
Inversion variants in human and primate genomes
- Claudia Rita Catacchio1,4,
- Flavia Angela Maria Maggiolini1,4,
- Pietro D'Addabbo1,
- Miriana Bitonto1,
- Oronzo Capozzi1,
- Martina Lepore Signorile1,
- Mattia Miroballo1,
- Nicoletta Archidiacono1,
- Evan E. Eichler2,3,
- Mario Ventura1 and
- Francesca Antonacci1
- 1Dipartimento di Biologia, Università degli Studi di Bari “Aldo Moro,” Bari 70125, Italy;
- 2Department of Genome Sciences, University of Washington School of Medicine, Seattle, Washington 98195, USA;
- 3Howard Hughes Medical Institute, University of Washington, Seattle, Washington 98195, USA
-
↵4 These authors contributed equally to this work.
Abstract
For many years, inversions have been proposed to be a direct driving force in speciation since they suppress recombination when heterozygous. Inversions are the most common large-scale differences among humans and great apes. Nevertheless, they represent large events easily distinguishable by classical cytogenetics, whose resolution, however, is limited. Here, we performed a genome-wide comparison between human, great ape, and macaque genomes using the net alignments for the most recent releases of genome assemblies. We identified a total of 156 putative inversions, between 103 kb and 91 Mb, corresponding to 136 human loci. Combining literature, sequence, and experimental analyses, we analyzed 109 of these loci and found 67 regions inverted in one or multiple primates, including 28 newly identified inversions. These events overlap with 81 human genes at their breakpoints, and seven correspond to sites of recurrent rearrangements associated with human disease. This work doubles the number of validated primate inversions larger than 100 kb, beyond what was previously documented. We identified 74 sites of errors, where the sequence has been assembled in the wrong orientation, in the reference genomes analyzed. Our data serve two purposes: First, we generated a map of evolutionary inversions in these genomes representing a resource for interrogating differences among these species at a functional level; second, we provide a list of misassembled regions in these primate genomes, involving over 300 Mb of DNA and 1978 human genes. Accurately annotating these regions in the genome references has immediate applications for evolutionary and biomedical studies on primates.
A long-standing question in evolutionary biology concerns the effect of inversions in shaping the genomic architecture of organisms. The most conspicuous differences between the human and chimpanzee karyotypes are nine pericentric inversions, suggesting that inversions occur quite frequently in primate chromosomal evolution (Yunis and Prakash 1982; Ventura et al. 2001, 2003, 2004, 2007, 2011; Carbone et al. 2002; Eder et al. 2003; Misceo et al. 2003, 2005; Kehrer-Sawatzki et al. 2005a,b; Cardone et al. 2006, 2007, 2008; Kehrer-Sawatzki and Cooper 2008; Stanyon et al. 2008; Capozzi et al. 2012). The main evolutionary importance of inversions comes from the fact that they suppress recombination in heterokaryotypes (Sturtevant 1917). As a consequence, inverted and noninverted segments can follow distinct evolutionary histories and accumulate variation independently, creating a genetic barrier to gene flux and contributing to speciation (Navarro et al. 1997; Farre et al. 2013). In addition to single-nucleotide variation, the two haplotypes can also harbor different segmental duplications (duplicated sequences ≥1 kb in length and showing ≥90% sequence identity) (Bailey et al. 2001). Differences in segmental duplication architecture can predispose one of the haplotypes to nonallelic homologous recombination (NAHR) leading to additional putative pathogenic rearrangements in subsequent generations (Lupski 1998). The best-known example of this effect in the human genome is the 900-kb polymorphic inversion on 17q21.31 (Stefansson et al. 2005). This locus occurs as two haplotypes, in direct and inverted orientation, not recombining over nearly 2 Mb, resulting in extended linkage disequilibrium (Skipper et al. 2004). The two haplotypes have different functional impacts: The direct haplotype is associated with neurological disorders such as Parkinson's disease (Tobin et al. 2008), while the inverted haplotype is enriched in European populations and predisposes to the 17q21.31 microdeletion in subsequent generations, as a result of NAHR between homologous segmental duplications (Koolen et al. 2008; Zody et al. 2008; Steinberg et al. 2012).
