
Parent-of-origin-dependent nucleosome organization correlates with genomic imprinting in maize
- Xiaomei Dong1,4,
- Jian Chen1,4,
- Tong Li1,4,
- En Li1,
- Xiangbo Zhang1,
- Mei Zhang1,2,
- Weibin Song1,
- Haiming Zhao1 and
- Jinsheng Lai1,3
- 1State Key Laboratory of Agrobiotechnology and National Maize Improvement Center, Department of Plant Genetics and Breeding, China Agricultural University, Beijing, 100193, People's Republic of China;
- 2Department of Biology, Stanford University, Stanford, California 94305, USA;
- 3Center for Crop Functional Genomics and Molecular Breeding, China Agricultural University, Beijing, 100193, People's Republic of China
-
↵4 These authors contributed equally to this work.
Abstract
Genomic imprinting refers to allele-specific expression of genes depending on their parental origin. Nucleosomes, the fundamental units of chromatin, play a critical role in gene transcriptional regulation. However, it remains unknown whether differential nucleosome organization is related to the allele-specific expression of imprinted genes. Here, we generated a genome-wide map of allele-specific nucleosome occupancy in maize endosperm and presented an integrated analysis of its relationship with parent-of-origin-dependent gene expression and DNA methylation. We found that ∼2.3% of nucleosomes showed significant parental bias in maize endosperm. The parent-of-origin-dependent nucleosomes mostly exist as single isolated nucleosomes. Parent-of-origin-dependent nucleosomes were significantly associated with the allele-specific expression of imprinted genes, with nucleosomes positioned preferentially in the promoter of nonexpressed alleles of imprinted genes. Furthermore, we found that most of the paternal specifically positioned nucleosomes (pat-nucleosomes) were associated with parent-of-origin-dependent differential methylated regions, suggesting a functional link between the maternal demethylation and the occurrence of pat-nucleosome. Maternal specifically positioned nucleosomes (mat-nucleosomes) were independent of allele-specific DNA methylation but seem to be associated with allele-specific histone modification. Our study provides the first genome-wide map of allele-specific nucleosome occupancy in plants and suggests a mechanistic connection between chromatin organization and genomic imprinting.
Genomic imprinting, an epigenetic phenomenon, primarily occurs in the endosperm of flowering plants in which a subset of genes is unequally expressed between two alleles depending on their parental origin (Kermicle 1970; Huh et al. 2008; Jahnke and Scholten 2009). Extensive transcriptomic studies have led to the identification of several hundred imprinted genes in a number of plant species including Arabidopsis thaliana, rice, maize, castor bean, sorghum, Arabidopsis lyrate, and Capsella rubella (Gehring et al. 2011; Hsieh et al. 2011; Luo et al. 2011; Waters et al. 2011; Wolff et al. 2011; Zhang et al. 2011, 2016; Xin et al. 2013; Xu et al. 2014; Hatorangan et al. 2016; Klosinska et al. 2016). Many reports from several plant species have suggested that allele-specific epigenetic modifications including DNA methylation (Kinoshita et al. 2004; Jullien et al. 2006; Tiwari et al. 2008; Hsieh et al. 2011; Wolff et al. 2011; Zhang et al. 2014), active and repressive histone modifications in endosperm possibly play important roles in imprinting regulation (Köhler et al. 2005; Haun and Springer 2008; Hsieh et al. 2011; Wolff et al. 2011; Du et al. 2014; Zhang et al. 2014; Dong et al. 2017). However, it remains unknown whether differential nucleosome organization is related to the monoallelic expression of imprinted genes in plants.
Nucleosomes, the fundamental structural units of chromatin, contain 147 bp of DNA wrapped around eight histone protein cores (Luger et al. 1997; Köhler et al. 2005; Haun and Springer 2008; Hsieh et al. 2011; Wolff et al. 2011; Du et al. 2014; Zhang et al. 2014). Nucleosome positioning in the genome is dynamic and can modulate the accessibility of DNA to transcription elements, thus regulating gene expression (Jiang and Pugh 2009; Bai and Morozov 2010; Chen et al. 2017). It has been demonstrated that the promoter regions of genes tend to be nucleosome depleted (Lee et al. 2007; Schones et al. 2008; Li et al. 2014). Well-phased nucleosomes were observed downstream from the transcriptional start site (TSS), particularly the first nucleosome, which has a close relationship with Pol II binding (Schones et al. 2008; Li et al. 2014). Although genome-wide nucleosome occupancy maps have been generated for Arabidopsis thaliana, rice, and maize (Chodavarapu et al. 2010; Fincher et al. 2013; Li et al. 2014; Wu et al. 2014; Liu et al. 2015; Rodgers-Melnick et al. 2016; Chen et al. 2017), there has been no report so far on the investigation of allele-specific nucleosome occupancy in any plant species.
