
4q-D4Z4 chromatin architecture regulates the transcription of muscle atrophic genes in facioscapulohumeral muscular dystrophy
- Alice Cortesi1,5,
- Matthieu Pesant1,5,
- Shruti Sinha1,5,
- Federica Marasca1,
- Eleonora Sala1,
- Francesco Gregoretti2,
- Laura Antonelli2,
- Gennaro Oliva2,
- Chiara Chiereghin3,4,
- Giulia Soldà3,4 and
- Beatrice Bodega1
- 1Istituto Nazionale di Genetica Molecolare “Romeo ed Enrica Invernizzi” (INGM), 20122, Milan, Italy;
- 2CNR Institute for High Performance Computing and Networking (ICAR), 8013, Naples, Italy;
- 3Department of Biomedical Sciences, Humanitas University, 20090, Pieve Emanuele, Milan, Italy;
- 4Humanitas Clinical and Research Center, 20089, Rozzano, Milan, Italy
-
↵5 These authors contributed equally to this work.
Abstract
Despite increasing insights in genome structure organization, the role of DNA repetitive elements, accounting for more than two thirds of the human genome, remains elusive. Facioscapulohumeral muscular dystrophy (FSHD) is associated with deletion of D4Z4 repeat array below 11 units at 4q35.2. It is known that the deletion alters chromatin structure in cis, leading to gene up-regulation. Here we show a genome-wide role of 4q-D4Z4 array in modulating gene expression via 3D nuclear contacts. We have developed an integrated strategy of 4q-D4Z4–specific 4C-seq and chromatin segmentation analyses, showing that 4q-D4Z4 3D interactome and chromatin states of interacting genes are impaired in FSHD1 condition; in particular, genes that have lost the 4q-D4Z4 interaction and with a more active chromatin state are enriched for muscle atrophy transcriptional signature. Expression level of these genes is restored by the interaction with an ectopic 4q-D4Z4 array, suggesting that the repeat directly modulates the transcription of contacted targets. Of note, the up-regulation of atrophic genes is a common feature of several FSHD1 and FSHD2 patients, indicating that we have identified a core set of deregulated genes involved in FSHD pathophysiology.
Among primate-specific macrosatellites, D4Z4 is a 3.3-kb unit tandem repeat duplicated on several chromosomes (Bakker et al. 1995; Deidda et al. 1995; Lyle et al. 1995; Bodega et al. 2006,2007) and, in particular, present as a polymorphic array of 11 to 100–150 copies at 4q35.2 (4q-D4Z4 array) in the general population (Hewitt et al. 1994). Reduction of 4q-D4Z4 array copy number below 11 units is associated with facioscapulohumeral muscular dystrophy (FSHD; MIM158900) (van Deutekom et al. 1993), one of the most common myopathies in humans with an overall prevalence of more than 1:10,000 (Sacconi et al. 2015). FSHD is characterized by progressive, often asymmetric, weakness and wasting of facial (facio), shoulder, and upper arm (scapulohumeral) muscles (Tawil and Van Der Maarel 2006), where fiber necrosis and degeneration give rise to muscle atrophy (Sacconi et al. 2015).
FSHD is a genetically variable disorder, mainly transmitted as an autosomal dominant trait, on a specific FSHD-permissive haplotype of Chromosome 4q, namely 4qA (Lemmers et al. 2002, 2007). This form accounts for ∼95% of the cases (FSHD1); however, ∼5% of the patients display FSHD lacking D4Z4 array contractions (FSHD2). FSHD2 is caused by mutations in SMCHD1, a member of the condensin/cohesin chromatin compaction complexes, which binds to the D4Z4 repeat array (Lemmers et al. 2012). Although in healthy individuals the 4q-D4Z4 array is characterized by highly methylated DNA, the contracted allele in FSHD1 and both the 4q-D4Z4 alleles in FSHD2 are hypomethylated (van Overveld et al. 2003; de Greef et al. 2009).
The highly heterogeneous FSHD clinical features suggest a strong epigenetic contribution to the pathology (Tawil et al. 1993; Cabianca and Gabellini 2010; Neguembor and Gabellini 2010; Lanzuolo 2012; Daxinger et al. 2015). It is described that the 4q-D4Z4 array is able to engage short- and long-range genomic contacts with several genes in cis (Petrov et al. 2006; Bodega et al. 2009; Himeda et al. 2014; Robin et al. 2015), concomitantly to the Polycomb group proteins' (PcG) binding and to histone deacetylation, resulting in an overall chromatin compaction. Instead, in the FSHD1 condition such interactions are lost, with the consequent alteration of the chromatin structure at the FSHD locus, leading to a more active chromatin state, which is responsible for the de-repression of the genes in cis (Gabellini et al. 2002; Jiang et al. 2003; Bodega et al. 2009; Zeng et al. 2009; Cabianca et al. 2012). In particular, one of the major players in FSHD pathogenesis is the transcription factor DUX4, encoded from the most telomeric D4Z4 repeat (Gabriëls et al. 1999; Dixit et al. 2007; Lemmers et al. 2010); DUX4 is normally silenced in somatic cells (Snider et al. 2010), but it has been found overexpressed in FSHD patients’ myotubes (MTs). DUX4 overexpression activates genes associated with RNA metabolism processes, stem cell and germ-line development, and MERVL/HERVL retrotransposons (Geng et al. 2012; Young et al. 2013; Rickard et al. 2015; Hendrickson et al. 2017), resulting in the induction of toxicity and apoptosis of muscle cells (Bosnakovski et al. 2008; Block et al. 2013).
Besides the established role of 4q-D4Z4 array in modulating the transcription of in cis genes, whether the repeat could also directly affect chromatin structure and gene expression of other loci via 3D physical contacts has not been investigated yet. Therefore, we have explored the 4q-D4Z4 chromatin architecture and possible alterations in FSHD.
Results
4q-specific D4Z4 interactome is deregulated in FSHD1 patients
Given the high duplication and sequence similarity of 4q35.2 with multiple regions of the genome, in particular with 10q26.3 (Bodega et al. 2006, 2007), we designed a 4q-specific 4C-seq (circular chromosome conformation capture sequencing) strategy to investigate its interactome. As 4C viewpoint (VP), we used the region nearby a single sequence length polymorphism (SSLP), present shortly upstream (∼3.5 kb) of the first D4Z4 repeat on 4q (4qA and 4qB) and 10q arrays (Lemmers et al. 2007); we performed paired-end sequencing, that allowed retrieval of the information of the SSLP variant (Read 1) and the interacting region (Read 2), assigning with high precision the allele origin of the D4Z4 interactome (Supplemental Fig. S1; Supplemental Table S1; see Methods; Supplemental Methods).
