
Distinct types of short open reading frames are translated in plant cells
- Igor Fesenko1,
- Ilya Kirov2,
- Andrey Kniazev1,
- Regina Khazigaleeva1,
- Vassili Lazarev3,4,
- Daria Kharlampieva3,
- Ekaterina Grafskaia3,4,
- Viktor Zgoda5,
- Ivan Butenko3,
- Georgy Arapidi1,3,
- Anna Mamaeva1,
- Vadim Ivanov1 and
- Vadim Govorun3
- 1Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry of the Russian Academy of Sciences, 117997 Moscow, Russian Federation;
- 2Laboratory of marker-assisted and genomic selection of plants, All-Russian Research Institute of Agricultural Biotechnology, 127550 Moscow, Russian Federation;
- 3Federal Research and Clinical Center of Physical-Chemical Medicine of Federal Medical Biological Agency, 119435 Moscow, Russian Federation;
- 4Moscow Institute of Physics and Technology (National Research University), 141701 Dolgoprudny, Moscow Region, Russian Federation;
- 5Laboratory of System Biology, Institute of Biomedical Chemistry, 119121 Moscow, Russian Federation
Abstract
Genomes contain millions of short (<100 codons) open reading frames (sORFs), which are usually dismissed during gene annotation. Nevertheless, peptides encoded by such sORFs can play important biological roles, and their impact on cellular processes has long been underestimated. Here, we analyzed approximately 70,000 transcribed sORFs in the model plant Physcomitrella patens (moss). Several distinct classes of sORFs that differ in terms of their position on transcripts and the level of evolutionary conservation are present in the moss genome. Over 5000 sORFs were conserved in at least one of 10 plant species examined. Mass spectrometry analysis of proteomic and peptidomic data sets suggested that tens of sORFs located on distinct parts of mRNAs and long noncoding RNAs (lncRNAs) are translated, including conserved sORFs. Translational analysis of the sORFs and main ORFs at a single locus suggested the existence of genes that code for multiple proteins and peptides with tissue-specific expression. Functional analysis of four lncRNA-encoded peptides showed that sORFs-encoded peptides are involved in regulation of growth and differentiation in moss. Knocking out lncRNA-encoded peptides resulted in a decrease of moss growth. In contrast, the overexpression of these peptides resulted in a diverse range of phenotypic effects. Our results thus open new avenues for discovering novel, biologically active peptides in the plant kingdom.
The genomes of nearly all organisms contain hundreds of thousands of short open reading frames (sORFs; <100 codons) whose coding potential has been the subject of recent reviews (Andrews and Rothnagel 2014; Couso 2015; Hellens et al. 2016; Couso and Patraquim 2017; Rothnagel and Menschaert 2018; Ruiz-Orera and Albà 2019). However, gene annotation algorithms are generally not suited for dealing with sORFs because short sequences are unable to obtain high conservation scores, which serve as an indicator of functionality (Ladoukakis et al. 2011). Nevertheless, using various bioinformatic approaches, sORFs with high coding potential have been identified in a range of organisms including fruit flies, mice, yeast, and Arabidopsis thaliana (Ladoukakis et al. 2011; Hanada et al. 2013; Aspden et al. 2014; Bazzini et al. 2014). The first systematic study of sORFs was conducted on baker's yeast, where 299 previously nonannotated sORFs were identified and tested in genetic experiments (Kastenmayer et al. 2006). Subsequently, 4561 conserved sORFs were identified in the genus Drosophila, 401 of which were postulated to be functional, taking into account their syntenic positions, low Ka/Ks (<0.1) values, and transcriptional evidence (Ladoukakis et al. 2011). In a recent study, Mackowiak and colleagues predicted the presence of 2002 novel conserved sORFs (from nine to 101 codons) in Homo sapiens, Mus musculus, Danio rerio, Drosophila melanogaster, and Caenorhabditis elegans (Mackowiak et al. 2015). The first comprehensive study of sORFs in plants postulated the existence of thousands of sORFs with high coding potential in Arabidopsis (Lease and Walker 2006; Hanada et al. 2007, 2013), including 49 that induced various morphological changes and had visible phenotypic effects.
Recent studies have pointed to the important roles of sORF-encoded peptides (SEPs) in cells (Magny et al. 2013; Nelson et al. 2016; D'Lima et al. 2017; Huang et al. 2017; Matsumoto et al. 2017; Rubtsova et al. 2018). However, unraveling the roles of SEPs is a challenging task, as is their detection at the biochemical level. In animals, SEPs are known to play important roles in a diverse range of cellular processes (Kondo et al. 2010; Magny et al. 2013). In contrast, only a few functional SEPs have been reported in plants, including POLARIS (PLS; 36 amino acids [aa]), EARLY NODULIN GENE 40 (ENOD40; 12, 13, 24, or 27 aa), ROTUNDIFOLIA4 (ROT4; 53 aa), KISS OF DEATH (KOD; 25 aa), BRICK1 (BRK1; 84 aa), Zm-908p11 (97aa), and Zm-401p10 (89 aa) (Andrews and Rothnagel 2014; Tavormina et al. 2015). These SEPs help modulate root growth and leaf vascular patterning (Chilley et al. 2006), symbiotic nodule development (Djordjevic et al. 2015), polar cell proliferation in lateral organs and leaf morphogenesis (Narita et al. 2004), and programmed cell death (apoptosis) (Blanvillain et al. 2011).
To date, functional sORFs have been found in a variety of transcripts, including untranslated regions of mRNA (5′ leader and 3′ trailer sequences), lncRNAs, and microRNA transcripts (pri-miRNAs) (Andrews and Rothnagel 2014; Laing et al. 2015; Lauressergues et al. 2015; Couso and Patraquim 2017; Brunet et al. 2019). Evidence for the transcription of potentially functional sORFs has been obtained in Populus deltoides, Phaseolus vulgaris, Medicago truncatula, Glycine max, and Lotus japonicus (Guillen et al. 2013). The transcription of sORFs can be regulated by stress conditions and depends on the developmental stage of the plant (De Coninck et al. 2013; Hanada et al. 2013; Rasheed et al. 2016). Indeed, sORFs might represent an important source of advanced traits required under stress conditions. During stress, genomes undergo widespread transcription to produce a diverse range of RNAs (Kim et al. 2010; Mazin et al. 2014); therefore, a large portion of sORFs becomes accessible to the translation machine for peptide production. Stress conditions can lead to the transcription of sORFs located in genomic regions that are usually noncoding (Giannakakis et al. 2015). Such sORFs appear to serve as raw materials for the birth and subsequent evolution of new protein-coding genes (Couso and Patraquim 2017; Ruiz-Orera and Albà 2019).