Genomic imprinting in plants is involved in the demethylation induced by DEMETER (DME), which is specifically expressed in the central cell of the female gametophyte and the vegetative cell of the male gametophyte (Choi et al. 2002; Schoft et al. 2011; Kawashima and Berger 2014). Several studies identified thousands of parent-of-origin-dependent differential methylated regions (pDMRs) where the maternal alleles are hypomethylated and the paternal alleles are hypermethylated in the endosperm (Ibarra et al. 2012; Rodrigues et al. 2013; Zhang et al. 2014). Active DNA demethylation also was directly reported in the central cell of Arabidopsis thaliana and rice, indicating that the DNA demethylation of maternal endosperm chromosomes observed in flowering plants is initiated in the central cell (Park et al. 2016). It is generally accepted that DNA methylation may impact nucleosome organization (Kelly et al. 2012; Yang et al. 2012; Collings et al. 2013; Portela et al. 2013). In addition, the process of DNA demethylation by DME involves a base-excision repair pathway in Arabidopsis (Choi et al. 2002; Ooi and Bestor 2008; Gehring et al. 2009), and the AP endonuclease-mediated DNA nicking activity that follows DME base excision might catalyze nucleosome sliding (Längst and Becker 2001). SSRP1, a component of the FACT histone chaperone, was demonstrated to potentially lead to a transition of chromatin state upon DNA demethylation in the central cell (Ikeda et al. 2011). However, there is still limited knowledge about the effect of the allele-specific DNA methylation pattern on nucleosome occupancy in plants.
Results
Identification of parent-of-origin-dependent nucleosomes in maize endosperm
To obtain the landscape of genome-wide parent-of-origin-dependent nucleosome occupancy, we generated MNase-seq libraries of 12-d after-pollination (DAP) maize dissected endosperm from the reciprocal crosses of B73 and Mo17 inbred. More than 700 million reads were produced for each sample, and more than half of them were uniquely mapped to the genome. The insert size distribution of the two libraries were highly similar with the major peak near the mononucleosomal DNA length (∼150 bp), according to the DNA electrophoretogram (Supplemental Fig. S1A,B), which reflected the expected digestion degree for the two libraries (Chen et al. 2017). In addition, the existence of the 10-bp periodicities of dinucleotides in two libraries was also observed (Supplemental Fig. S2), indicating the MNase-seq libraries accurately captured the mononucleosomal DNA fragment.
A total of 7,949,184 nucleosomes were identified in B73 × Mo17 (BM) and Mo17 × B73 (MB) endosperm samples all together by using DANPOS software (Chen et al. 2013). Among these nucleosomes identified, 1,475,669 (18.5%) nucleosomes covered SNPs between B73 and Mo17, and 996,273 (67.5%) nucleosomes covering SNPs had at least 10 reads on both B73 and Mo17 alleles, which were used for identifying allele-specific nucleosomes (Fig. 1A). We first calculated the ratio between the maternal and paternal alleles at each nucleosome site in endosperm tissue that is a triploid (2 maternal:1 paternal genomes) product of the double fertilization event in plant reproduction. As expected, most of the nucleosomes exhibited a maternal-to-paternal ratio of 2:1 in 12-DAP endosperm tissues (Fig. 1B). Only 14,568 nucleosomes showed significantly maternal bias, and 8610 nucleosomes showed significantly paternal bias (χ2 test; P-value <0.05) in both BM and MB endosperm, which were jointly called low-stringency parent-of-origin-dependent nucleosomes. Using a more stringent criterion that reads from the favorable allele were at least five times as many as that from the nonfavorable allele in both BM and MB endosperm, a total of 880 high-stringency parent-of-origin-dependent nucleosomes were identified, including 680 maternal specifically positioned nucleosomes (mat-nucleosomes) and 200 paternal specifically positioned nucleosomes (pat-nucleosomes) (Fig. 1A). Taken together, ∼2.3% of allelically analyzed nucleosomes showed significant parental bias. Figure 1C showed the regions around one mat-nucleosome and one pat-nucleosome. All SNPs in the mat-nucleosome and the pat-nucleosome exhibited significant parental bias, in contrasting to SNPs in the other nucleosomes showing a normal 2:1 ratio of maternal allele to paternal allele. To validate the accuracy of parent-of-origin-dependent nucleosomes identified above, RT-PCR amplicons from 12-DAP BM and MB endosperm for four parent-of-origin-dependent nucleosomes were sequenced. All the selected four nucleosomes exhibited the expected parental bias (Supplemental Fig. S3).
