as in macaques, our model of reproductive longevity overestimates the expected number of mutations per generation (∼53 vs. 36, using 7.8 yr as the average paternal age in macaques).
Much of the difference in the per-generation mutation rate between human and macaque can be attributed to the number of mutations predicted in the germline prepuberty: There are about half as many in macaques as in humans (25.4, 95% CI: [21.0, 29.7] in macaque and 44.1 [43.0, 45.2] in human). The rate at which mutations increase with paternal age after puberty was not significantly different between macaque (4.3 × 10−10 per bp per year, 95% CI [3.0, 5.5]) and human (3.4 × 10−10 per bp per year, [3.3, 3.5]; unequal variances t-test, P = 0.17). Though we have limited power to detect small differences in this rate between species (see Methods), even an age effect at the upper 95% CI bound in the macaque (i.e., the highest slope consistent with our data) would correspond to only approximately six more mutations over the average lifespan of macaques in our study.
We further used the regression model to estimate a per-year mutation rate for the macaque. Such values can be directly compared to substitution rates from phylogenetic studies. We calculated the per-year rate as a property of the species by predicting the mutation rate at the median age of reproduction. For a paternal age of 11 yr in macaques (Xue et al. 2016), our regression model predicts a rate of 0.65 × 10−9 per bp per year. This per-year rate is higher than the 0.43 × 10−9 per bp per year observed in humans (Jónsson et al. 2017), consistent with reports of a lower human per-year mutation rate (Scally and Durbin 2012; Ségurel et al. 2014; Besenbacher et al. 2019).
Sociability in male rhesus monkeys shows no connection with sire age
Sociability is a consistent personality dimension in humans that has also been identified in rhesus monkeys (Capitanio 1999). Low social ability in infant rhesus monkeys has been shown to predict poor adult social function (Sclafani et al. 2016), consistent with deficits in childhood social interaction and communication as risk factors for the development of autism spectrum disorder in humans (Ozonoff et al. 2010; Jones et al. 2014). We examined sociability across a sample of 203 male monkeys studied in adulthood to determine whether paternal age at conception was a significant contributor to low social function. These monkeys came from the same colony as those used for sequencing, but none of the individuals were the same.
In addition to sociability, we measured the frequency of eight behaviors associated with general social functioning, stratified by sex (Supplemental Table S1). We performed principal component analysis on these variables to reduce dimensionality and to extract a useful general score of social functioning from these behaviors (Supplemental Fig. S4). The first two principal components (PCs) explain >94% of the variance in these observations. Offspring social behavior PC1 explains the tendency for behaviors to be directed toward females versus males, while offspring social behavior PC2 explains overall contact and proximity with both sexes. Social behavior PC2 was significantly correlated with observer ratings of the sociability personality measure (Pearson's r = 0.37, P < 5 × 10−9).
We found no evidence for a relationship between paternal age and any measure of lowered social function (Fig. 4). Rather than a negative effect on sociability, there was a weak positive trend suggested between sociability and parental age at conception (sire age: r = 0.07, P = 0.21; dam age: r = 0.02, P = 0.09). Because there is a positive correlation between sire rank and sire age, we also calculated pairwise partial correlations between sire age and all measures of social functioning while attempting to control for sire rank. None of these correlations were significant (Supplemental Table S2).
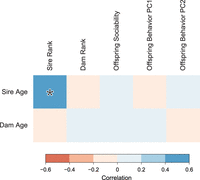
Correlations between parental age and behavioral traits in male rhesus monkeys. Boxes are shaded by the intensity of correlation in pairwise comparisons. Legend shows range of Pearson's correlation coefficient for each color. Significant correlation (P < 0.05) is highlighted with an asterisk.