The transcription of a sORF does not necessarily indicate that it fulfills any biological role, as opposed to being a component of the so-called translational noise (Guttman et al. 2013). According to ribosomal profiling data, thousands of lncRNAs display high ribosomal occupancy in regions containing sORFs in mammals (Ingolia et al. 2011; Aspden et al. 2014; Bazzini et al. 2014). However, lncRNAs can have the same ribosome profiling patterns as canonical noncoding RNAs (e.g., rRNA) that are known not to be translated, implying that these lncRNAs are unlikely to produce functional peptides (Guttman et al. 2013). In addition, identification of SEPs via mass spectrometry analyses has found many fewer peptides than predicted sORFs (Slavoff et al. 2013; Aspden et al. 2014). Thus, the abundance, lifetime, and other features of SEPs are generally unclear.
We performed a comprehensive analysis of the sORFs that have canonical AUG start codons and high coding potential in the Physcomitrella patens genome. The translation of tens of sORFs was confirmed by mass spectrometry analysis. From these, candidate lncRNA-encoded peptides were selected for further analysis, which provided evidence for their biological functions.
Results
Discovery and classification of potential coding sORFs in the moss genome
Our approach is summarized in Figure 1A. At the first stage of analysis, we used the sORF finder tool (Hanada et al. 2010) to identify single-exon sORFs starting with an AUG start codon and <300 bp long. This approach resulted in the identification of 638,439 sORFs with coding potential (CI index) in all regions of the P. patens genome.
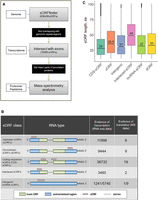
Several distinct types of sORFs are present in the moss genome. (A) Pipeline used in this study to identify coding sORFs. (B) Proposed classification of sORFs according to the types of encoding transcripts: upstream ORFs (uORFs) and downstream ORFs (dORFs) in the untranslated regions (UTRs) of canonical mRNAs; CDS-sORFs, which overlap with protein-coding sequences in alternative (+2 or +3) reading frames or are truncated versions of proteins generated by alternative splicing; interlaced-sORFs, which overlap both the protein-coding sequence and UTR on the same transcript; lncRNA-sORFs and intergenic sORFs, which are located on short nonprotein-coding transcripts. (C) Box plot of the length distribution of sORFs in different groups.
We selected 70,095 unique sORFs located on transcripts annotated in the moss genome (https://phytozome.jgi.doe.gov/pz/portal.html) and/or our data set (Fesenko et al. 2015) for further analysis, as well as those on lncRNAs from two databases—CANTATAdb (Szcześniak et al. 2016) and GreeNC (Paytuvi Gallart et al. 2016); sORFs located in repetitive regions were discarded (Supplemental Table S1). These selected sORFs, which were 33–303 bp long, were located on 33,981 transcripts (22,969 genes), with up to 28 sORFs per transcript (Supplemental Fig. S1A).
We then classified the sORFs based on their location on the transcript: 63,109 “genic-sORFs” (located on annotated transcripts but not on lncRNA); 1241 “intergenic-sORFs” (located on transcripts from our data set and not annotated in the current version of the genome); and 5745 “lncRNA-sORFs” (located on lncRNAs from CANTATAdb (Szcześniak et al. 2016), GreeNC (Paytuvi Gallart et al. 2016), or our data set (Fig. 1B; Fesenko et al. 2017). The genic-sORFs include 11,998 upstream ORFs (uORFs; for 5′ UTR location), 9443 downstream ORFs (dORFs; for 3′ UTR location), 36,732 coding sequence-sORFs (CDS-sORFs; sORFs overlapping with main ORFs [+1 frame] in noncanonical +2 and +3 reading frames), and 3485 interlaced-sORFs (overlapping with both the CDS and 5′ UTR or CDS and 3′ UTR on the same transcript) (Fig. 1B; Supplemental Fig. S1B).
As expected based on the sORF finder search strategy (Hanada et al. 2010), the sORF set was enriched in CDS-sORFs (52%, Fisher's exact test, P-value < 10−16), whereas dORFs, uORFs, and interlaced-sORFs were underrepresented (Fisher's exact test, P-value < 10−16) compared to a random exonic fragments set, which was used as a negative control. On average, CDS-sORFs (median size of 22 codons) were shorter than uORFs (median size of 35 codons; Mann–Whitney U test P-value < 10−16) and dORFs (median length 32 codons, Mann–Whitney U test P-value < 10−16). The median size of interlaced-sORFs was 49 codons, which is significantly longer than other genic-sORFs (Mann–Whitney U test P = 0.0021) (Fig. 1C).
To estimate the number of conserved transcriptable sORFs, we performed a TBLASTN search (e-value cutoff 0.00001) of each sORF sequence against the reconstructed genomes of three P. patens ecotypes, Villersexel, Reute, and Kaskasia, as well as the transcriptomes of 10 plant species (Supplemental Fig. S2). We found 5034 conserved sORFs with detectable homologous sequences in at least one species (Supplemental Fig. S3; Supplemental Table S1). A conservation analysis of the sORFs in the reconstructed P. patens ecotypes showed that 2.4% (1618) of the sORFs were lacking either the start or stop codons in at least one species. We then examined the differences in selection pressure at the amino acid level between different major groups of conservative sORFs (CDS-sORFs, uORFs, dORFs, lncRNA-sORFs, interlaced-sORFs) using the criterion of Ka/Ks. Higher retention rates were observed for uORFs and dORFs, whereas CDS-sORFs and lncRNA-ORFs were under strong positive selection (Supplemental Fig. S4). These observations are in agreement with the fact that some types of sORFs (for example, uORFs) play a regulatory role instead of being translated (Barbosa et al. 2013).
Experimental evidence for the translation of sORFs
Obtaining evidence for the translation of sORFs is an important step toward identifying functional SEPs. We analyzed the Kozak consensus sequences (Kozak 1986) surrounding sORF start codons. Kozak consensus sequence plays an important role in translation initiation (Kozak 1997). Depending on the presence of the purine in position −3 and the G in position +4 (where +1 is “A” in the “AUG” codon), the Kozak was considered to be “strong” (both are present), “medium” (one is present), or “weak” (neither are present) (Kozak 1997). According to our results, 41,816 (∼60%) of the predicted sORFs were surrounded by “strong” and “medium” Kozak sequences. These values were significantly smaller than those of annotated protein-coding ORFs (87%, Fisher's exact test P-value < 2.2 × 10−16).