With this approach, we probed the 4q-D4Z4 chromatin conformation in human primary muscle cells from two FSHD1 patients (FSHD1) and two healthy individuals (CN) (Supplemental Table S1) that did not differ for myoblast (MB) purity and differentiation efficiency (Supplemental Fig. S2). 4C-seq was performed on MBs to highlight differences that could precede any transcriptional effect, mediated also by DUX4 expression, in differentiated cells. Comparative analyses of 4C-seq samples showed high level of reproducibility and similarity both at the level of donor origin (fragend read count, CN or FSHD1) (Supplemental Fig. S3) and at the level of VP (called interacting regions, 4q vs. 10q) (Supplemental Fig. S4A,B). We identified 4q-D4Z4–specific cis interactions with FRG1, ZFP42, SORBS2, and FAM149A genes (Fig. 1A; Supplemental Fig. S4C–E), as already reported (Bodega et al. 2009; Robin et al. 2015), suggesting that our approach is robust in the detection of 4q-specific D4Z4 interactions.
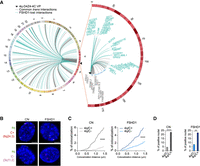
4q-D4Z4–specific 4C-seq highlights FSHD1 impaired interactome. (A, left) Circos plot (Krzywinski et al. 2009) depicting cis and trans 4q-D4Z4 interactions in CN (CN-3, CN-4) myoblasts (MBs) called by 4C-ker. Common interactions with FSHD1 (FSHD1-3, FSHD1-4) MBs are in gray, whereas interactions specifically lost in FSHD1 are highlighted in light blue. (Right) Zoomed-in Circos plot representation of common (gray) and FSHD1 lost (light blue) cis interactions on Chr 4. Genes are indicated for a region extending up to 4 Mb from the VP. Black triangles in Circos plots depict the VP localization. (B) Representative nuclei of 3D multicolor DNA FISH using probes mapping to 4q35.1 region (4q; green), a 4q-D4Z4–positive interacting region (8q24.3, C+; red), and a 4q-D4Z4 not interacting region (3q11.2, C−; magenta) in CN (CN-1, CN-2, CN-3, CN-4) and FSHD1 (FSHD1-1, FSHD1-2, FSHD1-3, FSHD1-4) MBs. Nuclei are counterstained with DAPI (blue). All images at 63× magnification. Scale bar, 5 µm. (C) Cumulative frequency distributions of distances (below 1.5 µm) between 4q and C+ and between 4q and C− in CN (dark and light gray; left) and FSHD1 (dark and light blue; right) MBs. n = 1296 (CN 4q/C+), 1708 (CN 4q/C−), 884 (FSHD1 4q/C+), and 1128 (FSHD1 4q/C−). P-values were calculated by unpaired one-tailed t-test with a confidence interval of 99%. Asterisks represent statistical P-values: for 4q/C+ versus 4q/C− in CN and FSHD1, P < 0.0001. (D) Percentage of nuclei positive for the interactions (under the cutoff of 1.5 µm). n = 427 (CN 4q/C−), 324 (CN 4q/C+), 282 (FSHD1 4q/C−), and 221 (FSHD1 4q/C+). P-values were calculated by Fisher's exact one-sided test with a confidence interval of 99%. Asterisks represent statistical P-values: for 4q/C− versus 4q/C+ in CN, P < 0.0001; for 4q/C− versus 4q/C+ in FSHD1, P = 0.0046.
We retrieved 244 and 258 4q-D4Z4 interacting regions for CN and FSHD1, respectively, and in particular among them, 175 for CN and 181 for FSHD1 were trans interactions. The resolution of trans interacting domains is ∼1 Mb (Supplemental Table S2). Moreover, 116 regions interacting in CN were specifically lost in FSHD1 cells, and the vast majority (101) were in trans (Fig. 1A; Supplemental Table S2).
3D multicolor DNA FISH was performed on the same and additional CN and FSHD1 donor MBs to validate 4C results, using a probe on a not duplicated region in 4q35.1 (Supplemental Fig. S5A; Tam et al. 2004) and a probe for a positive (C+, 8q24.3) or negative (C−, 3q11.2) 4q-D4Z4 interacting region (Fig. 1B; Supplemental Fig. S5B). We developed a novel algorithm (NuCLεD, or Nuclear Contacts Locator in 3D) (see Supplemental Methods) to automatically detect and localize fluorescent spots in 3D reconstructed nuclei. We observed that the 4q/C+ interaction had a higher frequency of contacts and a higher number of positive interacting nuclei compared with the 4q/C− interaction (Fig. 1C,D; Supplemental Table S3), with contact frequencies in the range of those estimated for long-range interactions (10%–20%) (Finn et al. 2019). Furthermore, 4q and C+ regions shared the same topological nuclear domain in both CN and FSHD1, whereas 4q and C− did not (Supplemental Fig. S5C,D; Supplemental Table S3). Same results were obtained in CN and FSHD1 MTs (Supplemental Fig. S5E–G; Supplemental Table S3). Additionally, with our 4C-seq approach we were also able to retrieve 4q allele–specific interactomes (4qA and 4qB), as well as 10q-D4Z4 interactomes (Supplemental Fig. S6; Supplemental Table S2; see Supplemental Material).
Overall, the 4q-D4Z4-4C-seq strategy allowed us to map genome-wide 4q-D4Z4 contacts and to highlight those deregulated in FSHD1.
Genes that show impaired 4q-D4Z4 interaction and activated chromatin state are enriched for atrophic transcriptional signature
To identify novel deregulated genes specific for the FSHD condition, we derived chromatin state changes in FSHD1 cells and intersected them with 4q-D4Z4 lost interactions in FSHD1, retrieving genes altered both at structural and chromatin levels.
To define the chromatin state, we generated or used available (ENCODE) ChIP-seq data sets for H3K36me3, H3K4me1, H3K27ac, H3K4me3, and H3K27me3 in CN and FSHD1 MBs and MTs (Supplemental Fig. S7A,B). The quality of ChIP-seq was validated on the same and additional CN and FSHD1 donor MBs and MTs (Supplemental Fig. S7C–H). Next, we identified 15 chromatin states using ChromHMM (Ernst and Kellis 2012), that were adopted for further downstream analyses (Fig. 2A; Supplemental Fig. S8A,B). Chromatin segmentation analysis revealed transitions at enhancers and promoters distinctive for FSHD1 cells (Supplemental Fig. S8C; see Supplemental Material).
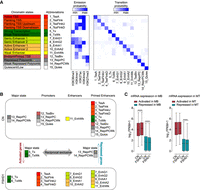
Chromatin segmentation analysis revealed chromatin state switches consistent with transcriptional changes in FSHD1 muscle cells. (A) ChromHMM 15-state model obtained with ChIP-seq data sets for H3K36me3, H3K4me1, H3K27ac, H3K4me3, and H3K27me3. Heatmaps display histone marks emission probabilities and transition probabilities between chromatin states. (B) Schematic representation of the strategy used to assign genes as activated or repressed in FSHD1. (C) Expression levels from RNA-seq data sets for FSHD1 activated and repressed genes in MBs (left) and MTs (right) in CN (CN-3, CN-4, and Yao's data sets C20, C21, C22) and FSHD1 (FSHD1-3, FSHD1-4, and Yao's data sets F4, F6) (Yao et al. 2014). Box-and-whisker plots show the median of matched expression values of each gene for CN and FSHD1, and whiskers extend to the 5 to 95 percentiles. P-values were calculated by paired two-tailed Wilcoxon matched-pair signed-rank test with a confidence interval of 99%. Asterisks represent statistical P-values: for CN versus FSHD1 activated in MBs, P < 0.0001; for CN versus FSHD1 repressed in MBs, P < 0.0001; for CN versus FSHD1 repressed in MTs, P < 0.0001.