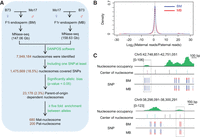
The identification of parent-of-origin-dependent nucleosomes. (A) Pipeline for identifying parent-of-origin-dependent nucleosomes. (B) Allele-specific analysis of nucleosomes in maize endosperm. The x-axis and y-axis represent the log2 of maternal reads divided by paternal reads for the allelically analyzed nucleosomes in endosperm from BM (blue lines) and MB (read lines). (C) Regions around maternal/paternal specifically positioned nucleosomes. Green lines indicate the levels of nucleosomes. The black, light blue, and pink lines indicate the center position of endosperm nucleosomes, allelically analyzed nucleosomes, and high-stringency allele-specific nucleosomes. The percentages of allelic reads of MNase-seq data for specific SNP sites are shown; red lines represent the paternal alleles, and blue lines represent the maternal alleles.
Most parent-of-origin-dependent nucleosomes are single isolated nucleosomes scattering along the chromosome
Analysis of the genome distributions showed that pat-nucleosomes preferentially located in genic regions, whereas most of the mat-nucleosomes were in intergenic regions (Supplemental Fig. S4). Similar to the typical open chromatin regions around the promoter of genes (Chen et al. 2017), parent-of-origin-dependent nucleosomes were found to display relative low nucleosome levels (Fig. 2A). Nucleosome depletion at promoters is important for the accessibility of transcription factors (Struhl and Segal 2013). Hence, these parent-of-origin-dependent nucleosomes might locate mostly on the open chromatin regions of various regulatory functions (Song et al. 2011). The intrinsic properties of poly (dA:dT) are important for nucleosome depletion, promoter accessibility, and transcriptional activity (Iyer and Struhl 1995; Segal and Widom 2009; Raveh-Sadka et al. 2012; Struhl and Segal 2013). We found that CG content of sequences around mat-nucleosomes/pat-nucleosomes were significantly lower than that of other allelically analyzed nucleosomes (Supplemental Fig. S5), indicating that the intrinsic DNA sequence properties could be involved in the occurrence of parent-of-origin-dependent nucleosomes.
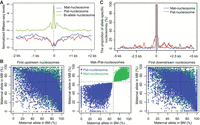
The characteristics of parent-of-origin-dependent nucleosomes. (A) The nucleosome levels around the region of parent-of-origin-dependent nucleosomes in endosperm (BM). Blue lines indicate the nucleosome levels around the region of maternally specific-positioned nucleosomes. Red lines indicate the nucleosome levels around the region of paternally specific-positioned nucleosomes. Green lines indicate the nucleosome levels around the region of 2000 random biallelic nucleosomes. Biallelic nucleosomes are nucleosomes that do not deviate significantly from the 2:1 ratio of maternal allele to paternal allele in each reciprocal hybrid. (B) Allele-specific analysis of parent-of-origin-dependent nucleosomes and their first upstream and downstream nucleosomes. The x-axis and y-axis represent the proportion of maternal reads for the nucleosomes that had at least one SNP and had at least 10 allelic reads in both reciprocal hybrids. The green points represent the allelic status of maternal specifically positioned nucleosomes (mat-nucleosomes) and their first upstream and downstream nucleosomes. The blue points represent paternal specifically positioned nucleosomes (pat-nucleosomes) and their first upstream and downstream nucleosomes. (C) The frequency of allele-specific nucleosomes at each base within ±5 kb of mat-nucleosomes (blue lines) or pat-nucleosomes (red lines) is shown. The gray rectangle represents the region of mat-nucleosomes and pat-nucleosomes.
We then investigated whether the parent-of-origin-dependent nucleosomes occupy as clusters or single isolated nucleosomes. We looked at the allelic status of the neighboring nucleosomes of the parent-of-origin-dependent nucleosomes identified. The neighboring (upstream or downstream) nucleosomes around 87.4%/85.9% of low-stringency mat-/pat-nucleosomes exhibited a typical maternal-to-paternal ratio of 2:1 (Fig. 2B). Similarly, the neighboring nucleosomes around 86.2%/69.2% of high-stringency mat-/pat-nucleosomes exhibited a maternal-to-paternal ratio of 2:1. In addition, we observed a sharply decreased proportion of the allele-specific nucleosome around the high-stringency mat-nucleosomes/pat-nucleosomes (Fig. 2C) and a relatively long average distance between any neighboring mat-nucleosome (140.2 kb) and pat-nucleosome (239.4 kb) (Supplemental Fig. S6). These results together suggested that parent-of-origin-dependent nucleosomes tend to be isolated single nucleosomes that scatter along the chromosomes.