We then verified the translation of our predicted sORFs using mass spectrometry (MS) analysis. Taking into account the shortage of proteomic methods for identifying small proteins or peptides, in the current study, we generated two data sets: the “peptidomic” data set—endogenous peptides extracted from three types of moss cells: gametophores, protonemata, and protoplasts; and the “proteomic” data set—tryptic peptides generated in a standard proteomic pipeline (Supplemental Table S2). All data sets were mapped with MaxQuant (Tyanova et al. 2016) against a custom database containing our sORFs together with nuclear, chloroplast, and mitochondrial moss protein sequences (see details in the Methods). Peptide spectrum matches (PSMs) were identified at 1% FDR, and ambiguous peptides were filtered out. This resulted in 1177 PSMs corresponding to 296 distinct peptide sequences in the peptidomic data set and 920 PSMs corresponding to 532 peptide sequences in the proteomic data set. To generate a high-confidence sORF candidate set, we increased our acceptance threshold to a minimum posterior error probability (PEP) of 0.01 and Andromeda score of higher than 60. The final set underwent a manual inspection of spectra. As a result, we confirmed the translation of 46 sORFs: 17 in gametophores, 29 in protonemata, and 14 in protoplasts (“confident sORFs”) (Fig. 2A; Supplemental Table S3). The length of these small protein-coding sORFs ranged from 14 to 99 aa, which were generally longer than untranslatable sORFs (Mann–Whitney U test P-value = 5.33 × 10−6) (Fig. 2B). We observed that PSMs supporting SEP identifications had lower average quality than those mapped to the protein sequences (Supplemental Fig. S5A,B). This finding is in agreement with data obtained for the animal kingdom (Slavoff et al. 2013; Mackowiak et al. 2015). The quality of spectra and the values of PSMs supporting the expression of SEPs were better in the “peptidomic” data set (Supplemental Fig. S5C). Also, translatable sORFs were longer for those identified in the peptidomic data set (Supplemental Fig. S5D). Approximately 63% of the translated sORFs (29 sORFs) contained “strong” and “medium” Kozak elements, which is similar to the results obtained for all predicted sORFs (∼60%). This result suggests that translation initiation may differ for sORFs and protein-coding ORFs.
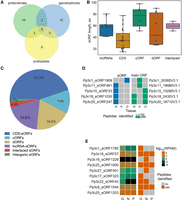
Moss contains tens of small protein-coding sORFs. (A) Venn diagram showing the distribution of the identified translatable sORFs among three types of moss cells. (B) Length distribution of various groups of small protein-coding sORFs. (C) Distribution of small protein-coding sORFs based on the suggested classification. (D) Binary heat map showing evidence of translation for sORFs and proteins in multicoding genes in three moss tissues. G, N, and P correspond to gametophores, protonemata, and protoplasts, respectively. (E) Heat map showing expression levels (log10[RPKM]) for the lncRNAs (left) carrying sORFs (lncRNA-sORFs) and binary heat map showing evidence of translation (determined as whether a peptide was identified [brown] or not [gray] in MS data) for the corresponding lncRNA-sORFs (right) in three moss tissues: gametophores (G), protonemata (N), and protoplasts (P).
The most prominent group of small protein-coding sORFs consisted of CDS-sORFs (19 sORFs, 41.3%) (Fig. 2C). Also, the translation of uORFs (six sORFs, 13%) and dORFs (nine sORFs, 19.6%) was confirmed by our analysis. Based on our MS data, we identified seven loci with at least two translated ORFs (annotated as main ORF and sORF), including five CDS-sORFs, that represent putative multicoding genes (Fig. 2D; Supplemental Table S4). Some of the putative multicoding genes were translated simultaneously with protein-coding ORFs in the same type of moss cell (e.g., Pp3c11_sORF461), while others showed different patterns of sORF and main ORF translation (e.g., Pp3c1_sORF1909). These findings indicate that small protein-coding CDS-sORFs are expressed simultaneously with main ORFs and the translation of sORFs and proteins located together in the same locus might be regulated in a tissue-specific manner.
The translation of nine sORFs located on lncRNAs was also detected by our analysis. The level of transcription of some lncRNAs (according to the previous data [Fesenko et al. 2017] and Phytozome 12.0 expression atlas) and evidences of translation for the corresponding lncRNA-sORFs are shown in Figure 2E. Three of these SEPs, Pp3c18_sORF57 (40 aa), Pp3c9_sORF1544 (41 aa), and Pp3c25_sORF1000 (61 aa), were common to all three cell types and were confirmed by several unique endogenous peptides (Fig. 2E). These data may point to biological significance for the peptides translated from these sORFs rather than the sORFs having regulatory functions in the translation of the main ORF. To explore this notion, we investigated the functions of four SEPs encoded by lncRNAs (see below).
Most small protein-coding sORFs are not evolutionarily conserved
Analysis of the evolutionary conservation of sORFs is often a key step in revealing biologically active sORFs (Andrews and Rothnagel 2014). To investigate whether the trend in small protein-coding sORF evolution differs from that of the other sORFs, we estimated the number of species in which homologs can be found and the selection pressure (Ka/Ks) on translatable sORFs on an evolutionary timescale using the transcriptomes of the 10 above-mentioned species. Overall, we found that five sORFs had evidence of translation and conservation in at least one species, and four of them were under negative selection (Ka/Ks << 1). Thus, analysis of sORF sequence conservation showed that only 11% of our small protein-coding sORFs have a signature of conservation between species.
Alternative splicing regulates the number of sORFs in protein-coding transcripts
Alternative splicing (AS) is a universal process among eukaryotic organisms, and more than 50% of P. patens genes are alternatively spliced (Chang et al. 2014; Wu et al. 2014; Fesenko et al. 2017). AS events may lead to the specific gain, loss, or truncation of different groups of sORFs located on the transcripts of the same gene. We found 6092 alternatively spliced sORFs (AS-sORFs) belonging to transcripts from 4389 genes. CDS-sORFs were significantly overrepresented (Supplemental Fig. S6) while interlaced-sORFs, uORFs, and dORFs were significantly underrepresented among AS-sORFs compared to the control set of random exonic fragments. We found that approximately half of the entire set of AS-sORFs (48%, 2933) underwent complete excision (complete sORF removal from an isoform) (Fig. 3). The complete excision of sORFs occurred significantly more frequently in uORFs (57% of all AS-sORFs) than in the other AS-sORF groups (20%–44% of all AS-sORFs, Fisher's exact test P-value < 10−6). Among small protein-coding AS-sORFs, we found three affected by stop codon excision. Two of the translatable AS-sORFs were affected by start codon excision and one had undergone complete excision. We then randomly selected 13 different AS-sORFs with/without evidence of translation and searched for the corresponding isoforms in the transcriptomes of three types of moss cells. RT-PCR analysis revealed the transcription of these isoforms, confirming that they could indeed be translated (Supplemental Fig. S7). Moreover, some sORFs contained isoforms showing tissue-specific transcription. These observations led to the hypothesis that the translation of sORFs is regulated by AS.