To identify the genes that specifically switched to activated or repressed chromatin state in FSHD1, we designed the strategy shown in Figure 2B. Activated genes were defined as those that showed a transition toward a more active state (considering the coverage of the gene body, promoter, and enhancer regions), and repressed genes are those that showed an opposite change (Fig. 2B; Supplemental Table S4; see Methods). To verify the reliability of this approach, we inspected the expression level of these genes by analyzing RNA-seq data sets performed on the corresponding cell lines and additional publicly available RNA-seq data sets (Yao et al. 2014). The activated or repressed chromatin state switches were associated with higher or lower mRNA expression levels, respectively, in FSHD1 compared with CN (Fig. 2C; Supplemental Table S4).
We next sought genes that had lost the interaction with 4q-D4Z4 and also showed chromatin deregulation in FSHD1. We observed that 28% (450/1614) of genes that had lost contact with 4q-D4Z4 in FSHD1 were mainly activated (FSHD1 lost-activated genes, 71%, 319/450), whereas a minority of them (FSHD1 lost-repressed genes, 29%, 131/450) were repressed (Fig. 3A; Supplemental Fig. S9A–C; Supplemental Table S5; see Supplemental Material). Only few of these FSHD1-altered genes were regulated by DUX4 (Supplemental Figs. S9D, S10; Supplemental Table S6; see Supplemental Material).
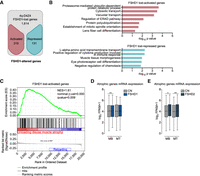
Genes that have lost the interaction with 4q-D4Z4 have a more active chromatin state and are enriched for muscle atrophy signature in FSHD muscle cells. (A) Flowchart of filtering steps to identify FSHD1-altered genes. Genes within lost 4q-D4Z4 interactions were filtered as activated (red) or repressed (blue) in FSHD1. (B) Gene Ontology analysis (Biological Processes) of FSHD1 lost-activated and lost-repressed genes. Bars correspond to −log10 of the P-value. (C) Gene set enrichment analysis (GSEA) results of the 319 FSHD1 lost-activated genes performed on expression data from unloading-induced muscle atrophy subjects (Reich et al. 2010). Genes up-regulated in atrophic condition are depicted in red, whereas genes not enriched are depicted in blue. (NES) normalized enrichment score. (D) Expression levels from RNA-seq data sets for atrophic genes (Reich et al. 2010) in CN (CN-3, CN-4, and Yao's data sets C20, C21, C22) and FSHD1 (FSHD1-3, FSHD1-4, and Yao's data sets F4, F6) (Yao et al. 2014) MBs and MTs. Box-and-whisker plots show the median of matched expression values of each gene for CN and FSHD1, and whiskers extend to the 5 to 95 percentiles. P-values were calculated by paired two-tailed t-test with a confidence interval of 99%. Asterisks represent statistical P-values: for CN versus FSHD1 in MBs, P = 0.0099. (E) Expression levels from RNA-seq data sets for atrophic genes (Reich et al. 2010) in CN (CN-3, CN-4, and Yao's data sets C20, C21, C22) and FSHD2 (Yao's data sets F12, F14, F20) (Yao et al. 2014) MBs and MTs. Box-and-whisker plots show the median of matched expression values of each gene for CN and FSHD2, and whiskers extend to the 5 to 95 percentiles. P-values were calculated by paired two-tailed t-test with a confidence interval of 99%. Asterisks represent statistical P-values: for CN versus FSHD2 in MBs, P = 0.0251; for CN versus FSHD2 in MTs, P = 0.0041.
We performed Gene Ontology (GO) analyses on the FSHD1-altered genes and found that FSHD1 lost-activated genes were enriched in GO terms linked to protein catabolic processes and, in particular, with protein ubiquitination/degradation pathways (Fig. 3B; Supplemental Table S5) that are highly relevant to the FSHD-associated atrophic phenotype (Tawil and Van Der Maarel 2006; Sacconi et al. 2015; Statland and Tawil 2016). Similar analysis on 10q-D4Z4 FSHD1-altered genes did not reveal GO terms related to atrophy (Supplemental Fig. S11A,B).
Indeed, we executed gene set enrichment analysis (GSEA) and further showed that 4q-D4Z4–specific lost-activated genes in FSHD1 were enriched for genes up-regulated in the atrophic condition (Fig. 3C; Supplemental Fig. S11C–E; Supplemental Table S5). The FSHD1 lost-activated genes included in the atrophic data set displayed higher expression levels both in several FSHD1 (Fig. 3D), as well as in FSHD2 (Fig. 3E) RNA-seq data sets, revealing that the epigenetic and transcriptional deregulation of this core set of genes represents a novel transcriptional signature that is common among different FSHD patients.
4q-D4Z4 lost interacting FBXO32 gene is deregulated in FSHD patients at the chromatin and transcriptional level
To finely dissect how the 4q-D4Z4 lost interactions could influence the up-regulation of muscle atrophy genes in the FSHD1 condition, we further investigated the regulation of FBXO32 (also known as ATROGIN1), which is one of the top-enriched genes identified by GSEA and also one of the major player in different atrophy-related conditions, in human and mouse (Gomes et al. 2001; Lecker et al. 2004; Sandri et al. 2004; Bodine and Baehr 2014). We verified by 3D multicolor DNA FISH the loss of FBXO32 (8q24.13)/4q-D4Z4 interaction on several FSHD1 donors compared with CN, extending our analysis also to FSHD2 (Fig. 4A; Supplemental Fig. S12A,B), and observed a decrease in FBXO32/4q-D4Z4 interaction frequency in FSHD1 and FSHD2 MBs (Fig. 4B,C; Supplemental Table S3). Similar results were obtained in FSHD1 MTs, although with a smaller difference (Supplemental Fig. S12C–E; Supplemental Table S3).