Discussion
Both the rate and the spectrum of mutations are intimately linked with life history (Walter et al. 2004; Goldmann et al. 2016; Rahbari et al. 2016), complicating comparisons across studies that report point estimates. We discovered a significant paternal age effect on mutation rate in rhesus macaques, consistent with findings from other direct estimates of the mutation rate in primates. The overall per-generation mutation rate in the macaque is substantially lower than has been found in humans and other great apes but similar to the rate in owl monkeys (Thomas et al. 2018). Our analysis indicates that this lower value compared to the apes is largely due to a younger age at reproduction and a lower number of mutations before puberty, with little effect from differences in the rate at which mutations accumulate after puberty. While the effect of maternal age was positive, it was not significant. Nevertheless, the ratio of male-to-female mutations, α ≈ 3, closely matches the value described in human studies (Jónsson et al. 2017; Gao et al. 2019). The similarity in α-values, coupled with the small but significant effect of maternal age on mutation rate in humans, suggests that a maternal age effect might be detected in macaques with larger sample sizes.
In contrast to the per-generation rate, our estimate of the per-year mutation rate in macaques is 1.5 times higher than the estimate in humans, similar to the higher rate found in chimpanzees, gorillas, and orangutans (Besenbacher et al. 2019). Estimates from phylogenetic studies, however, indicate only a 30% higher per-year substitution rate in macaques compared to humans (Kim et al. 2006; Elango et al. 2009). This discrepancy is consistent with a recent, and perhaps ongoing, slowdown in the mutation rate on the human lineage (Goodman 1985; Li and Tanimura 1987; Yi 2013). That is, if a lower per-year mutation rate evolved sometime after the human-chimpanzee divergence, then substitution rate comparisons will underestimate the degree to which the rate has decreased.
Despite accounting for the number of mutations transmitted with paternal age, a model that adjusts for reproductive longevity (Thomas et al. 2018) does not account for all differences in mutation rate between macaques and humans. The biggest difference between these species appears to be the number of mutations present at puberty, before active spermatogenesis begins. It is not clear, however, what changes have occurred before puberty to lower the mutation rate in macaques. Though our data suggest that the mutation rate per-cell division postpuberty is the same between species, it is possible that there are differences between species in the error-prone divisions of early embryogenesis (Huang et al. 2014; Rahbari et al. 2016; Ju et al. 2017). Under such a model, the decreased number of mutations before puberty in the macaque may be explained by a lower number of postzygotic mutations, a process that is not modeled well by mutation rates during spermatogenesis. In any case, the evolution of life-history appears to have played a large role in shaping differences in the per-generation mutation rate between human and macaque.
With the large effect of paternal age on mutation rates within species, differences in key parameters of spermatogenesis—including timing of cell division, cell cycle length, and efficiency—have been hypothesized to explain variation in mutation and substitution rates between species (Wilson Sayres et al. 2011; Thomas and Hahn 2014; Amster and Sella 2016; Moorjani et al. 2016; Scally 2016). The seminiferous epithelial cycling time is one such parameter of particular interest because it makes straightforward predictions about mutation rate variation between species. This time describes the period between successive waves of spermatogenesis in the testis and is known to be 34% shorter in the macaque than in humans (de Rooij et al. 1986). All things being equal, the shorter time between cycles suggests that male macaques should accumulate mutations postpuberty at a higher rate than male humans. However, our results reveal little difference between macaques and humans in how mutation accumulation scales with paternal age. The absence of a proportionate effect on mutation rate brings into question the long-held assumption that spermatogonial stem cells (SSCs) are all actively dividing (Drost and Lee 1995). If division by only a fraction of SSCs are needed to replenish the seminiferous epithelium each cycle, a shorter cycling time would not require a commensurate increase in the number of stem cell divisions (Ehmcke and Schlatt 2006; Scally 2016). Cell proliferation experiments in the macaque suggest that the fraction of SSCs actively dividing varies under endocrine control (Simorangkir et al. 2009; Plant 2010). Relaxing the assumption that spermatogonial stem cells are all actively dividing may also help to explain the reported disconnect between the male-to-female ratio of germline divisions and the ratio of X-to-autosome substitutions (Wilson Sayres and Makova 2011; Ségurel et al. 2014; Gao et al. 2016; Scally 2016).