The mat-nucleosomes and pat-nucleosomes have different allele-specific methylation patterns
Parent-of-origin-dependent nucleosomes can result from either nucleosome eviction or addition on one specific allele. To explore a potential mechanism for the occurrence of parent-of-origin-dependent nucleosomes, we compared the nucleosome levels between endosperm and seedling tissues in the region covered by the parent-of-origin-dependent nucleosomes identified in this study. We found that the nucleosome levels in the region of pat-nucleosomes in endosperm were significantly lower than those in seedling tissue (Supplemental Fig. S7). On the contrary, the nucleosome levels in the region of mat-nucleosomes in endosperm are significantly higher than those in seedling tissue (Supplemental Fig. S7). The results indicated that the occurrence of pat-nucleosomes might be mainly due to the specific eviction of nucleosomes positioned at maternal alleles while the occurrence of mat-nucleosomes might be mainly due to the specific addition of nucleosomes positioned at maternal alleles.
Analysis of whole-genome bisulfite sequencing data from BM and MB endosperm samples revealed that the DNA methylation levels on the maternal alleles were significantly lower than those on paternal alleles at the region of pat-nucleosomes (Fig. 3A). However, most regions of the mat-nucleosomes mainly exhibited hypermethylation on both alleles, and a small part (∼15%) of the region of mat-nucleosomes exhibited hypomethylation on both alleles (Fig. 3B). About 70% of pat-nucleosomes were significantly associated with CG_pDMRs (pDMRs at CG context) identified previously (Zhang et al. 2014), compared with 2.8% of mat-nucleosomes and 1.5% of allelically analyzed nucleosomes that overlapped with CG_pDMRs (Fig. 3C). The result suggested that the mechanism for the occurrence of the pat-nucleosomes could be different than that of the mat-nucleosomes. For the pat-nucleosomes, it is possible that a DNA methylation–dependent mechanism contributed to the specific eviction of nucleosomes positioned at maternal alleles, resulting in nucleosomes specifically positioned at paternal alleles.
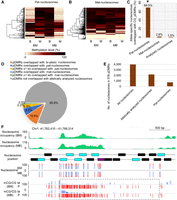
Relationship between DNA methylation and parent-of-origin-dependent nucleosomes. (A,B) Heatmaps of CG methylation levels between alleles of B73 and Mo17 reciprocal crosses at paternal specifically positioned nucleosomes (pat-nucleosomes) and maternal specifically positioned nucleosomes (mat-nucleosomes): (B) B73 allele; (M) Mo17 allele; (BM) B73 × Mo17; (MB) Mo17 × B73. (C) Association of paternal specifically positioned nucleosomes (pat-nucleosomes) and maternal specifically positioned nucleosomes (mat-nucleosomes) with CG_pDMRs. The proportions of pat-nucleosomes, mat-nucleosomes, and all allelically analyzed nucleosomes overlapped with CG_pDMRs are shown on the y-axis. (*) Significant association of pat-nucleosomes with CG_pDMRs (Fisher's exact test; P-value <2.2 × 10−16) compared with that of mat-nucleosomes. (D) The proportion of 6910 CG_pDMRs overlapped with parent-of-origin-dependent nucleosomes. (E) The allelic status of all nucleosomes in 578 CG_pDMRs overlapped with pat-nucleosomes. (F) The integrated view of nucleosome occupancy in one CG_pDMR. The normalized nucleosome levels of endosperm (BM) are plotted in green. The black, blue, and purple rectangles indicate the position of endosperm nucleosomes, allelically analyzed nucleosomes, and high-stringency allele-specific nucleosomes. The percentages of allelic reads of MNase-seq data for specific SNP sites are shown, with red lines for the paternal allele and blue lines for the maternal allele. The DNA methylation level for specific SNP sites are shown for both maternal and paternal alleles, with red lines for the paternal allele (P) and blue lines for the maternal allele (M). The dotted rectangle highlights the pDMRs identified in this region.