FBXO32 gene has a deregulated chromatin structure, and it is overexpressed in FSHD1 and FSHD2 muscle cells. (A) Representative nuclei of 3D multicolor DNA FISH using probes mapping to 4q35.1 region (4q; green) and FBXO32 (red) in CN (CN-1, CN-3, CN-4), FSHD1 (FSHD1-1, FSHD1-3, FSHD1-4), and FSHD2 (FSHD2-1, FSHD2-2) MBs. Nuclei are counterstained with DAPI (blue). All images are at 63× magnification. Scale bar, 5 µm. (B) Cumulative frequency distribution of distances (below 1.5 µm) between 4q and FBXO32 in CN (gray), FSHD1 (blue), and FSHD2 (dark blue) MBs. n = 3652 (CN), 2464 (FSHD1), and 1020 (FSHD2). P-values were calculated by unpaired one-tailed t-test with a confidence interval of 99%. Asterisks represent statistical P-values: for CN versus FSHD1, P = 0.0473; for CN versus FSHD2, P = 0.0036. (C) Percentage of nuclei positive for the interactions (under the cutoff of 1.5 µm). n = 913 (CN), 616 (FSHD1), and 255 (FSHD2). (D) 4C normalized coverage tracks at the FBXO32 locus for FBXO32-4C VP in CN (CN-3, CN-4; gray) and FSHD1 (FSHD1-3, FSHD1-4; blue). (E, top) Schematic representation of the FBXO32 locus and HindIII sites. (Middle) Chart showing the frequencies of 3C interaction between FBXO32 promoter and the indicated HindIII restriction sites (sites 4–32), using the same bait of the 4C VP (light gray vertical bar) in CN (gray) and FSHD (blue). n = 3 (CN) and 3 (FSHD1). SEM is indicated. P-values were calculated by two-way ANOVA followed by Bonferroni posttest correction. Asterisks represent statistical P-values: for P19, P25, and P30 CN versus FSHD1, P < 0.001; for P32 CN versus FSHD1, P < 0.01. (Bottom) 4C normalized coverage tracks as well as ChromHMM chromatin states tracks at the FBXO32 locus for FBXO32-4C VP in CN (gray) and FSHD1 (blue). The arrow represents the promoter region; enhancers are highlighted in yellow. (F) Bar plot showing enrichment of RNA Pol II at FBXO32 promoter (left) and an intragenic region (right) assessed by ChIP-qPCR experiment in CN (gray) and FSHD1 (blue) MBs. Results are presented as a percentage of input. n = 2 CN (CN-3, CN-4) and 2 FSHD1 (FSHD1-3, FSHD1-4). SEM is indicated. P-values were calculated by unpaired one-tailed t-test with a confidence interval of 99%. Dots represent the values of each replicate; asterisks represent statistical P-values: for FBXO32 intragenic region CN versus FSHD1, P = 0.0050. (G) Expression levels of FBXO32 gene during CN (gray), FSHD1 (blue), and FSHD2 (dark blue) differentiation. (MT2) Myotubes day 2, (MT4) myotubes day 4, (MT6) myotubes day 6. Data were normalized on GAPDH expression and on MBs. n = 4 CN (CN-1, CN-2, CN-3, CN-4), 4 FSHD1 (FSHD1-1, FSHD1-2, FSHD1-3, FSHD1-4), and 2 FSHD2 (FSHD2-1, FSHD2-2). SEM is indicated. P-values were calculated by two-way ANOVA followed by Bonferroni posttest correction. Dots represent the values of each replicate; asterisks represent statistical P-values: for MT4, CN versus FSHD1, P < 0.0290; CN versus FSHD2, P < 0.0001.
FBXO32 belongs to the category of activated genes in FSHD1, with the appearance of primed enhancers specifically in FSHD1 condition (Fig. 2B; Supplemental Fig. S13A; Supplemental Table S4). Therefore, we further investigated whether the FBXO32 locus could display distinct chromatin loops at the level of enhancers-promoter in FSHD1. We performed 4C-seq (Fig. 4D; Supplemental Fig. S13B–D; Supplemental Tables S1, S2), showing interaction peaks between enhancers-promoter with higher normalized 4C reads coverage in FSHD1 (Fig. 4E). These results were further corroborated in chromatin conformation capture (3C) experiments (Fig. 4E), suggesting a strengthening of enhancers-promoter contacts at the FBXO32 locus in FSHD1 cells.
In line with this observation, the binding of RNA Pol II at the FBXO32 promoter and an intragenic region was increased in FSHD1 MBs respect to CN (Fig. 4F; Supplemental Fig. S14A), and the FBXO32 expression was up-regulated in several FSHD1 donor muscle cells during differentiation, a trend that is also observed in FSHD2 (Fig. 4G). Finally, the FBOX32 expression is not dependent on DUX4, as it is not affected by DUX4 overexpression (Supplemental Fig. S14B–D; see Supplemental Material), and DUX4 ChIP-seq peaks (Geng et al. 2012) are absent in the FBXO32 gene region (Supplemental Fig. S14E).
Ectopic 4q-D4Z4 array restores the expression of FSHD1-lost interacting genes
To further investigate whether the 4q-D4Z4 array could directly modulate the expression of interacting genes, we transfected CN and FSHD1 MBs with a BAC containing a wild-type D4Z4 array (BAC 4q-D4Z4n from 4q35.2 region) (see Methods) in parallel with a control BAC (Ctrl BAC; unrelated and not interacting region); transfection efficiency was comparable among the BACs and ranging ∼45% (Supplemental Fig. S14F–H; see Supplemental Methods).
We observed that the ectopic 4q-D4Z4 array was in close spatial proximity to the endogenous 4q region in 70% of the analyzed nuclei (Fig. 5A,B). The interaction frequency with FBXO32 is similar to the endogenous locus (∼20% of analyzed nuclei) (Fig. 5C), indicating that the 4q-D4Z4 BAC occupies the same nuclear topological domain of the endogenous 4q region. We then assessed the effect of 4q-D4Z4 BAC transfection on the expression level of a subset of genes that had lost 4q-D4Z4 interactions in FSHD1. We observed that the transcription of FSHD1 lost-activated genes was reduced (FBXO32, TRIB3, and ZNF555) (Fig. 5D), whereas a lost-repressed gene was up-regulated (LZTS3) (Fig. 5E), and no effect was detected for not interacting genes (FOXO3 and MYOG) (Fig. 5F).