While our finding of a linearly increasing number of mutations with paternal age in the macaque is consistent with a replication-dependent model of mutagenesis, a role for damage-dependent mutagenesis cannot be ruled out. For instance, a model of mutagenesis that relies solely on environmental damage could explain why differences in the seminiferous epithelial cycling time between species have no effect on the rate of mutation accumulation with paternal age. However, such a model is difficult to reconcile with the differences in substitution rates seen across primates. A damage-dependent model also need not be independent of the rate of cell division, as both the male mutation bias and paternal age effect can be explained if the accumulation of spontaneous mutations relies on the rate of cell division (Gao et al. 2016; Seplyarskiy et al. 2019). In such a model, the repair of DNA lesions is limited by the time between replications: Rapidly dividing cells such as spermatocytes accumulate a greater number of mutations because there is less time for lesions to be repaired. The replication process instead ensures that such lesions appear as mutations in future generations. The presence of a paternal age effect on mutations at CpG sites (e.g., Supplemental Fig. S3), despite their ostensibly replication-independent origin, is better explained by a model of unrepaired damage before replication.
Previous studies have found that both the number of de novo mutations and the risk of neurodevelopmental disorders increase with paternal age in humans (Kong et al. 2012). We find no link between paternal age and negative social behavioral outcomes in offspring, despite an increasing number of mutations with paternal age in the rhesus macaque. We must acknowledge that social behavior is a complex human construct that our assay is unlikely to fully capture. Furthermore, differences in sociability are only a single component in the complex syndromes that constitute neurodevelopmental disorder. One explanation for the slightly positive trend we observe between parental age and sociability may be from younger parents in our behavioral sample. Whereas advanced paternal age is associated with extreme deficits in sociability and neurodevelopmental disorder in the offspring, very young parental age has been associated with reductions to offspring social development (McGrath et al. 2014; Janecka et al. 2017a), though these may be due to the environmental effects of early life rather than a genetic effect (Tung et al. 2016). Alternatively, the absence of an age effect on sociability would be consistent with the hypothesis that the increased risk of neurodevelopmental disorder with paternal age in humans is not primarily driven by de novo mutations (Hultman et al. 2011; Gratten et al. 2016). While this latter hypothesis does not exclude a role for inherited genetic factors in the development of such disorders, it posits no direct role for the elevated de novo mutation rate in the higher risk of disorders observed in offspring of older fathers.
Methods
Sequencing
Genomic DNA isolated from blood samples (buffy coats) from 32 Indian-origin rhesus macaques from the California National Primate Research Center (Univ. of California at Davis) was used to perform whole-genome sequencing. These WGS libraries were sequenced on an Illumina HiSeq X instrument to generate 150-bp paired-end reads (see Supplemental Methods for additional details).
Mapping and variant calling
BWA-MEM version 0.7.12-r1039 (Li 2013) was used to align the Illumina sequencing reads to the rhesus macaque reference assembly Mmul_8.0.1 (GenBank accession GCA_000772875.3) and to generate BAM files for each of the 32 individuals. Picard MarkDuplicates version 1.105 (https://broadinstitute.github.io/picard/) was used to identify and mark duplicate reads. Single nucleotide variants were called using GATK version 3.6 (Van der Auwera et al. 2013) following best practices. We also applied an alternative pipeline, FreeBayes version v0.9.21-19-gc003c1e (Garrison and Marth 2012), to get an independent estimate of de novo mutations. See Supplemental Materials for details on how both methods were run.
Filtering candidate mutations
Our initial list of candidates from GATK variant calls included 300,065 Mendelian violations (MVs) among the 14 trios. The initial list from FreeBayes calls included 23,240 MVs. In the next steps, we progressively applied filters to improve the accuracy of identifying actual de novo mutations. To be specific, we
-
Removed candidate sites that had fewer than 20 reads or more than 60 reads. Sites with too many reads may represent problematic repetitive regions (Li 2014).
-
Removed candidate sites that were not homozygous reference in both parents or appeared as a variant in an unrelated trio. This step reduces the chances that a segregating variant was miscalled as homozygous reference in a parent.
-
Restricted candidate sites to those with high genotype quality (GQ > 70) in both parents and offspring (see Supplemental Fig. S5).
-
Removed heterozygotes in the offspring that did not have at least one alternate read on both the forward and reverse strand (i.e., ADF > 0 and ADR > 0). We also removed candidate sites where homozygote calls in the parent had more than one alternate read on either strand (AD < 2). These alternate allelic depths were evaluated before genotype calling to minimize genotyping errors from local realignment (Karczewski et al. 2019).