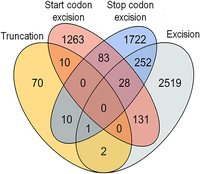
Venn diagram showing the number of AS-sORFs influenced by different AS events.
The formation of a premature termination codon (PTC) as a result of alternative splicing events might lead to mRNA decay (Ge and Porse 2014; Karousis et al. 2016) and rapid nonsense-mediated decay (NMD)-coupled degradation of sORF-encoded peptides (Popp and Maquat 2013). Using recently published transcriptomic data from moss NMD-deficient mutants (Lloyd et al. 2018), we investigated whether our translatable sORFs were present on NMD-targeted transcripts. Only one CDS-sORF (Pp3c7_sORF1583) was potentially present on such transcripts. Therefore, it is difficult to judge if AS-sORFs can trigger NMD-dependent transcript degradation.
Thus, our analysis demonstrated that AS might regulate the excision of sORFs from the transcriptome of P. patens, preventing AS-sORF translation by start or stop codon as well as complete sORF excision.
The sequence similarity analysis reveals sORFs with high identity to coding genes
Competitive inhibitors of protein–protein interactions (PPI) are referred to as microProteins (miPs) or small interfering peptides (siPEPs) and can be generated by alternative splicing or evolutionarily generated by domain loss (Seo et al. 2011; Staudt and Wenkel 2011; Eguen et al. 2015). Using BLASTP (e-value < 10−6) similarity searches, we identified 363 sORFs resulting from AS events that partially overlapped with the main ORF, thereby generating truncated versions of the proteins (cis-sORFs) (Supplemental Table S5). We found that 60 cis-sORFs harbored intrinsically disordered regions (IDRs) (van der Lee et al. 2014), while 30 contained parts of 28 different domains (Supplemental Table S5). However, we did not identify small protein-coding cis-sORFs in our data set. It could be explained by a significant overlap with the protein sequences, whereas we filtered out the “ambiguous” PSMs.
We then identified 272 sORFs that shared similarity with annotated proteins but were located on other transcripts (trans-sORFs) (see in Supplemental Table S5). Trans-sORFs may have originated through the divergence of ancient paralogous genes, which occurred after the paleo duplication of the moss genome (Rensing et al. 2007, 2008). In fact, 159 (58.5%) trans-sORFs shared similarity to genes from at least one species. In addition, all of these trans-sORFs are under strong purifying selection (Ka/Ks << 1).
Several distinct clusters with sORF-encoded peptides sharing similarity with more than four proteins from distinct genes were detected (Supplemental Fig. S8). Each cluster encompasses genes from different protein families, including one containing leucine-rich repeat and zinc-finger domains involved in protein–protein and protein–nucleic acid interactions, respectively. We examined the coexpression data and compared the distribution of correlation coefficient values between potential SEPs and their targets with those from randomly selected pairs (10 iterations) of genes. On average, these sORF-protein pairs had higher correlation coefficients than randomly selected gene pairs (Wilcoxon rank-sum and Kolmogorov-Smirnov tests P-value < 0.05), implying that sORF-bearing and target genes are frequently coexpressed.
SEPs regulate moss growth
Despite the recent finding that 10% of overexpressed intergenic sORFs have clear phenotypes in Arabidopsis (Hanada et al. 2013), the functions of most sORFs and SEPs in plants are generally unknown. Known bioactive SEPs in plants are encoded by sORFs located on short nonprotein-coding transcripts, which can be referred to as lncRNAs (Rohrig et al. 2002; Chilley et al. 2006). In this context, it would be important to determine how many plant lncRNAs encode peptides, as well as the biological functions of these SEPs. Our pipeline allowed us to identify translated sORFs, including those encoded by lncRNAs. Some of these lncRNA-sORFs showed tissue-specific transcription and translation patterns, while others were expressed in all types of moss cells (Fig. 2E). We reasoned that stably expressed lncRNA-sORFs can produce peptides that play fundamental roles in various cellular processes. To explore this hypothesis, we examined the impact of lncRNA-sORF overexpression and knockout on moss morphology using four lncRNAs-sORFs: Pp3c9_sORF1544, Pp3c25_sORF1253, Pp3c25_sORF1000, and Pp3c18_sORF57 (Fig. 2E). The translation of these SEPs was confirmed by several unique peptides, and they contained “strong” and “medium” Kozak elements. We obtained multiple independent mutant lines for each of these lncRNAs-sORFs (Supplemental Figs. S9–S12). Both the overexpression and knockout of sORFs resulted in morphological changes, implying that these peptides play a role in growth and development of P. patens (Figs. 4, 5; Supplemental Table S6).
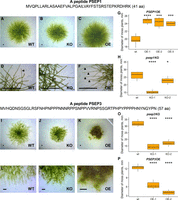
Morphology of wild-type and sORF-encoded peptide mutant lines. The phenotypes of psep1 KO and PSEP1 OE lines grown on BCD medium with 0.5% glucose: (A,D) wild type; (B,E) knockout of PSEP1; (C,F) overexpression of PSEP1. (G) Diameter of moss plants with overexpression of PSEP1 (Supplemental Fig. S14A–D; Supplemental Table S6). (H) Diameter of moss plants with knockout of PSEP1 (Supplemental Fig. S13A–C; Supplemental Table S6). The phenotypes of psep3 KO and PSEP3 OE lines grown on BCD medium: (I,L) wild type; (J,M) knockout of PSEP3; (K,N) overexpression of PSEP3. (O) Diameter of moss plants with knockout of PSEP3 (Supplemental Fig. S13G–I; Supplemental Table S6). (P) Diameter of moss plants with overexpression of the PSEP3 (Supplemental Fig. S14E–H; Supplemental Table S6). Scale bar: 0.5 mm. P-value was calculated by Student's unpaired t-test. (****) P-value < 0.0001, (***) P-value < 0.001, (*) P-value < 0.05.