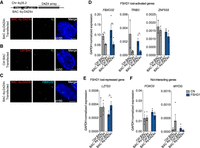
Ectopic 4q-D4Z4 array restores the expression of FSHD1-lost interacting genes. (A, top) Representation of the BAC containing a 4q upstream region and wild-type D4Z4 (BAC 4q-D4Z4n) (see Methods). (Bottom) Representative nucleus of 3D multicolor DNA FISH using probes for the transfected BAC backbone (red) and 4q35.1 region (4q; green) in MBs transfected with BAC 4q-D4Z4n. Nuclei are counterstained with DAPI (blue). All images are at 63× magnification. Scale bar, 5 µm. (n) Number of nuclei analyzed. (B) Representative nucleus of 3D multicolor DNA FISH using probes for the transfected BAC backbone (red) and 4q35.1 region (4q; green) in MBs transfected with Ctrl BAC (RP11-2A16, representative of an unrelated and not interacting genomic region, Chr 17q21.33). Nuclei are counterstained with DAPI (blue). All images are at 63× magnification. Scale bar, 5 µm. (n) Number of nuclei analyzed. (C) Representative nucleus of 3D multicolor DNA FISH using probes for the transfected BAC backbone (red) and FBXO32 region (FBXO32; light blue) in MBs transfected with BAC 4q-D4Z4n. Nuclei are counterstained with DAPI (blue). All images are at 63× magnification. Scale bar, 5 µm. (n) Number of nuclei analyzed. (D) Bar plots showing expression levels of FBXO32, TRIB3, and ZNF555 (FSHD1 lost-activated genes) in CN (gray) and FSHD1 (blue) MBs transfected with Ctrl BAC and BAC 4q-D4Z4n. Data were normalized on GAPDH expression. n = at least three (with the exception of TRIB3 and ZNF555 CN Ctrl BAC, n = 2). SEM is indicated. P-values were calculated by paired one-tailed t-test with a confidence interval of 99%. Dots represent the values of each replicate; asterisks represent statistical P-values: for FBXO32 Ctrl BAC versus BAC 4q-D4Z4n in CN, P = 0.0182; for FBXO32 Ctrl BAC versus BAC 4q-D4Z4n in FSHD1, P = 0.0073; for TRIB3 Ctrl BAC versus BAC 4q-D4Z4n in FSHD1, P = 0.0281. (E) Bar plot showing expression levels of LZTS3 (FSHD1 lost-repressed gene) in CN (gray) and FSHD1 (blue) MBs transfected with Ctrl BAC and BAC 4q-D4Z4n. Data were normalized on GAPDH expression. n = 3 (with the exception of CN Ctrl BAC, n = 2). SEM is indicated. P-value was calculated by paired one-tailed t-test with a confidence interval of 99%. Dots represent the values of each replicate; asterisks represent statistical P-values: for Ctrl BAC versus BAC 4q-D4Z4n in FSHD1, P = 0.0296. (F) Bar plots showing expression levels of FOXO3 and MYOG (not interacting genes) in CN (gray) and FSHD1 (blue) MBs transfected with Ctrl BAC and BAC 4q-D4Z4n. Data were normalized on GAPDH expression. n = at least three. SEM is indicated. Dots represent the values of each replicate.
Collectively, these results show that the 4q-D4Z4 array directly modulates the transcription of its interacting targets, suggesting a simultaneous fine-tuning of genes that occupy the same topological domain.
Discussion
Here, we sought to identify the mechanism by which the contraction of the tandem repeat D4Z4 on Chromosome 4 contributes to FSHD pathogenesis, using an integrated multiomics approach (4C-seq, ChIP-seq, and RNA-seq).
We found that the 4q-D4Z4 interactome is altered in FSHD1 patients. In particular, the normal 4q-D4Z4 array contacts several regions in a peripheral nuclear domain, controlling their transcription (Fig. 6A). In FSHD1 patients, the shortened and hypomethylated 4q-D4Z4 array causes an impairment of the chromatin conformation, which results in the loss of contacts with atrophic genes, with their consequent chromatin structure alteration and transcriptional up-regulation (Fig. 6B). In this regard, it is already shown that chromatin topological structures predominantly consist of simultaneous multiplex chromatin interactions with high heterogeneity between individual cells (Jiang et al. 2016; Zheng et al. 2019). Indeed, we show that an ectopic wild-type 4q-D4Z4 array has the ability to get in close spatial proximity to the endogenous locus, resulting in the restoration of the expression of multiple targets, opening the possibility for further mechanistic studies on the dynamics of 3D interactions.
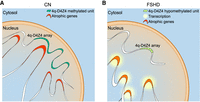
Model of 4q-D4Z4–mediated regulation of atrophic genes transcription. (A) 4q-D4Z4 array is interacting with a subset of atrophic genes, organizing their chromatin structure and keeping on hold their transcription in healthy donor muscle cells. (B) In FSHD1 patients’ muscle cells, the deleted and hypomethylated 4q-D4Z4 array causes an impairment of D4Z4 interactome, leading to a chromatin switch toward an active state (mainly enhancer and promoter regions), which in turn results in the transcriptional up-regulation of the atrophic genes.
We propose that the genetic deletion of 4q-D4Z4 array in FSHD1 patients leads to a rewired interactome that may represent an additional component of FSHD pathophysiology.
Because we discovered that the subset of genes losing contact with the 4q-D4Z4 array in FSHD1 mainly show chromatin state switches toward activation, we hypothesize that this might be consistent with a broader derepression occurring at the 4q-D4Z4 array, such as lesser PcG recruitment together or not with an enhanced activity of Trithorax complex, as already shown in cis (Cabianca et al. 2012). Of note, SMCHD1 protein, mutated in FSHD2 patients (Lemmers et al. 2012), is involved in higher-order chromatin organization of the inactive X Chromosome (Jansz et al. 2018; Wang et al. 2018). We could hypothesize that this architectural protein could have a central role in regulating 4q-D4Z4 interactions and that its altered function in FSHD1 (owing to the contraction and hypomethylation of the array) and FSHD2 patients (owing to its mutation) could explain the common atrophic signature. Indeed, FSHD-associated atrophy is one of the main signs of the disease (Tawil and Van Der Maarel 2006; Lanzuolo 2012; Sacconi et al. 2015), for which a direct link with the genetic defect remained elusive until now.
We identified a core set of impaired atrophic genes that is aberrantly transcribed in FSHD1 muscle cells used in this study and in other FSHD1 cells (Yao et al. 2014). Furthermore, they are also deregulated in FSHD2 muscle cells, indicating that the atrophic signature is a common trait in FSHD pathology. We further investigated FBXO32 gene regulation, which was one of the top ranking genes; although it was already described as overexpressed in muscle biopsies derived from FSHD1 fetuses and adults (Broucqsault et al. 2013), here we linked its transcriptional deregulation to the reduction in FBXO32/4q-D4Z4 interaction. This was predominantly observed in FSHD MBs compared with MTs, in line with previous reports that changes in 3D structure precede changes in gene expression (Hug et al. 2017; Krijger and de Laat 2017; Cheutin and Cavalli 2018), and already shown also for the FRG1 gene (Bodega et al., 2009).
Few atrophic genes (i.e., FBOX32 and TRIM63) were already reported as up-regulated in FSHD1 MTs and muscle biopsies and under the control of DUX4 transcription factor (Vanderplanck et al. 2011). However, DUX4-dependent FBOX32 regulation has been debated (Tsumagari et al. 2011; Homma et al. 2012), and also, our results did not confirm this finding (Supplemental Fig. S14B–E). Here we show that the atrophic genes chromatin structure (chromatin state and 3D organization) is already altered in FSHD1 MBs, foreseeing another layer of transcriptional regulation that could precede DUX4 action on its targets. We propose that the D4Z4 deregulated interactome and DUX4 overexpression could act in concert to cause the complex cascade of molecular events that lead to FSHD manifestation.