-
Removed heterozygotes in the offspring that were called with an allelic depth of <35% alternate reads (Supplemental Fig. S6).
The same filters were applied to MVs from both variant calling pipelines, except for GQ which is not calculated by default in FreeBayes. To evaluate the sensitivity of our mutation calls to the GQ filter, we re-estimated the mutation rate (accounting for callability; see below) at several filter limits (Supplemental Fig. S5). After applying the above filters to the set of MVs from both pipelines, we found: 269 overlapping candidates, 44 unique to FreeBayes, and 38 unique to GATK. Our subsequent analyses use the set of mutations from the GATK calls, but estimated mutation rates are similar between the two pipelines (Table 1).
Estimating the fraction of callable sites
To calculate a mutation rate while considering differences in coverage and filtering, we adapted the strategy from Besenbacher et al. (2019). Raw counts of de novo mutations were converted into a mutation rate by dividing by the total number of callable sites. Estimates
of site callability, the probability that a true de novo mutation would be correctly called as such at a given site x, are factored into estimates of the mutation rate by using the following equation:
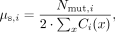 where μs,i is the per-site per-generation mutation rate for trio i, Nmut,i is the number of de novo mutations identified in trio i, and Ci(x) is the callability of site x in that trio. We take x to be a site from the set of all haploid sites with depth between 20 and 60. This strategy assumes that the ability to correctly
call each individual in the trio is independent, allowing us to estimate Ci(x) as
where μs,i is the per-site per-generation mutation rate for trio i, Nmut,i is the number of de novo mutations identified in trio i, and Ci(x) is the callability of site x in that trio. We take x to be a site from the set of all haploid sites with depth between 20 and 60. This strategy assumes that the ability to correctly
call each individual in the trio is independent, allowing us to estimate Ci(x) as
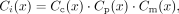 where Cc, Cp, and Cm are the probability of calling the child, father, and mother correctly in trio i. We estimate each of these by considering the proportion of sites that pass our set of filters in a set of high-confidence
calls from each trio. For heterozygous calls in the child, we estimate
where Cc, Cp, and Cm are the probability of calling the child, father, and mother correctly in trio i. We estimate each of these by considering the proportion of sites that pass our set of filters in a set of high-confidence
calls from each trio. For heterozygous calls in the child, we estimate
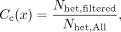 taking Nhet,All to be the number of variants where one parent is homozygous for the reference allele and one parent is homozygous for the
alternate allele with high confidence. Nhet,filtered is the number of heterozygote calls in the child remaining after applying all filters (including ADF > 0, ADR > 0, and alternate
allele depth > 35%).
taking Nhet,All to be the number of variants where one parent is homozygous for the reference allele and one parent is homozygous for the
alternate allele with high confidence. Nhet,filtered is the number of heterozygote calls in the child remaining after applying all filters (including ADF > 0, ADR > 0, and alternate
allele depth > 35%).
Similarly, we estimate parental callability as (in the case of the father)
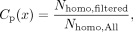 where Nhomo,All is the number of variants where both parents are homozygous for the reference allele with high confidence and Nhomo,filtered is the number of homozygous calls in the child that pass all filters (including AD < 2). Sampled sites were restricted to
those where the variant was present no more than once across all individuals (i.e., allelic count, AC < 2).
where Nhomo,All is the number of variants where both parents are homozygous for the reference allele with high confidence and Nhomo,filtered is the number of homozygous calls in the child that pass all filters (including AD < 2). Sampled sites were restricted to
those where the variant was present no more than once across all individuals (i.e., allelic count, AC < 2).
We estimated callability for each individual, Cc, Cp, and Cm, from a random sample of 250,000 sites across the genome that matched the respective criteria for each trio. The de novo callability, Ci(x), calculated for each trio from this strategy is listed in Table 1. We used our previously published data set on mutation rates in the owl monkey (Thomas et al. 2018) to test this approach. The new pipeline with callability produced an estimate of the per-generation mutation rate for the owl monkey that was within 1% of the original estimate.