About half of CG_pDMRs were associated with pat-nucleosomes
Although several thousand pDMRs have been identified in maize endosperm (Zhang et al. 2014), little is known about their status of chromatin accessibility. Here, we investigated the relationship between CG_pDMRs and parent-of-origin-dependent nucleosomes. As observed, among the 6910 CG_pDMRs, there are 1317 (19.1%) CG_pDMRs including at least one allelically analyzed nucleosome, and 28.6% (377/1,317) of CG_pDMRs included pat-nucleosomes, but only 0.99% (13/1,317) of CG_pDMRs were associated with mat-nucleosomes (Fig. 3D). If we extended to the ±1 kb regions flanking 1317 CG_ pDMRs, a total of 578 (43.8%) were found to be associated with nucleosomes exhibiting paternal bias (Fig. 3D). Overall, based on our current analysis, about half of maternal demethylation events were associated with events of maternal nucleosome depletion.
As discussed earlier, parent-of-origin-dependent nucleosomes are mostly isolated single nucleosomes; therefore, not all the nucleosomes in any CG_ pDMRs were paternal specifically positioned. We analyzed the allelic status of all nucleosomes in 578 CG_pDMRs associated with pat-nucleosomes identified above. A total of 3982 nucleosomes were positioned in the 578 CG_pDMRs (about seven nucleosomes in one pDMR on average), of which 2165 nucleosomes can be analyzed for their status of allele-specific occupancy, but only 734 nucleosomes displayed paternal bias (about one pat-nucleosomes in one pDMR on average) (Fig. 3E). Figure 3F displays the integrated view of nucleosome occupancy in a region of CG_pDMR. The CG_pDMRs with a length of ∼3 kb displayed maternal hypomethylation and paternal hypermethylation. A total of 16 nucleosomes were positioned in the region, but only one out of the seven allelically analyzed nucleosomes showed paternal bias.
The nucleosomes in the promoter regions of imprinted genes and imprinted noncoding RNAs exhibit parental bias
To investigate the roles of nucleosome organization in the regulation of genomic imprinting, we investigated the relationship between parent-of-origin-dependent nucleosomes and imprinted transcripts. A total of 142 imprinted genes, including 28 MEGs and 70 PEGs that were constitutively expressed (con-MEGs/con-PEGs), 24 MEGs and 20 PEGs that were primarily expressed in endosperm (endo-MEGs/endo-PEGs) in 12-DAP endosperm have been identified previously (Dong et al. 2017). Because con-MEGs seem to be expressed in the specific compartments that are underrepresented in typical endosperm sample (Dong et al. 2017), we did not explore the relationship between parent-of-origin-dependent nucleosomes and con-MEGs. By looking at the distribution of parent-of-origin nucleosomes around genes, we found that parent-of-origin-dependent nucleosomes were significantly associated with imprinted genes, as compared with nonimprinted genes (P-value <2.2 × 10−16, Fisher's exact test) (Fig. 4A), genes expressed in endosperm and genes nonexpressed in endosperm (P-value <2.2 × 10−16, Fisher's exact test) (Supplemental Fig. S8). Further, the parent-of-origin-dependent nucleosomes tend to locate around the TSS regions of imprinted genes (Fig. 4A). More importantly, nucleosomes positioned around TSS of endo-MEGs showed paternal bias, whereas nucleosomes positioned around TSS of PEGs were maternal bias, which indicated that stronger nucleosome occupancy exhibited in the nonexpressed allele in promoter of imprinted genes (Fig. 4A). Indeed, of the 24 endo-MEGs, we could track the allelic status of at least one nucleosome positioned in the promoter (−300 to +200 bp from TSS) of eight endo-MEGs, and nucleosomes positioned at the promoter of five (62.5%) endo-MEGs exhibited paternal bias (Supplemental Fig. S9A,B). We also could track the allelic status of nucleosomes positioned at the promoter of 26 con-PEGs and seven endo-PEGs. Nucleosomes positioned at the promoter of 14 (53.8%) con-PEGs and five (71.4%) endo-PEGs exhibited maternal bias (Supplemental Fig. S9A,B). In contrast, only 0.66% (3/451) of nonimprinted genes harbored parent-of-origin-dependent nucleosomes at promoter regions. The extent of the association of parent-of-origin nucleosomes and imprinted genes were underestimated because not all of the nucleosomes in promoter could be analyzed allelically. The distance of parent-of-origin-dependent nucleosomes to TSS of imprinted genes is concentrated on −100 ∼ 0 bp, which indicated that parent-of-origin-dependent nucleosomes is the first nucleosome upstream of TSS (−1 nucleosome) (Supplemental Fig. S9C). In addition, we also looked at the relationship between 35 maternally expressed noncoding RNAs (MNCs) and the parent-of-origin-dependent nucleosomes. Although we could not accurately define the TSS of MNCs, we also found that only pat-nucleosomes were associated with MNCs and these pat-nucleosomes were located at the 5′ end of MNCs (Supplemental Fig. S10). Collectively, our results indicated that differential nucleosome occupancy between alleles is associated with allelic expression of imprinted genes and imprinted noncoding RNAs.