DNA repetitive elements are involved in a plethora of regulatory mechanisms, such as nuclear structure organization and spatiotemporal gene expression regulation (Gregory 2005; de Laat and Duboule 2013; Bodega and Orlando 2014). Additionally, recent studies have highlighted the contribution of satellite repeats in shaping 3D genome folding and function, as evidenced for pericentromeric satellites (Politz et al. 2013; Wijchers et al. 2015) and the DXZ4 macrosatellite (Giacalone et al. 1992; Rao et al. 2014; Deng et al. 2015; Darrow et al. 2016; Giorgetti et al. 2016). Our study is the first demonstration of a role of DNA repetitive elements in the alteration of genomic architecture in the context of a human genetic disease. It further corroborates the concept that perturbations of the 3D genome structure are involved in various diseases (Krijger and de Laat 2016; Lupiáñez et al. 2016), such as cancers (Corces and Corces 2016; Rivera-Reyes et al. 2016; Achinger-Kawecka and Clark 2017) and developmental defects (Woltering et al. 2014; Lupiáñez et al. 2015; Woltering and Duboule 2015).
Our work highlights a novel role of DNA repeats in orchestrating gene transcription by shaping 3D genomic and chromatin architecture. We propose that perturbation of this DNA repeat-mediated regulatory network may be important in other complex genetic and epigenetic diseases.
Methods
Cell cultures
Although it was not always possible to ascertain the status of the muscle origin used in this study (see Supplemental Table S1, sheet Cell Line Information), the majority of the cells used derived from the quadriceps, which in general was asymptomatic. Human primary MB cell lines from healthy donors (CN) and patients affected by FSHD1 or FSHD2 were obtained from the Telethon BioBank of the C. Besta Neurological Institute (Milan, Italy) and the Fields Center for FSHD of the Rochester Medical Center Department of Neurology (New York), whereas human immortalized MB cell lines from healthy donors and FSHD1 patients were obtained from the University of Massachusetts Medical School Wellstone Center for FSH Muscular Dystrophy Research, Wellstone Program & Dept. of Cell & Developmental Biology (Worcester, MA). Details of all cell lines are reported in Supplemental Table S1; details on media preparation and FACS analysis for Desmin staining are provided in Supplemental Methods.
4C-seq assay
The 4C assay was performed as previously described (Splinter et al. 2012) with minor modifications. A paired-end 4q-D4Z4–specific 4C-sequencing strategy was developed, in which one 4C primer was designed to read the SSLP sequences located shortly upstream (∼3.5 kb) of the first D4Z4 repeat on 4q or 10q-D4Z4 arrays (Lemmers et al. 2007) and the second 4C primer reads into the captured sequence ligated to the “bait” fragment. (Supplemental Fig. S1; Supplemental Table S1). Two donor muscle cell lines of CN (CN-3, CN-4) and FSHD1 (FSHD1-3, FSHD1-4) human primary MB (3.5 × 106 per sample) nuclei were processed. Five biological replicates (start to finish experiments) for each cell line were performed (Supplemental Fig. S3A). For FBXO32 4C-seq, we designed specific 4C primers as indicated in Supplemental Table S1. Two donor muscle cell lines of CN (CN-3, CN-4) and FSHD1 (FSHD1-3, FSHD1-4) human primary MB (3.5 × 106 per sample) nuclei were processed. From one to two biological replicates (start to finish experiments) for each cell line were performed. HindIII and DpnII were used for enzymatic digestions. 4C samples were amplified using the bait and the SSLP-specific primers. 4C-sequencing libraries were prepared with 4C templates using the NEBNext ultra DNA library prep kit for Illumina, according to the manufacturer's protocol, cleaned with Agencourt AMPure XP PCR purification, and sequenced on the Illumina NextSeq 500. For more details, see Supplemental Methods.
4C-seq analysis
The paired-end 4q-D4Z4–specific 4C-seq reads were demultiplexed based on the 4C bait reading primer that included the restriction site sequence. All reads were then trimmed, and read pairs belonging to 4q-D4Z4, 10q-D4Z4, and 4q alleles were identified using the SSLP reading mate (Read 1) (Supplemental Fig. S1; Supplemental Table S1), where no mismatch for the genotype sequence was allowed. Read pairs reading into the captured sequences ligated to the bait (Read 2) from the biological replicates of each donor muscle cell line were then pooled and mapped with Bowtie 2 (Langmead and Salzberg 2012). To find chromosome-wide interacting domains, 4C-ker (Raviram et al. 2016) was used. The reproducibility between donor muscle cell lines and the quality of sequencing were assessed using Pearson correlation and cis/overall ratio (see Supplemental Methods). High-frequency interactions for each VP were intersected using BEDTools v2.2.4 (Quinlan and Hall 2010), and overlapping regions between donor muscle cell lines (CN-3 vs. CN-4 and FSHD1-3 vs. FSHD1-4) after removing overhangs were considered as high-confidence interacting domains. The interacting genes were defined as those that fall within the coordinates of these domains. Comparative analyses were performed between the 4q-D4Z4 allele interactomes and also between the 4q and 10q-D4Z4 interactomes (see Supplemental Methods).
The paired-end FBXO32 4C-seq reads were demultiplexed based on the 4C bait reading primer that includes the restriction site sequence. All reads were trimmed, and reads from the biological replicates of each donor muscle cell line were pooled and then mapped with Bowtie 2 (Langmead and Salzberg 2012). The reproducibility between donor muscle cell lines and the quality of sequencing were assessed using Pearson correlation and cis/overall ratio. Cis-interacting domains were identified using 4C-ker (Raviram et al. 2016), and high-confidence interacting domains were selected. Full lists of interactions are available in Supplemental Table S2. For more details, see Supplemental Methods.
ChIP-seq and ChIP-qPCR experiments
ChIP experiments were performed as previously described (Bodega et al. 2017) with minor modifications. The same donor muscle cell lines used for 4C-seq analysis of CN (CN-3, CN-4) and FSHD1 (FSHD1-3, FSHD1-4) human primary MBs and MTs day 4 (3.5 × 106 per sample) were processed for ChIP-seq analysis. For ChIP-seq and ChIP-qPCR, chromatin was immunoprecipitated with anti-H3K36me3 (ab9050, Abcam), anti-H3K4me1 (07-436, Millipore), anti-H3K27ac (07-360, Millipore), anti-H3K4me3 (07-473, Millipore), and anti-H3K27me3 (07-449, Millipore), as well as anti-RNA polymerase II CTD repeat YSPTSPS (phospho S5) antibody (4H8; ab5408, Abcam). ChIP-sequencing libraries were prepared using the NEBNext ultra DNA library prep kit for Illumina according to the manufacturer's protocol, cleaned with Agencourt AMPure XP PCR purification, and sequenced on the Illumina NextSeq 500 or HiSeq 2000. For ChIP-qPCR experiments, qRT-PCR analysis was performed on a StepOnePlus real-time PCR system, using power SYBR Green qPCR master mix. The relative enrichment obtained by using all the antibodies was quantified after normalization for input chromatin. Primers used are reported in Supplemental Table S6. For more details, see Supplemental Methods.