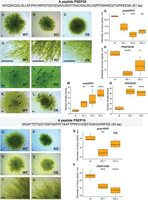
Morphology of wild-type and sORF-encoded peptide mutant lines. The phenotypes of psep25 KO and PSEP25 OE lines grown on BCDAT medium: (A,D) wild type; (B,E) knockout of PSEP25; (C,F) overexpression of PSEP25. (G) Diameter of moss plants with knockout of PSEP25 (Supplemental Fig. S13J–M; Supplemental Table S6). (H) Diameter of moss plants with overexpression of PSEP25 (Supplemental Fig. S14M–P; Supplemental Table S6). (I,J,M) Number of leafy shoots in wild-type and three psep25 KO lines. (K,L,N) Number of leafy shoots in wild-type and two PSEP25 OE lines on BCD medium. Arrows show young leafy gametophores. The phenotypes of psep18 KO and PSEP18 OE lines grown on BCD medium with 0.5% glucose: (O,R,T) wild type; (P) knockout of PSEP18. (Q) Diameter of moss plants with knockout of PSEP18 (Supplemental Fig. S16A–C; Supplemental Table S6). (S,U) Overexpression of PSEP18. (V) Diameter of moss plants with overexpression of PSEP18 (Supplemental Fig. S16D–F; Supplemental Table S6). Scale bar: 0.5 mm. P-value was calculated by Student's unpaired t-test. (****) P-value < 0.0001, (***) P-value < 0.001, (**) P-value < 0.01, (*) P-value < 0.05.
Overexpression of a 41-aa peptide (PSEP1, Physcomitrella patens sORF encoded peptide 1) encoded by the lncRNA-sORF Pp3c9_sORF1544 resulted in longer caulonema cells (filaments implicated in a rapid radial extension of the protonemal tissues) compared to the wild-type and psep1 knockout lines (Fig. 4A–F,G; Supplemental Figs. S13A–F, S14A–D). Rapid growth in the PSEP1 overexpressing lines (OE) was accompanied by earlier aging and cell death (Supplemental Fig. S15). In contrast, there was a small but significant difference in growth rate between the wild-type and psep1 mutant lines grown on solid media and in the liquid culture without glucose (Fig. 4H; Supplemental Fig. S13A–F).
The lines with a knockout in a 57-aa peptide (psep3 KO) encoded by conservative lncRNA-sORF Pp3c25_sORF1253 displayed a decrease in growth rate and altered filament branching (Fig. 4I–O). In the wild-type moss plants, a pale-green diffuse network of caulonemal filaments surrounded the central zone (principally chloronemata), while psep3 KO mutant lines displayed short lateral filaments on medium without glucose and ammonium tartrate, which favors chloronemal growth (Supplemental Fig. S13G–I). Overexpression of PSEP3 (PSEP3 OE) resulted in a significant decrease in growth rate compared to the wild type (Fig. 4P). Moreover, much of the PSEP3 OE protonemal tissue grown on medium without ammonium tartrate turned brown (Fig. 5K–N; Supplemental Fig. S14E–L).
Similar to the results for the psep3 knockout, knocking out a 61-aa peptide (psep25 KO) encoded by conserved lncRNA-sORF Pp3c25_sORF1000 also resulted in a decrease in growth rate and altered protonemal architecture on medium without glucose but supplemented with ammonium tartrate (Fig. 5A–G; Supplemental Fig. S13J–M). PSEP25-overexpressing mutant lines displayed a slight decrease in growth rate compared to the wild type (Fig. 5H) and almost no morphological differences in protonemal tissue structure (Supplemental Fig. S14M–P). In contrast to the PSEP25 OE lines, psep25 knockouts had a significant increase in the number of leafy shoots on medium without glucose but supplemented with ammonium tartrate, which usually reduces gametophore development (Fig. 5I–M). However, PSEP25 OE mutant lines displayed an increase in the number of leafy shoots compared to the wild type on solid medium without ammonium tartrate (Fig. 5K–N).
Knocking out a 40-aa peptide (psep18 KO) encoded by lncRNA-sORF Pp3c18_sORF57 showed a slight decrease in moss plant diameter on medium with glucose and without ammonium tartrate (Fig. 5O–Q; Supplemental Fig. S16A–C). However, only one psep18 knockout line (KO-1) (Fig.5Q) with deletion of a start codon had a significant decrease in growth rate compared to the wild type. Taking into account that sORFs can trigger NMD in lncRNAs (Ruiz-Orera and Albà 2019), this fact requires more detailed investigation. PSEP18 OE lines displayed a significant decrease in growth rate compared to the wild type on medium with glucose (Fig. 5R–V; Supplemental Fig. S16D–F). We did not observe any changes in protonemal architecture and in the number of leafy shoots or filament branching in both overexpressing mutant lines and knockouts.
Taken together, our findings suggest that lncRNA-sORFs can influence growth and development in moss.
Discussion
Although functionally characterized SEPs have been shown to play fundamental roles in key physiological processes, sORFs are arbitrarily excluded during genome annotation. Given the difficulty in identifying translatable, functional sORFs, we know little about their origin, evolution, and regulation in the genome. In the present study, we investigated the abundance, evolutionary history, and possible functions of sORFs in the genome of the model moss Physcomitrella patens. The use of an integrated pipeline that includes transcriptomics, proteomics, and peptidomics data allowed us to identify tens of small protein-coding sORFs in three types of moss cells.
sORFs with high coding potential are not conserved among genomes
Although analyzing the conservation of short amino acid sequences is not trivial (Moyers and Zhang 2016), hundreds of conserved sORFs have recently been identified in plants, yeast, and animals (Ladoukakis et al. 2011; Hanada et al. 2013; Mackowiak et al. 2015; Brunet et al. 2019). The number of sORFs conserved in the plant kingdom is undoubtedly underestimated due to the low sensitivity of tools used for conservation analysis and the limited number of available sequenced genomes from closely related species. Our pipeline allowed us to identify 5034 conserved sORFs among the transcriptomes of 10 different plant species, five of which showed evidence of translation according to our MS data. Three of five conserved sORFs belonged to lncRNAs. These data are in line with a previously published study showing that a large fraction of small ORFs in the mouse genome evolves neutrally (Ruiz-Orera et al. 2018). We also found that uORFs and dORFs were significantly underrepresented among the sORFs that are conserved in the closest related species. We even detected rapid inactivation of uORFs and dORFs in the reconstructed genomes of three P. patens ecotypes due to disruptions in the start or stop codons (47% of the total disrupted sORFs). As the occurrence of sORFs downstream from or upstream of the main ORF can be deleterious to its translation or induce nonsense-mediated decay (NMD), we cannot rule out the possibility that this may cause strong selection pressure and the rapid elimination of uORFs and dORFs (Iacono et al. 2005; Neafsey and Galagan 2007; Johnstone et al. 2016; Ruiz-Orera and Albà 2019). Taken together, these findings suggest that sORFs located in untranslated regions of mRNAs are evolving rapidly and may play regulatory roles rather than encoding bioactive peptides.