Despite their importance in human disease and genome evolution, inversions represent relatively unexplored forms of structural variation mainly due to the lack of high-throughput techniques for detecting them. Most inversions described to date between human and nonhuman primate genomes result from laborious and target-based chromosomal studies in cytogenetics that led to the identification of several large inversion variants (Ventura et al. 2001, 2003, 2004, 2007, 2011; Carbone et al. 2002; Eder et al. 2003; Misceo et al. 2003, 2005; Goidts et al. 2004; Kehrer-Sawatzki et al. 2005a,b; Cardone et al. 2006, 2007, 2008; Kehrer-Sawatzki and Cooper 2008; Stanyon et al. 2008; Capozzi et al. 2012).
A major breakthrough in the discovery of inversions came with the introduction of paired-end sequencing and mapping (Newman et al. 2005; Tuzun et al. 2005; Kidd et al. 2008). Nevertheless, a huge limitation of this method is related to the genome architecture associated with inversions. The majority of the inversions described in the human genome to date are flanked by high-identity segmental duplications (Kidd et al. 2008; Sanders et al. 2016) or inverted repeats (Vicente-Salvador et al. 2017), causing problems for inversion discovery using paired-end mapping (Alkan et al. 2011).
Exploiting new technologies for DNA sequencing, researchers have significantly improved the reference genome assemblies for a number of primates in the last decade (The Chimpanzee Sequencing and Analysis Consortium 2005; Rhesus Macaque Genome Sequencing and Analysis Consortium 2007; Locke et al. 2011; Scally et al. 2012; Gordon et al. 2016). Access to these genome sequences has increased the ability to carry out basic comparative and structural genomics analyses in these species. For instance, Feuk et al. (2005) identified 1576 submicroscopic inversions at a genome-wide level through computational comparisons of genome sequences between human and chimpanzee. However, only 27 predicted inversions were experimentally validated to distinguish real inversions from false positives. In this study, we took advantage of the recent reference genome releases and compared the net alignments for the most recent builds of human and nonhuman primate genome assemblies, including chimpanzee, gorilla, orangutan, and macaque genomes. Given the extensive karyotypic diversity compared to the other apes, gibbons were excluded from the analysis (Jauch et al. 1992; Muller et al. 2003; Capozzi et al. 2012; Carbone et al. 2014). Our study shows that comparison of independently assembled primate genomes with high-quality sequence is a good alternative to overcome some of the limitations of paired-end mapping. We identified, validated, and studied the evolutionary history of genomic inversions, and discovered regions that are misassembled in one or more reference genomes. Our study emphasizes the importance of improving the quality of primate assemblies to the current level of the human reference in order to facilitate additional comparative analyses and to fully enable the use of these organisms in biomedical research.
Results
Sequence alignments between human and primate genome assemblies
Net alignments between human (GRCh38/hg38) (Schneider et al. 2017), chimpanzee (Pan_tro 3.0/panTro5) (Kuderna et al. 2017), gorilla (gorGor4.1/gorGor4) (Scally et al. 2012), orangutan (WUGSC 2.0.2/ponAbe2) (Locke et al. 2011), and macaque (BCM Mmul_8.0.1/rheMac8) (Zimin et al. 2014) genomes were downloaded from the UCSC Genome Browser (University of California, Santa Cruz; http://genome.ucsc.edu/) and filtered for those longer than 100 kb and containing <90% of repeats or segmental duplications. After filtering, 38 alignments in inverted orientation were identified between human and chimpanzee, 28 between human and gorilla, 36 between human and orangutan, and 112 between human and macaque genomes (Supplemental Table S1). Multiple flanking alignments were manually inspected and merged into a single inversion call (Supplemental Table S2). After merging, 136 regions (156 inversion calls) were identified as potentially inverted in one or more species. These include 18 regions inverted between human and chimpanzee, 11 between human and gorilla, 13 between human and orangutan, and 78 between human and macaque. Sixteen regions were found to be inverted between human and more than one species: one in common between chimpanzee and orangutan; two in common between gorilla and orangutan; nine in common between orangutan and macaque; one in common between chimpanzee and macaque; one in common between chimpanzee, orangutan, and macaque; one in common between gorilla, orangutan, and macaque; and one in common between all four primates. These inversions range in size from 103 kb to 91 Mb and are distributed throughout all autosomes, with the highest number mapping on Chromosome 7.