ChIP-seq analysis
We generated ChIP-seq data sets for CN MBs and MTs day 4 for the following histone marks: H3K36me3, H3K4me3, and H3K27me3. H3K36me3, H3K4me1, H3K27ac, H3K4me3, and H3K27me3 data sets were generated for FSHD1 MBs and MTs day 4. The following already published ChIP-seq data sets from ENCODE were used: H3K4me1 of human skeletal MBs (ENCSR000ANI), H3K27ac of human skeletal MBs (ENCSR000ANF), H3K4me1 of human skeletal MTs (ENCSR000ANX), and H3K27ac of human skeletal MTs (ENCSR000ANV). Reads were mapped with Bowtie 2 (Langmead and Salzberg 2012) on quality-checked (FastQC v0.11.2) and trimmed reads (Trimmomatic v0.32) (Bolger et al. 2014). For visualization of ChIP-seq tracks of independent samples, reads were normalized using bins per million mapped reads (BPM), same as TPM in RNA-seq, and, to ensure fair comparison between all data sets, were further normalized to the respective input to produce coverage files reporting the log2 ratio of normalized read number between samples and inputs using bamCompare module. Quality and reproducibility assessment were performed using deepTools2 package (Ramírez et al. 2016). For more details, see Supplemental Methods. Details on DUX4 ChIP-seq analysis is provided in Supplemental Methods.
Chromatin state analysis
We used ChromHMM (Ernst and Kellis 2012) with default parameters to derive genome-wide chromatin states maps of CN and FSHD1 MBs and MTs. We used the five histone marks H3K36me3, H3K4me1, H3K27ac, H3K4me3, and H3K27me3, as well as the respective input files, and binarized the data with BinarizeBed. We chose 15 states as the optimal number according to the maximal informative annotated genomic features and minimal redundancy. Subsequent functional annotations were attributed to each state, choosing names and a color code for visualization according to the Roadmap Epigenomics Consortium nomenclature (Roadmap Epigenomics Consortium et al. 2015). The total number of derived chromatin features was similar between the samples (CN MB: 503,507; FSHD1 MB: 565,653; CN MT: 609,058; FSHD1 MT: 581,946). We performed overlap enrichment of the 15 chromatin states with known genome organization features (Supplemental Fig. S8A,B) and intersected chromatin states and genes bodies retrieved from GENCODE version 19 using BEDTools v2.2.4 (Quinlan and Hall 2010). Calculations of pairwise Jaccard were performed with BEDTools.
Chromatin state switch analysis
To define whether CN and FSHD1 cells showed differences at gene chromatin state level, we took CN data as reference to search for specific switches in FSHD1. We intersected chromatin states retrieved from gene bodies in CN MBs, CN MTs, FSHD1 MBs, and FSHD1 MTs. We postulated that a given state in a particular condition (CN MB or MT) should intersect another state in the other condition (FSHD1 MB or MT) in a reciprocal manner. We thus performed BEDTools intersect using –f .60 –r, thus requiring that at least 60% of a state in CN MBs or MTs recovered a state in FSHD1 MBs or MTs in a reciprocal manner. In this way, identical states in the CN versus FSHD1 comparison (conserved states in the CN/FSHD1 comparison), as well as different states in the CN versus FSHD1 comparison (switching states in the CN/FSHD1 comparison), were retrieved. We focused on chromatin state switches between conditions (Supplemental Table S4) and added directionality to the chromatin state switches (that we chose to be active or repressive switches). We grouped the states into three main categories: promoters, enhancers, and enhancer priming. The states involved in each group, as well as the definition of the directional switches they are involved in, are summarized in Figure 2B. For each gene, we also summarized all chromatin states expressed as a percentage of coverage across the gene body and defined the state with the highest coverage as being the “major state” for a given gene. We grouped those major states into the two categories of active and repressed as indicated in Figure 2B. To obtain the genes showing directional switches, genes activated should display one of the following features: (1) major state transition from repressed to active or (2) major active state with at least one additional chromatin state switch toward activation. On the contrary, genes repressed should display either (1) transition from an active to a repressed major state or (2) major repressive state with at least one additional chromatin state switch toward repression as defined in Figure 2B.
RNA-seq assay and data analysis
RNA-seq studies were performed on the same donor muscle cell lines used for 4C-seq and ChIP-seq analyses of CN (CN-3, CN-4) and FSHD1 (FSHD1-3, FSHD1-4) human primary MBs and MTs day 4. Briefly, total RNA was isolated using the miRNA tissue kit on an automated Maxwell RSC extractor, following the manufacturer's instructions. RNA integrity was assessed on TapeStation. Subsequently, RNA for each donor muscle cell line was used to generate single-end 75-bp sequencing libraries with the TruSeq stranded mRNA library prep kit, according to the manufacturer's protocol. Sequencing was performed on a NextSeq 500.
In addition, a published RNA-seq data set, GSE56787 (Yao et al. 2014), consisting of human primary healthy and FSHD1 and FSHD2 MBs/MTs obtained from the University of Rochester bio-repository (http://www.urmc.rochester.edu/fields-center) was also analyzed.
All FASTQ files were analyzed with FastQC v0.11.2. Adapters were removed, and trimming was performed with Trimmomatic with standard parameters. Reads mapping to the reference genome GRCh37/hg19 were performed with STAR 2.3.0e (Dobin et al. 2013). The reference annotation used was GENCODE v19, and normalized fragments per kilobase of transcript per million mapped reads (FPKM) values were obtained with Cuffdiff (Trapnell et al. 2013). Normalized FPKM were log2 transformed, and a value of one was added to all FPKM values to finally obtain log2 (1 + FPKM) values used in downstream analyses (Supplemental Tables S4, S5). For more details, see Supplemental Methods.
GO analysis
GO analysis was performed on protein-coding genes with the Cytoscape v3.2.0 (Shannon et al. 2003) plug-in ClueGO v2.1.5 (Bindea et al. 2009). Statistically enriched biological processes (updated on 04/18/2016) were functionally grouped according to their k-score, and the most significant GO term of each group was used to summarize GO term for the group. Full lists of GO terms and associated genes are available in Supplemental Table S5.
GSEA
GSEA was performed as described previously (Subramanian et al. 2005). The gene set was represented by the 319 lost-activated genes or by the 131 lost-repressed genes (genes from Fig. 3A; Supplemental Table S5). We tested if those genes were significantly enriched in a gene expression data set associated with a skeletal muscle atrophic condition (disuse muscle atrophy, GSE21496) (Supplemental Table S5; Reich et al. 2010). We also tested the association of our gene sets with a gene expression data set from skeletal muscle hypertrophy (GSE12474) (Supplemental Table S5; Goto et al. 2011). GSEA was performed on those data sets with the ranking metric Signal2noise with 1000 phenotype permutations for statistical assessment of enrichment.