In recent studies, thousands of alternative proteins were experimentally detected in human cell lines (Vanderperre et al. 2013; Samandi et al. 2017; Brunet et al. 2019). In P. patens, we found tens of thousands of sORFs (CDS-sORFs) that overlapped with the CDS of protein-coding genes. The evolution of CDS-sORFs is undoubtedly an expensive process for the cell, as these elements may be located in regions encoding protein domains and influence the structure and function of the protein encoded by the main ORF (Cherry 2010). We found both CDS-sORFs originated from regions associated with known protein domains and CDS-sORFs from disordered regions, with higher conservation for CDS-sORFs originated from protein domain-encoding regions. These results indicate that the evolution of CDS-sORFs depends on their locations inside main CDS sequence. However, whether sORFs are preferentially generated in fast-evolving regions of proteins or whether the selective pressure on sORFs leads to changes in protein-coding sequences is still unknown.
Analysis of sORF translation: approaches that make sense
It was recently suggested that sORFs are randomly generated in a genome (Couso and Patraquim 2017). Assuming that the average length of a sORF is ∼60 bp and that sORFs do not overlap, these elements occupy a substantial portion of the moss genome. This raises the question: To what extent are sORFs present in the transcriptome and the proteome of a cell? According to ribosome profiling data from a wide variety of species, sORFs translation appears to occur in a pervasive manner (Ingolia et al. 2011; Guttman et al. 2013; Bazzini et al. 2014; Couso and Patraquim 2017). However, ribosome-profiling data alone are not sufficient to classify transcripts as coding or noncoding (Guttman et al. 2013). Mass spectrometry studies have thus far confirmed the presence of a few dozen SEPs in the peptidomes of animal cells (Slavoff et al. 2013; Prabakaran et al. 2014; Mackowiak et al. 2015; Ma et al. 2016; Tharakan et al. 2019). Comparisons of ribosome profiling and mass spectrometry results have led to the conclusion that MS detects peptides arising from the most highly translated sORFs (Aspden et al. 2014; Bazzini et al. 2014). However, a recent study showed that there are no technical obstacles to the detection of sORF-encoded peptides by mass spectrometry (Verheggen et al. 2017).
In previous studies, only standard proteomics analysis was used to identify SEPs. We reasoned that analyzing endogenous peptide pools instead of tryptic peptides has some disadvantages in terms of SEP identification: (1) Standard proteomic approaches are not suitable for the isolation and analysis of small and low-abundance peptide molecules; and (2) SEPs are shorter than standard proteins and it is unlikely that more than one tryptic fragment will be detected in a single proteomic experiment. Moreover, peptidomic approaches can theoretically be used to identify full-length SEPs in a cell. We did not observe any significant overlap between the sORFs detected using proteomic and peptidomic approaches. Thus, our study demonstrates the advantage of using complementary approaches for building a complete list of SEPs.
According to our MS data, the translation patterns of most small protein-coding sORFs tend to be tissue-specific (Fig. 2A). We suggest that the slight overlap in tissue-specific expression among SEPs from various types of moss cells could be due to either specific SEP post-translational modification (PTM) patterns, tissue-specific transcription of sORFs, or the limitations of mass spectrometry in detecting low-abundance or modified sORF-encoded peptides. According to our results, alternative splicing is an additional mechanism that controls tissue-specific sORF expression in plant cells. Also, the number of sORFs that were commonly translated between two types of moss cells was higher for related cell types: protonemata and gametophores (two growth stages) as well as protonemata and protoplasts (protoplasts were generated from the protonemata). These observations indicate tissue-specific characteristics of SEPs translation and modification rather than a technical limitation in detection.
Functionality of SEPs
We identified tens of small protein-coding sORFs representing multiple sORF types and suggested various functions for the types of sORFs. Clear evidence of transcription and translation points to a possible biological significance of the small protein-coding sORFs that we identified here. Based on our results (evolution, alternative splicing analysis), we suggest that the majority of uORFs play regulatory roles instead of having peptide-encoding functions.
In contrast, CDS- and lncRNA-sORFs have greater potential to encode bioactive peptides, as they are more highly conserved, frequently contain known protein domains and, according to the MS data, produce peptides. We identified 19 small protein-coding CDS-sORFs in our data set, seven of which were translated simultaneously with previously annotated longer protein-coding ORFs. This finding is in agreement with a recent study on mammals, reporting that a gene MIEF1 translational product is not the canonical protein, but the small 70-aa alternative MiD51 protein is (Delcourt et al. 2018).
One possible role for CDS-sORFs that are similar to known proteins is to mimic the similar protein to interfere with its function. MiPs (or siPEPs) are important modulators of protein–protein and protein–DNA interactions that, for example, prevent the formation of functional protein complexes (Seo et al. 2013; Graeff et al. 2016). We found that ∼30% of cis-SEPs harbor protein domains such as protein kinase domains and MYB-like DNA-binding domains or IDRs. Also, some sORFs with disordered regions might mediate protein–protein or protein–nucleic acid interactions, as suggested previously (Mackowiak et al. 2015). However, we failed to identify the translation of such sORFs using stringent identification criteria in our mass spectrometry analysis. Therefore, this point requires further confirmation.
The transcription of the noncoding portions of the genome into lncRNAs is thought to give rise to the translation of sORFs located within them. Nevertheless, the functions of these peptides are unclear and require more detailed investigation. According to our results, knocking out the selected lncRNA-encoded peptides was not lethal in moss but did influence moss growth and development. All SEP knockouts showed a decrease in growth rate compared to the wild-type plants. In contrast, we found that plants overexpressing lncRNA-encoded peptides showed more phenotypic differences compared to the wild-type plants and knockouts. We observed both a significant increase in growth rate (PSEP1 OE) and in the number of leafy shoots (PSEP25 OE) and a decrease in growth rate in PSEP3 OE, PSEP18 OE, and PSEP25 OE lines. The differences between the wild-type and mutant lines often appeared only under certain growth conditions—solid or liquid media with/without glucose or tartrate ammonia. These data may point to a tight regulation of lncRNA-encoded peptide translation in cells. In light of these findings, we hypothesized that lncRNA-encoded peptides may not be vital but may be important for survival under certain conditions by serving as raw material for the evolution. According to the recently proposed classification of small ORFs, lncRNA-sORFs used in our functional analysis may be referred to as both lncORFs and short CDSs (Couso and Patraquim 2017). Both short CDSs and lncRNAs have a median size of 79 aa and 24 aa in animal genomes, respectively (Couso and Patraquim 2017). We suggest that the differences in types of predicted sORFs between plant and animal genomes require further investigation. Our results lay the groundwork for the systematic analysis of functional peptides encoded by sORFs.