Inversion maps of large known genomic inversions
All previously reported inversions larger than 5 Mb were used to draw ideograms for each chromosome in all the species analyzed (Fig. 1; Supplemental Fig. S1; Ventura et al. 2007, 2011; Cardone et al. 2008). Each colored block (synteny block) represents a region that is inverted in at least one of the species analyzed, but the order of the markers within the block is conserved in all the different species. Previous comparative studies focused on autosomal variants only, and the sex chromosomes were neglected. As a consequence, we excluded the X and Y Chromosomes from our analysis. We uploaded the inversion calls obtained from the net alignments comparison on the UCSC Genome Browser and highlighted the synteny blocks with the same color used to generate the ideograms (Fig. 1; Supplemental Fig. S1). This information was used in order to understand the relative orientation of the predicted inversions with respect to larger cytogenetic inversions.
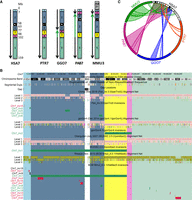
Inversions map of human Chromosome 7. (A) Ideograms for human Chromosome 7 (HSA7) and its chimpanzee (PTR7), gorilla (GGO7), orangutan (PAB7), and macaque (MMU3) homologs. Ideograms only show previously reported inversions larger than 5 Mb. Synteny blocks, distinguished by colors and numbers, represent regions that are inverted in at least one of the species analyzed, but the order of the markers within the block is conserved in all of the different species. Green arrows indicate inverted blocks with respect to human orientation. (B) UCSC Genome Browser view of Chromosome 7 net alignments and inversions predicted in this study between human and nonhuman primate genomes. Regions called to be inverted in this study are shown as green, red, and black horizontal bars and represent real, false, and not determined (ND) inversions, respectively. Synteny block colors are consistent with panel A and allow for comparison of regions called to be inverted in this study with previously identified inversions. For example, Chr7_inv1 corresponds to an inversion involving synteny blocks 2 (blue) and 3 (yellow) that was previously reported in orangutan and macaque (shown in panel A). (C) Circos diagram (Krzywinski et al. 2009) reporting all validated evolutionary inversions between human Chromosome 7 and its primate homologs.