Phasing mutations
We traced the parent of origin for de novo mutations that were transmitted to the third generation. This was accomplished by tracking their inheritance on haplotype blocks that we assembled from phase-informative sites. These informative sites were biallelic and had genotypes that were different between grandparents, heterozygous in the next generation, and not heterozygous in both the third-generation proband and the other parent. These phase-informative sites could be traced unambiguously to one of the grandparents. Sites were assembled into haplotype blocks under the assumption that multiple recombination events in a single meiosis were unlikely to occur within a 1-Mb interval (Rogers et al. 2006; Huang et al. 2009; Smeds et al. 2016; Xue et al. 2016).
Mutation rates with parental age
We estimated the effect of parental age on the mutation rate with a Poisson regression, modeling the number of mutations for
trio i as
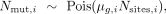 where μg,i is the per-generation mutation rate in trio i and Nsites,i =
where μg,i is the per-generation mutation rate in trio i and Nsites,i = 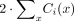 , the diploid callable genome size for trio i (see Supplemental Methods).
, the diploid callable genome size for trio i (see Supplemental Methods).
We compared our Poisson regression model for the mutation rate in rhesus macaque to a model of the mutation rate in human. Since maternal age was not significantly correlated with mutations in macaque, we omitted the variable in the comparison between species. This yielded the following regression coefficients: βN = 2.43 × 10−9, 95% CI [1.47, 3.38], βP = 4.27 × 10−10 [3.01, 5.53] for macaque and βN = 2.56 × 10−9 [2.26, 2.86], βP = 3.37 × 10−10 [3.28, 3.47] for human. To test whether coefficients in this regression were significantly different, we used an unequal variances t-test and conducted simulations to determine its power to detect differences in the age effect between species.
We predicted the number of mutations at different paternal ages using the above regression model and a diploid genome size (i.e., Nsites,i) approximated by twice the UCSC golden path length for each species (https://genome.ucsc.edu). If the same number of mutations accumulate before puberty in rhesus macaque as in human and the rate of accumulation after puberty is the same, our sample of macaques should have the same number of mutations as an average 17.8-yr-old human. This is based on the mean paternal age of 7.8 yr among macaques in our data set and an age of 3.5 yr for male puberty in macaques (Plant et al. 2005), resulting in 4.3 yr of postpuberty mutation accumulation. Together with the 13.5 yr to reach male puberty in humans, the corresponding age for the human model becomes 17.8 yr (= 13.5 + 4.3).
Collecting sociability data from captive rhesus monkeys
We observed 203 male rhesus monkeys at the California National Primate Research Center, in cohorts of 5–8 animals (mean age = 6.9 yr, range = 4.0 to 19.2 yr), unobtrusively in their half-acre outdoor enclosures across four summers. Observations were conducted over the course of 8 d within a 2-wk period and consisted of two 10-min sessions per day. Using focal animal sampling (Altmann 1974), behavioral observers recorded the frequencies of the following behaviors directed at other animals: approach (locomotion to within arm's reach), proximity (being within arm's reach for at least 3 sec), contact (physical, nonaggressive contact between animals), and grooming (picking with fingers and/or licking another animal's hair). Following completion of behavioral observations on each cohort, the observer rated each animal using a seven-point Likert-type scale on three trait adjectives. Previous work (Capitanio and Widaman 2005) had demonstrated these ratings form a scale that reflects the personality characteristic Sociability (see Supplemental Methods for details).
Data access
All sequencing data generated in this study have been submitted to the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession numbers SRR10693549–SRR10693581.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was funded by the Precision Health Initiative of Indiana University. Behavioral assessments (thanks to K. Bone for collecting these data) were made possible by funding from California National Primate Research Center (CNPRC) base grant P51 OD011107 and National Institutes of Health (NIH) grants R37 AG033590 and R24 OD010962. We also thank the production staff of the Human Genome Sequencing Center and its Director, Richard Gibbs. Three reviewers gave valuable feedback that greatly improved the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.255174.119.
- Received July 29, 2019.
- Accepted May 21, 2020.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