The possible evolution of noncoding portions of the genome into protein-coding genes is also a subject of intensive debate (Carvunis et al. 2012; McLysaght and Guerzoni 2015; Couso and Patraquim 2017; Ruiz-Orera and Albà 2019). According to our data, putative homologous sORFs tended to differ in length in most cases. Thus, we suggest that most sORFs expanded during evolution, providing support for the notion that they function as raw materials for selection; however, this point requires further confirmation.
Methods
Physcomitrella patens growth conditions
Physcomitrella patens subsp. patens (“Gransden 2004”, Frieburg) protonemata were grown on BCD medium supplemented with 5 mM ammonium tartrate (BCDAT) or 0.5% glucose during a 16-h photoperiod at 25°C in 9-cm Petri dishes (Nishiyama et al. 2000). For all analyses, the protonemata were collected every 5 d. The gametophores were grown on ammonium tartrate-free BCD medium under the same conditions, and 8-wk-old gametophores were used for analysis. Protoplast was prepared from protonemata as described previously (Fesenko et al. 2015).
For morphological analysis, protonemal tissues 2 mm in diameter were inoculated on BCD and BCDAT 9-cm Petri dishes. For growth rate measurements, photographs were taken at 7-d intervals over 42 d. Protonemal tissues and cells were photographed using a Microscope Digital Eyepiece DCM-510 attached to a Stemi 305 stereomicroscope or Olympus CKX41.
Identification of coding sORFs in the P. patens genome
To identify sORFs with high coding potential, the sORFfinder (Hanada et al. 2010) tool was utilized. Intron sequences and CDS were used as negative and positive sets, respectively. Additional details are described in the Supplemental Methods. To select for sORFs that are transcribed, located in the exons of transcripts, and have introns, a BED file was generated using a Python script (GffParser.py) and intersected with exon positions extracted from a gff3 file of P. patens genome annotations. To identify intergenic-sORFs, the BED file was also intersected with transcribed regions determined based on our RNA-seq data (Fesenko et al. 2017). Using an R script, sORFs fully overlapping with exons were removed; 75,685 sORFs remained after this step. Identical sORFs were removed from the data set. In addition, sORFs overlapping repetitive regions identified by RepeatMasker (Tempel 2012), as well as sORFs comprising parts of annotated P. patens main and alternative protein isoforms, were also removed from the data set, resulting in a final data set of sORFs comprising 70,095 sequences.
sORF classification
The step-by-step procedure performed for sORF classification is illustrated in Supplemental Figure S17. In the first step, lncRNA-sORFs were identified by searching for identical sORFs in known lncRNA databases, including CANTATAdb (Szcześniak et al. 2016), GreeNC (Paytuvi Gallart et al. 2016), and our previously published moss data set (Fesenko et al. 2017). After this sORF BED file was intersected with the latest moss genome annotation V3.3 (Lang et al. 2018), the locations of the sORFs on transcripts were determined, resulting in the further classification of genic-sORFs into uORFs, dORFs, CDS-sORFs, and interlaced-sORFs. sORFs were denoted as upstream or downstream if they were fully separated from the longer protein-coding ORF as previously described (Calviello et al. 2016; Samandi et al. 2017).
Because alternative splicing leads to inaccuracy in genome annotation, the locations of a subset of genic-sORFs cannot be unambiguously classified, as they can be located in different regions in different isoforms of the same gene. All sORFs located on transcripts that were not annotated in the P. patens genome V3.3 but were identified using our RNA-seq data were classified as intergenic-sORFs. To detect alternatively spliced sORFs, a BED file with sORF locations was intersected with a BED file containing intron coordinates for all isoforms. Those sORFs that overlapped for both exons (see above) and introns were classified as AS-sORFs.
Evolutionary conservation analysis
The transcriptomes of nine plant species were downloaded from Phytozome v12: Sphagnum fallax (release 0.5), Marchantia polymorpha (release 3.1), Selaginella moellendorffii (release 1.0), Spirodela polyrhiza (release 2), Arabidopsis thaliana (TAIR 10), Zea mays (Ensembl-18), Oryza sativa (release 7), Volvox carteri (release 2.1), and Chlamydomonas reinhardtii (release 5.5). The transcriptome of Ceratodon purpureus was de novo assembled using Trinity (Haas et al. 2013). To identify transcribed homologous sequences, TBLASTN (word size = 3) was performed using sORF peptide sequences as queries and the transcriptome sequences of the above-mentioned species as subjects. The following cutoffs parameters were used to distinguish reliable alignments: e-value <10−6 and query coverage >60%. Our e-value cutoff was obtained by applying a multiple comparison correction (Bonferroni correction) of 0.05, which is commonly used in biological experiments.
Pairwise Ka/Ks ratios were calculated using the codeml algorithm with PAML software (Yang 2007). The calculation procedure, which was facilitated using a custom-made Python script (protein_Ka_Ks_codeml.py), included alignment extraction from the TBLASTN output, PAL2NAL (Suyama et al. 2006) correction of the nucleotide alignment using the corresponding aligned protein sequences, and calculation of Ka/Ks ratios using codeml. The script implements packages from Biopython (Cock et al. 2009). To estimate homologous sORF lengths, a Python script (sORF_completeness_v2.0.py) was designed. Additional details are described in the Supplemental Methods.
Gene Ontology (GO) term enrichment analysis
GO enrichment analysis was performed using the topGO bioconductor R package using the Fisher's exact test in conjunction with the “classic” algorithm (false discovery rate [FDR] < 0.05). GO terms assigned to P. patens genes were downloaded from Phytozome. Only GO terms containing more than five genes in a background data set were considered in the enrichment analysis. Redundant GO terms were removed using the web-based tool REVIGO (Supek et al. 2011).
Peptide and protein extraction
Endogenous peptide extraction was conducted as described previously (Fesenko et al. 2015). Proteins were extracted as described previously (Fesenko et al. 2016). Additional details are described in the Supplemental Methods.