Inversion validations
First, we compared our inversion calls to previously reported inversions (Feuk et al. 2005; Ventura et al. 2007, 2011; Cardone et al. 2008; Antonacci et al. 2009; Nuttle et al. 2016) and confirmed the inverted orientation of 39 events (Fig. 1; Supplemental Fig. S1; Supplemental Table S3). These correspond to the large known inversions shown by the green arrows in the chromosome ideograms in Figure 1A and Supplemental Figure S1 and that were also reidentified in the current study. Then, we tested 30 inversions using fluorescence in situ hybridization (FISH) in multiple human HapMap cell lines and primate species cell lines where the regions were called to be inverted (Supplemental Tables S3, S4). Owing to limitations in resolution, FISH analysis allowed us to validate only inversions larger than 500 kb. In particular, we used interphase three-color FISH for inversions between 500 kb and 2 Mb in size and metaphase two-color FISH for inversions larger than 2 Mb (Supplemental Table S5; Supplemental Fig. S2). Testing the inversion in multiple individuals allowed us to investigate if the inversion was polymorphic in the species analyzed. However, we only tested two individuals (four chromosomes) per primate species, and therefore, we were unable to define if an inversion was polymorphic for allele frequencies lower than 25%. FISH experiments showed that 16 calls were not inverted and therefore represent an orientation error in the assembly of the primate genomes analyzed, while 19 inversion calls were confirmed and were further studied in order to understand the evolutionary history of the inversion events (see “Evolutionary analyses”). One region of 846 kb on human Chromosome 1 (Chr1_inv3, GRCh38/hg38 Chr 1: 147,079,442–147,925,603), called to be inverted in chimpanzee and orangutan (calls hg38_panTro5_22 and hg38_ponAbe2_22), was confirmed as inverted by FISH in 10 human cell lines compared to the reference genome (GRCh38/hg38), suggesting that this is either an error in the human assembly or the reference genome represents the minor allele. FISH results showed that all primate individuals tested are inverted compared to human except for gorilla; therefore, this region represents a real evolutionary inversion event but assembled in the wrong orientation in chimpanzee, gorilla, and orangutan genomes (Supplemental Table S3). Four inversions were polymorphic in one or more primate individuals (Supplemental Table S4), while one inversion mapping on 10q11.22 was polymorphic just in humans with an inverted allele frequency of 37% (Supplemental Fig. S3).
Next, we investigated the BAC-end sequence (BES) pair mapping profiling of the predicted inversions by downloading the BES from all primate species analyzed and mapping them to the human reference GRCh38/hg38. BACs spanning inversions are discordant since they have end pairs that map abnormally far apart and have ends that are incorrectly oriented when mapped to the human reference genome sequence (Tuzun et al. 2005). Fifty-six of the putative inversion sites had BACs spanning at least one breakpoint. Of these, 24 showed discordant clones supporting the inversion and 31 events were false positives since only concordant clones from the primate species detected to be inverted mapped at the putative inversion breakpoints (Supplemental Tables S3, S6; Supplemental Fig. S4).
As a more direct means of validation, we selected 29 BAC clones for complete BAC-insert sequencing with Illumina sequencing as previously described (Supplemental Tables S3, S6; Supplemental Fig. S4; Steinberg et al. 2012). Sequencing was 100% consistent with our BES pair mapping profiling and FISH validation analysis (Supplemental Table S3).
In total, we investigated 126 inversion calls (109 human loci) through the literature (33/109 loci), experimental analyses (70/109 loci), and a combination of both (6/109 loci) and found that 83 calls (67/109 loci) represent real inversions. These include 14 calls between human and chimpanzee, nine between human and gorilla, 22 between human and orangutan, and 38 between human and macaque (Supplemental Table S3). Almost half of these loci (28/67 human regions) have been identified as inverted for the first time in this study. The remaining 39 events (39/126 inversion calls, 31%) were determined to be errors in the primate reference genomes, where the sequence was likely assembled in the wrong orientation (Supplemental Tables S3, S7).
Evolutionary analyses
We determined whether the 67 regions inverted in one or more primate species represent the derived or ancestral state based on comparisons with outgroup nonhuman primate species. To do so, we used previously published data (Feuk et al. 2005; Ventura et al. 2007, 2011; Cardone et al. 2008; Antonacci et al. 2009; Nuttle et al. 2016) for 33 regions, experimental analyses for 28 newly identified loci, and a combination of both for six regions for which published data was available for just a subset of primates.
We tested for the presence of 30 larger inversions by FISH analysis of cell lines from multiple individuals of chimpanzee (Pan troglodytes), gorilla (Gorilla gorilla), orangutan (Pongo pygmaeus), and macaque (Macaca mulatta). When necessary, marmoset (Callithrix jacchus) was used as an outgroup species (Supplemental Tables S3, S4).