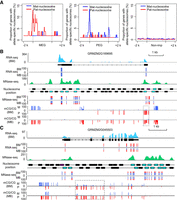
Relationship between imprinted genes and parent-of-origin-dependent nucleosomes. (A) The distribution of parent-of-origin nucleosomes at imprinted genes/nonimprinted genes and its up and down 2-kb regions. The blue line represents the maternal specifically positioned nucleosome (mat-nucleosome), and the red line represents the paternal specifically positioned nucleosome (pat-nucleosome). Nonimprinted genes were expressed in endosperm and did not deviate significantly from the 2:1 ratio of maternal allele to paternal allele in both reciprocal hybrids. (B,C) The investigated view of expression, DNA methylation, and nucleosome occupancy at GRMZM2G169695 (B) and GRMZM2G045503 (C). The expression levels of transcribed regions are shown in light blue. The normalized nucleosome levels of endosperm (BM) are plotted in green. The black, blue, pink, and purple rectangles indicate the position of endosperm nucleosomes, allelically analyzed nucleosomes, low-stringency allele-specific nucleosomes, and high-stringency allele-specific nucleosomes at genes. The percentages of allelic reads of RNA-seq and MNase-seq data for specific SNP sites are shown, with red lines for the paternal allele and blue lines for the maternal allele. The DNA methylation level for specific SNP sites are shown for both maternal and paternal alleles, with red lines for the paternal allele (P) and blue lines for the maternal allele (M). The dotted rectangles highlight the pDMRs identified in this region.
Figure 4B,C displayed the profiles of nucleosomes at the GRMZM2G169695 (MEG) and GRMZM2G045503 (PEG), respectively. As is shown, one pat-nucleosome is located upstream TSS of GRMZM2G169695. All SNPs within the pat-nucleosome exhibited significant paternal bias. One mat-nucleosome is located upstream TSS of GRMZM2G045503. All SNPs within the mat-nucleosome exhibited significant maternal bias. Results of RT-PCR amplicons sequencing from 12-DAP BM and MB endosperm for the two parent-of-origin-dependent nucleosomes at TSS of GRMZM2G169695 and GRMZM2G045503 also exhibited expected parental bias (Supplemental Fig. S3). Integrating the allele-specific DNA methylation, nucleosome occupancy, and gene expression, we found the pat-nucleosome overlapped with CG_ pDMRs at GRMZM2G169695 (Fig. 4B), but mat-nucleosomes are positioned in the hypomethylated promoter of GRMZM2G045503 (PEG) (Fig. 4C).
Discussion
Single nucleosome rearrangements could be correlated with the chromatin status and transcriptional activity
Nucleosome positioning as a component of overall chromatin organization is known to be critical for gene transcription. Although a genome-wide allele-specific chromatin accessibility map had recently been generated using DNase-seq and ATAC-seq in mammal (Lu et al. 2016; Wu et al. 2016), a global pattern of allele-specific nucleosome occupancy has not been reported in any other high eukaryotic species. Here, we reported the generation of MNase-seq data of maize endosperm from the reciprocal crosses of B73 and Mo17 inbred to obtain a genome-wide allele-specific nucleosome occupancy map and an integrated analysis together with allele-specific DNA methylation profiling data and imprinted gene expression.
It was reported that chromatin remodeling in response to physiological perturbation was typically associated with the eviction, appearance, or repositioning of one or two nucleosomes in the promoter, rather than broader region-wide changes (Lu et al. 2016). By comparing nucleosome occupancy in mouse and human embryonic stem cells, induced-pluripotent stem cells, and differentiated cell types using MNase-seq, a majority of rearrangements also were observed in a single nucleosome (West et al. 2014). Consistent with the above studies, we observed that chromatin remodeling between two alleles was typically associated with the rearrangements of only one/two nucleosomes in the promoter of imprinted genes. It had also been demonstrated in mouse that most imprinted genes showed allelically biased DNase I-hypersensitive sites (DHSs) in the promoter regions of genes in preimplantation embryos (Lu et al. 2016). Therefore, it is reasonable to think that the rearrangement of only one or two nucleosomes might be sufficient to alter the chromatin accessibility in the promoter of imprinted genes. Of note, the majority of parent-of-origin-dependent nucleosomes existed as isolated single nucleosomes, in promoter and other genomic regions, and the regions of parent-of-origin-dependent nucleosomes may be the open chromatin regions with notable lower nucleosomes levels. Thus, single nucleosome gain or loss could be enough to induce functional chromatin status change. This type of nucleosomal rearrangement is highly specific, which must require a dedicated targeting mechanism. The factors involved in the recognition and remodeling of this type of nucleosome rearrangement would be a fascinating topic to address in the future. Several studies reported that open chromatin regions induced by 5-AzaCdR treatment, regions of DHSs, or distal nucleosome-depleted regions (NDRs) were preferentially enriched with TF motifs (Pandiyan et al. 2013; Lu et al. 2016; Guo et al. 2017). We speculated that transcription factors could work in conjunction with other known chromatin remodelers to result in the preferential targeting of remodelers to the regions of parent-of-origin-dependent nucleosomes, and hence, chromatin opening.