Mass spectrometry analysis and peptide identification
Mass spectrometry analysis was performed using three biological and three technical repeats for the proteomic and peptidomic data sets (Supplemental Table S2). Analysis was performed on two different mass spectrometers: a TripleTOF 5600+ mass spectrometer with a NanoSpray III ion source (ABSciex) and a Q Exactive HF mass spectrometer (Q Exactive HF Hybrid Quadrupole-Orbitrap mass spectrometer, Thermo Fisher Scientific). Additional details are described in the Supplemental Methods.
All data sets were searched individually with MaxQuant v1.5.8.3 (Tyanova et al. 2016) against a custom database containing 32,926 proteins from annotated genes in the latest version of the moss genome (V3.3) (Lang et al. 2018), 85 moss chloroplast proteins, 42 moss mitochondrial proteins, and 70,052 predicted sORF peptides (Supplemental Code). MaxQuant's protein FDR filter was disabled, while 1% FDR was used to select high-confidence PSMs, and ambiguous peptides were filtered out. Moreover, any PSMs with Andromeda scores of less than 30 were discarded (to exclude poor MS/MS spectra). For the data set of endogenous peptides (named “peptidomic”) (Supplemental Table S2), the parameter “Digestion Mode” was set to “unspecific” and modifications were not permitted. All other parameters were left as default values. For the data set of tryptic peptides (named “proteomic”), the parameter “Digestion Mode” was set to “specific” (the Trypsin/P), MaxQuant's protein FDR filter was disabled, and the peptide FDR remained at 1%. All other parameters were left as default values. Features of the PSMs (length, intensity, number of spectra, Andromeda score, intensity coverage, and peak coverage) were extracted from MaxQuant's msms.txt files. Annotated spectra for identified sORFs were exported from MaxQuant (Supplemental Fig. S18) and manually inspected.
To filter out MS peptides that do not provide unambiguous evidence of sORF peptide expression, we assessed the number of times a peptide occurred in the whole moss genome by searching for exact matches to the MS peptides in the six-frame translated genome (see Supplemental Methods).
RT-PCR analysis of AS-sORFs
Total RNA from gametophores, protonema, and protoplasts was isolated as previously described (Cove et al. 2009). RNA quality and quantity were evaluated via electrophoresis in an agarose gel with ethidium bromide staining. The precise concentration of total RNA in each sample was measured using a Quant-iT RNA Assay kit, 5–100 ng on a Qubit 3.0 (Invitrogen) fluorometer. The cDNA for RT-PCR was synthesized using an MMLV RT kit (Evrogen) according to the manufacturer's recommendations employing oligo(dT)17 -primers from 2 µg total RNA after DNase treatment. The primers were designed using Primer-BLAST (Ye et al. 2012; Supplemental Table S7). The minus reverse transcriptase control (-RT) contained RNA without reverse transcriptase treatment to confirm the absence of DNA in the samples. The RT-PCR products were resolved on a 1.5% agarose gel and visualized using ethidium bromide staining.
Generation of overexpression and knockout lines
To obtain PSEP1 (Pp3c9_sORF1544), PSEP3 (Pp3c25_sORF1253), PSEP25 (Pp3c25_sORF1000), and PSPE18 (Pp3c18_sORF57) overexpression lines, PCR was carried out using genomic DNA as a template and the PEP4f, PEP4r, pep3FXho, pep3RNhe, pep25FXho, pep25RNhe, pep18FXho, and pep18RNhe primers, respectively (Supplemental Table S7). Amplicons were cloned into the pPLV27 vector (GenBank JF909480) using the ligation-independent cloning (LIC) procedure (Aslanidis and de Jong 1990; De Rybel et al. 2011). The resulting plasmids were named pPLV-Hpa-4FR (PSEP1), pPLV-Hpa-3FR (PSEP3), pPLV-Hpa-25FR (PSEP25), and pPLV-Hpa-18FR (PSEP18) and used for transformation. Additional details are described in the Supplemental Methods.
psep1 (sORF Pp3c9_sORF1544), psep3 (Pp3c25_sORF1253), psep25 (Pp3c25_sORF1000), and psep18 (sORF Pp3c18_sORF57) knockout lines were created using the CRISPR/Cas9 system (Collonnier et al. 2017). The coding sequences were used to search for CRISPR RNA (crRNA) preceded by a Streptococcus pyogenes Cas9 PAM motif (NGG) using the web tool CRISPR DESIGN (http://crispr.mit.edu/). The crRNA closest to the translation start site (ATG) was selected for cloning (Supplemental Table S7).
Protoplasts were transformed using a PEG transformation protocol (Schaefer and Zryd 1997). Additional details are described in the Supplemental Methods. The plasmids pACT-CAS9 (for CAS9 expression) and pBNRF (resistance to G418) were kindly provided by Dr. Fabien Nogué. Independent knockout and overexpression mutant lines have been obtained (Supplemental Figs. S9–S12).
The ploidy level of the PSEP1 overexpression and psep1 knockout lines was estimated using flow cytometry. Protoplasts were fixed in cold 70% methanol, washed in TBS with 0.1% Triton X-100, then washed with TBS and stained with 500 ng/ml DAPI. The fluorescence was analyzed with a flow cytometer NovoCyte (ACEA Biosciences) and Novoexpress data software. Fluorescence was excited at 405 nm, and detection was at 445/45 nm.
Software availability
All data were analyzed using Python (http://www.python.org, v 3.5), and R (R Core Team 2017). All scripts are available at Zenodo (doi: 10.5281/zenodo.1160331) and are maintained in the GitHub code repository: https://github.com/Kirovez/Scripts_ sORFs_MS and as Supplemental Code.
Data access
All raw mass spectrometry data from this study have been submitted to the ProteomeXchange Consortium via the PRIDE (Vizcaíno et al. 2016) partner repository with the data set identifiers PXD007922, PXD007923, and PXD007973.
Acknowledgments
We thank Dr. James Lloyd for his assistance in NMD-targeted transcript analysis. This work was supported by the Russian Science Foundation (project No. 17-14-01189). Some of the mass spectrometric measurements were performed using the equipment of the “Human Proteome” Core Facility of the Orekhovich Institute of Biomedical Chemistry (Russia) which is supported by the Ministry of Education and Science of the Russian Federation.
Author contributions: I.F. and I.K. conceived and designed experiments. A.K. and A.M. performed moss transformation experiments. R.K., V.L., D.K., E.G., V.Z., I.B., and A.M. performed the proteomics analyses. I.F., I.K., and G.A. performed the statistical and bioinformatics analyses. I.F., I.K., V.I., and V.G. wrote the manuscript with input from all authors. I.F. supervised the project. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.253302.119.
- Received June 4, 2019.
- Accepted August 1, 2019.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