For all these larger inversions and to resolve the status of the smaller inversions, we analyzed the BES pair mapping profiling in the different primate species analyzed and used marmoset as an outgroup when necessary. Of the 142 clones spanning the breakpoints of the validated inversions, 59 were fully sequenced with Illumina (Fig. 2; Supplemental Fig. S4; Supplemental Tables S3, S6).
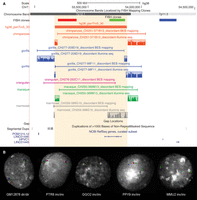
Experimental validation of Chr7_inv10 inversion. (A) UCSC Genome Browser view of Chr7_inv10 (hg38_panTro5_30), exclusively predicted in the chimpanzee lineage. BES pair mapping of primate clones and their Illumina sequencing (reads in the colored frames) show that all primates analyzed carry the inverted orientation. Discordant clones spanning the inversion breakpoints appear to be discontinuous due to the presence of the inversion. (B) The same inversion has been further validated by FISH in human (GM12878), chimpanzee (PTR8), gorilla (GGO2), orangutan (PPY9), and macaque (MMU2) individuals using the FISH clones shown in panel A.
In total, we were able to successfully determine the lineage specificity of 60 out of 67 inversions (Supplemental Table S3). Of these, three inversions occurred in the great ape ancestor, six in the human and chimpanzee ancestor, six are human-specific, seven occurred in the chimpanzee lineage, six in the gorilla lineage, three are orangutan-specific, 15 occurred in the macaque lineage, and two in the Old World monkey ancestor. The remaining 12 inversions occurred in the human–African great ape ancestor, with two (Chr1_inv3; Chr15_inv1) in direct orientation (compared to the ancestral orientation) in chimpanzee. Here, either the region flipped back to the ancestral orientation in the chimpanzee lineage or the chimpanzee configuration may represent a case of incomplete lineage sorting (Fig. 3).
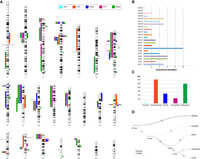
Map of primate inversions and assemblies errors. (A) All inversions discovered and validated between human, chimpanzee, gorilla, orangutan, and macaque chromosomes are shown on the left side of the chromosome ideograms. In particular, previously reported inversions reidentified in the current study are represented by colored blocks with a diagonal pattern, while novel inversions are depicted with solid color blocks. Errors in human and nonhuman primate assemblies are shown on the right side of the chromosome ideograms. (B) The horizontal bar chart shows the number of inversions per human chromosome. (C) Megabases (Mb) of assembly errors are shown for each species. (D) All inversions for which the lineage specificity has been determined are mapped on a phylogenetic tree (Sudmant et al. 2013) in which the branch thickness is proportional to the number of inversions. (HSA) Homo sapiens, (PTR) Pan troglodytes, (GGO) Gorilla gorilla, (PPY) Pongo pygmaeus, (MMU) Macaca mulatta.
Since duplications play a pivotal role in origin of inversions, we compared the extent of segmental duplication at the breakpoints of the inversions among the different primate species. Of the great ape inversions, 82% (32 of 39) map to segmentally duplicated regions of the genome, compared to the inversion events identified in macaque that overlap with segmental duplications at their breakpoints in only 41% of the cases (seven out of 17) (Supplemental Table S3). Sequence analyses of the remaining macaque inversions show that 47% (eight out of 17) contain SINEs or LINEs at their breakpoints (Supplemental Table S8). We annotated the breakpoint ranges of each real inversion, based on the different validation methods used (i.e., previous studies, FISH, BES pair mapping, Illumina sequencing of BAC clones) (Supplemental Table S9).
Of the 67 inverted regions, 36 have breakpoints overlapping with 81 human (RefSeq curated) genes (Supplemental Table S10). We performed a Gene Ontology analysis and found that these genes belong to functional groups related to drug metabolism (cytochrome P450), receptors (G protein-coupled receptors and olfactory receptors), DNA-binding proteins (ZFP14 zinc finger protein), and transport proteins.