3D multicolor DNA FISH
To produce probes for 3D multicolor DNA FISH, we used the following BAC DNA clones (BACPAC Resources Program, CHORI): CH16-77M12 (D4Z4, containing wild-type D4Z4 array), RP11-279K24 (4q35.1), RP11-846C19 (C-, 3q11.2, pericentromeric region), RP11-115K4 (C+, 8q24.3, subtelomeric region), RP11-288G11 (10q26.3), and RP11-174I12 (FBXO32, 8q24.13, mid q arm region). Probes used for 3D multicolor DNA FISH in transfection experiments presented in Figure 5, A through C, were produced from PCR designed on the pTARBAC6 backbone of the transfected CH16-291A23 BAC (BAC 4q-D4Z4n), on the pBACe3.6 backbone of the transfected RP11-2A16 BAC (Ctrl BAC), on a 35-kb genomic region of 4q35.1 (4q), and on a 35-kb genomic region of FBXO32. Primers used are reported in Supplemental Table S6. One to three micrograms of BAC DNA or pooled PCR products was labeled with bio-dUTP, dig-dUTP, or cy3-dUTP through nick translation. The 3D multicolor DNA FISH assay was performed as previously described (Cremer et al. 2008) with minor adaptations. One to three donor muscle cell lines of CN and FSHD human primary MBs or MTs day 4 were processed for each experiment. An eclipse Ti-E (Nikon Instruments) microscope was used to scan the nuclei, with an axial distance between 0.2- and 0.25-μm consecutive sections. To automatically analyze 3D multicolor DNA FISH in fluorescence cell image z-stacks, we developed a tool in MATLAB. The tool, which we named Nuclear Contacts Locator in 3D (NuCLεD), is capable of automatically detecting and localizing fluorescent 3D spots in cell image stacks. Measurements retrieved are shown in Supplemental Table S3. Details on the 3D multicolor DNA FISH protocol and NuCLεD algorithm description are provided in Supplemental Methods.
Chromatin conformation capture
The 3C assay was performed as previously described (Cortesi and Bodega 2016) with minor adaptations. Two donor muscle cell lines of CN (CN-3, CN-4) and FSHD1 (FSHD1-3, FSHD1-4) human primary MBs (3.5 × 106 per sample) nuclei were processed. One to two biological replicates for each cell line were performed. Digestion was performed using HindIII. A reference template was generated by digesting, mixing, and ligating a BAC covering the genomic region of interest (FBXO32 region, RP11-174I12). 3C templates and the reference template were used to perform PCR analysis with DreamTaq DNA Polymerase using primers designed around the HindIII restriction sites present at FBXO32 region and, as bait primer, the same of FBXO32 4C (Supplemental Table S6) on a Veriti 96-well thermal cycler. The PCR products were densitometrically quantified using ImageJ software. Data are presented as the ratio of amplification obtained with 3C templates in respect to the reference template. For more details, see Supplemental Methods.
BAC transfection
BAC transfections were performed as previously described (Montigny et al. 2003) with minor adaptations. CN and FSHD1 human primary and immortalized MBs were plated. The following day, BAC DNA (RP11-2A16, as control BAC, representative of an unrelated and not interacting genomic region, Chr 17q21.33; CH16-291A23, containing at least 15 units of D4Z4 repeat, as derived by EcoRI and KpnI digestions) were diluted in Opti-MEM with the addition of P3000 reagent. Lipofectamine 3000 reagent was diluted in Opti-MEM. After 5 min, BAC DNA (plus P3000) and Lipofectamine preparations were gently mixed and incubated for 20 min at room temperature. Transfection complexes were then added to the cells and incubated for 48 h at 37°C. The primer pairs used for PCR or qRT-PCR amplifications are shown in Supplemental Table S6. For more details on transfection efficiency and DNA extraction, see Supplemental Methods.
Statistics and bioinformatics
To determine the significance between two groups, we used a Wilcoxon matched-pair signed-rank test, Student's t-test, or Fisher's exact test, as reported in figure legends; exact P-values and exact types of tests used are specified in figure legends. For correlation analysis, we used Pearson correlation; the exact values are specified in the figures. Multiple comparisons were performed by two-way ANOVA followed by Bonferroni posttest correction; exact P-values and types of tests used are specified in figure legends.
For all statistical tests, the 0.05 level of confidence was accepted for statistical significance; statistical significance is denoted by asterisks in figures, where single asterisk represents P-value <0.05, double asterisk represents <0.01, triple asterisk represents <0.001, and quadruple asterisk represents <0.0001.
All reads were assessed for quality using FastQC and were processed using Trimmomatic. They were aligned to the human genome (hg19) using either Bowtie 2 (Langmead and Salzberg 2012) or STAR 2.3.0e (Dobin et al. 2013). Aligning to GRCh38 is expected to provide similar results as only a small number of bases change genome-wide; the major difference between the releases is in centromere assembly (Guo et al. 2017), which is not the focus of our study.
Data access
Circular chromosome conformation capture and sequencing data (4C-seq) and chromatin immunoprecipitation and RNA sequencing data (ChIP-seq and RNA-seq) for the human samples from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under accession number SRP117155.
Acknowledgments
We thank C. Lanzuolo, D. Gabellini, V. Ranzani, M.R. Matarazzo, and E. Battaglioli for their stimulating discussions and constructive criticism of this manuscript. We thank M. Mora from the Telethon BioBank of the C. Besta Neurological Institute, R. Tawil from the Fields Center for FSHD of the Rochester Medical Center Department of Neurology, and J. Chen from the University of Massachusetts Medical School Wellstone Center for FSH Muscular Dystrophy Research, Wellstone Program & Department of Cell & Developmental Biology for providing the human myoblasts. We acknowledge the scientific and technical assistance of the INGM Imaging Facility (Istituto Nazionale di Genetica Molecolare “Romeo ed Enrica Invernizzi” [INGM], Milan, Italy), in particular C. Cordiglieri, for assistance during 3D multicolor DNA FISH images acquisition. pCMV-Mock and pCMV-DUX4 plasmids were a kind gift of D. Gabellini. This work has been supported by the following grants to B.B.: EPIGEN Italian flagship program, Association Française contre les Myopathies (AFM-Telethon, grant 18754), and Giovani Ricercatori, Italian Ministry of Health (GR-2011-02349383).
Author contributions: A.C., M.P., and S.S. designed and performed experiments, analyzed the data, and wrote the manuscript. F.M. and E.S. performed experiments. F.G., L.A., and G.O. developed the new algorithm for 3D multicolor DNA FISH image analysis. C.C. and G.S. performed RNA-seq library preparation, NGS data processing, and sequencing. B.B. conceived this study, designed experiments, analyzed the data, and wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.233288.117.
-
Freely available online through the Genome Research Open Access option.
- Received December 4, 2017.
- Accepted May 13, 2019.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