The association between parent-of-origin-dependent nucleosomes and the expression of imprinting genes raises a question about whether differential chromatin accessibility between maternal and paternal alleles is the cause or the consequence of monoallelic expression. As a matter of fact, the relationship of nucleosome occupancy and transcriptional activity can be quite complex. It was reported that the dynamic change of nucleosome occupancy was linked to the transcription process (Huebert et al. 2012). However, specific nucleosomal rearrangements at promoters were also observed when there was no apparent transcriptional change (Shivaswamy et al. 2008). A recent work in mouse indicated that imprinting information has already been “written” in the form of chromatin accessibility during early preimplantation development before the onset of parental allelic expression (Lu et al. 2016). Hence, whether or not there is causal relationship between parental-specific nucleosomes in imprinted genes and their monoallelic expression in plant remains to be resolved in the future.
DNA demethylation induced by DME-like factor plays an important role in the nucleosome eviction
Many studies have shown that DME or DME-like glycosylase can induce DNA demethylation in the central cell of higher plants and lead to the presence of large number of pDMRs in endosperm (Ibarra et al. 2012; Rodrigues et al. 2013; Xu et al. 2014; Zhang et al. 2014). However, the functional consequence of most of pDMRs is unclear, except for some pDMRs associated with imprinted genes or imprinted noncoding RNAs (e.g., Zhang et al. 2014). In this study, we observed that most (70%) of pat-nucleosomes were located in pDMRs, whereas only 43.8% of pDMRs contained pat-nucleosomes. Moreover, there were no methylation differences between the two alleles of the mat-nucleosomes we identified. Thus, we proposed that demethylation of the maternal allele might be required for the occurrence of pat-nucleosomes, and the demethylation likely happens before the eviction of maternal nucleosome. To some extent, our hypothesis is consistent with a previous report that loss of DNA methylation can lead the loss of nucleosomes (Pandiyan et al. 2013). Nevertheless, there was also report that the base excision reaction (BER), the process required for demethylation induced by DME, could be intimately connected with the structural chromatin changes (Choi et al. 2002; Ooi and Bestor 2008; Gehring et al. 2009). However, our observation that the entire region of pDMRs (with length up to several kilobases) were overlapped with only one or two pat-nucleosomes, and about half of pDMRs do not include pat-nucleosomes, again supports the notion that maternal nucleosomes eviction is more likely occurring after demethylation, rather than induced directly by the BER. Of course, it is very likely that DNA demethylation by DME-like glycosylase is not enough for nucleosome eviction. Additional factor(s) are possibly needed to accurately guide the nucleosome remodeling factors to specific sites of particular nucleosomes. In summary, our result demonstrated that DNA demethylation induced by DME-like factor could be functionally linked with allele-specific nucleosome eviction.
Open chromatin regions tend to be genetic regulatory elements including promoters, enhancers, insulators, and locus control regions (Boyle et al. 2008). Compared with only <2% of demethylated regions induced by 5-AzaCdR treatment located in open chromatin regions (Pandiyan et al. 2013), we found that about half of pDMRs were associated with pat-nucleosomes in 12-DAP endosperm, which indicated that the DME-like glycosylase tends to remove methylation in regions with potential regulatory elements. We also investigated the difference between the CG_pDMRs overlapping with pat-nucleosomes and those not overlapping with pat-nucleosomes. Although the CG_pDMRs overlapping with pat-nucleosomes and those not overlapping with pat-nucleosomes have very similar genome distributions (Supplemental Fig. S11), the average length of the CG_pDMRs including pat-nucleosomes is significantly longer than that of CG_pDMRs not including pat-nucleosomes (Supplemental Fig. S12), indicating that longer regions of demethylation might be more favorable for the access of nucleosome remodeling factors. Notably, we found only CG_pDMRs including pat-nucleosomes were associated with the MEGs and MNCs, suggesting that the CG_pDMRs including pat-nucleosomes are more likely functional in 12-DAP endosperm. Of course, it is also possible that the CG_pDMRs nonoverlapping with pat-nucleosomes in 12-DAP endosperm might be overlapping with pat-nucleosomes in the central cell or in earlier developmental stages of endosperm.
Maternal specifically positioned nucleosomes might be associated with the changes of histone modifications
In contrast to the involvement of DNA demethylation for the occurrence of pat-nucleosomes, we observed that the occurrence of mat-nucleosomes was independent of DNA methylation. Interestingly, a recent study in mouse showed that maternal allele-specific H3K27me3 plays an important role in formation of paternal allele-specific DNase I hypersensitive sites, rather than maternal allele-specific DNase I hypersensitive sites (Inoue et al. 2017), consistent with our result that mat-nucleosomes and pat-nucleosomes are induced by different mechanisms. In the study, we observed that the mat-nucleosome positioned in the promoter of con-PEGs and endo-PEGs. In our previous works (Dong et al. 2017), we found that H3K4me3 modifications at con-PEGs and endo-PEGs showed significant paternal bias. It has also been suggested that chromatin modification by H3K4me3 may facilitate nucleosome eviction or repositioning in the −1 nucleosome region (Schones et al. 2008; Yang et al. 2012). Thus, we hypothesized that the changes in histone modifications might accelerate one or limited nucleosomes remodeling in the promoter of PEGs. However, most of the mat-nucleosomes were shown to be in intergenic regions. We speculated that other histone modifications or histone variants might also be involved in contributing to the occurrence of maternally specific-positioned nucleosomes, which can be tested in the future when additional allele-specific histone modifications data become available.
Methods
Tissue collection
The maize (Zea mays) inbred lines B73 and Mo17 were grown in fields at the experiment station of China Agricultural University, Beijing. Three ears of reciprocal crosses of B73 and Mo17 were collected after 12 d pollination. Endosperm tissues were collected from three different ears by manual dissection from whole kernels and were immediately frozen in liquid nitrogen.
Library construction for MNase-seq
The MNase-seq libraries of BM and MB endosperm were constructed according to the protocol described in our previous study (Chen et al. 2017). Notably, the appropriate levels of 1 unit of MNase were chosen for BM and MB endosperm samples, respectively. The MNase-seq libraries were sequenced to generate 100-nt paired-end reads on the Illumina HiSeq platform. The samples of reciprocal crosses, BM and MB, are serving as internal replicates to identify the parent-of-origin-dependent nucleosomes.
Defining nucleosome occupancy
The raw reads of MNase-seq data were aligned to the B73 reference genome (RefGen_v2) allowing two mismatches with Burrows–Wheeler Aligner (BWA) (Li and Durbin 2010). Only uniquely aligned paired reads were used for further analysis. The in vivo nucleosome occupancy of shoot and endosperm (BM and MB) was calculated using DANPOS (Chen et al. 2013), with the parameters set as -s 1, -a 1, -m 1, -p 1.0 × 10−5, -t 1.0 × 10−5.
Identification of parent-of-origin-dependent nucleosomes occupancy in maize endosperm
The number of reads containing the B73 or Mo17 allele was summed for all SNPs (Schnable et al. 2009; Lai et al. 2010; Jiao et al. 2012) within the same nucleosomes. The nucleosomes that contain at least 10 reads that could be assigned to a particular allele were used to perform χ2 test (relative to the expected 2 maternal:1 paternal) ratio. The nucleosomes showing significant parental bias (χ2 test; P-value <0.05) in both BM and MB endosperm were jointly called low-stringency parent-of-origin-dependent nucleosomes. The nucleosomes in which reads from the active allele were more than five times than that from the silenced allele in both BM and MB endosperm were identified as high-stringency parent-of-origin-dependent nucleosomes.
Data access
Raw reads as FASTQ files generated in this study have been submitted to the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession number SRP107032.
Acknowledgments
Research is supported by the National Natural Science Foundation of China (31421005, 91635303), National Key Research and Development Program (2016YFD0100802, 2016YFD0101803), 948 Project (2016-X33), and National Postdoctoral Program for Innovative Talents (BX201600149).
Author contributions: X.D., J.C., and J.L. designed the experiments. X.D. and J.C. collected all the materials. J.C. performed MNase-seq. X.D. and T.L. completed the bioinformatics analyses of all data. X.D. and J.L. wrote the manuscript. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.230201.117.
-
Freely available online through the Genome Research Open Access option.
- Received September 17, 2017.
- Accepted May 31, 2018.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