Identification of human polymorphic inversions through comparison with previous studies
We compared our predicted inversion calls with previously identified human polymorphic inversions and found a match for seven regions. These include a 5.1-Mb inversion on Chromosome 4 and a 3.8-Mb inversion on Chromosome 8, both of which occurred in the human–chimpanzee common ancestor and were previously shown to be predisposing to further rearrangements associated with complex neurological disorders (Giglio et al. 2001, 2002; Antonacci et al. 2009; Sanders et al. 2016); a 2-Mb inversion on Chromosome 7 occurring in the great ape common ancestor, predisposing to the deletion in Williams-Beuren syndrome (Osborne et al. 2001; Schubert 2009; Sanders et al. 2016); a 735-kb inversion on Chromosome 7 occurring specifically in the human lineage (Feuk et al. 2005; Sanders et al. 2016); and two inversions of 287 kb and 1.3 Mb mapping on Chromosome 10 and Chromosome 16 (Sanders et al. 2016), respectively, that we were not able to validate. However, these last two inversions were predicted just in chimpanzee and therefore might have occurred in the human–chimpanzee ancestor (Table 1).
Inversions polymorphic in human and/or corresponding to sites of recurrent rearrangements associated with human disease
Errors in human and nonhuman primate reference genome assemblies
Our validations by FISH, BES pair mapping, and BAC clone Illumina sequencing (see “Inversion validations”) identified 39 regions that represent genome assembly errors in one or more primate species (Supplemental Tables S3, S7). Moreover, by studying the evolutionary history of the 67 validated inversions (see “Evolutionary analyses”), we were able to identify 14 regions that are in direct orientation in some primate assemblies but were actually inverted in the species analyzed (Supplemental Table S7). An example is an inversion on Chromosome 15q13.1 (Chr15_inv1 inversion) identified in chimpanzee through alignments in inverted orientation between human and chimpanzee. Further evolutionary analyses by FISH showed that this region is also inverted in orangutan and macaque, although the genome assemblies for this region are in the same orientation as in human (Supplemental Tables S3, S4).
Finally, we searched for previously reported inversions in human and nonhuman primates and identified five regions inverted in one or more species (Antonacci et al. 2010; Ventura et al. 2011) but in direct orientation in their reference genome assemblies (Supplemental Table S7). Additional misassemblies identified through FISH experiments, BES pair mapping, and Illumina sequencing of BAC clones include 13 breakpoint errors of inversion calls and three inversions made up of two or more calls interrupted by an interval of sequence in direct orientation (Supplemental Table S7; Supplemental Fig. S5).
In total, we detected 74 regions (Fig. 3), including over 300 Mb of sequence within human, great ape, and macaque reference genome assemblies involved in sequence orientation errors, overlapping with 1978 human RefSeq curated genes.
Discussion
We have generated the first genome-wide map of intermediate-scale inversion variants between human, great ape, and macaque genomes by comparing the net alignments for the most recent builds of these primate genome assemblies. We initially identified 136 human regions potentially inverted between human and either chimpanzee, gorilla, orangutan, or macaque genomes. Combining the literature, FISH analyses, BES pair mapping, and complete sequencing of primate clones for 120 inversion calls showed that 83 (69%: corresponding to 67 human loci) are real events. Each verified inversion was additionally tested in multiple chimpanzee, gorilla, orangutan, and macaque individuals, allowing us to determine the ancestral state of each event and to also verify the inversion status in species where the assembly did not show the presence of the inversion. This allowed us to extend the number of validated primate inversions from 83 to 105 (Fig. 3). With our experimentally verified inversions, we more than doubled the number of events known between human, great ape, and macaque genomes. We identified 28 new regions between 103 kb and 5 Mb that are inverted in one or more primate species (Table 1; Fig. 3) and determined the ancestral orientation for 23 of these loci (Table 1; Supplemental Table S3).
In total, our inversions range in size from 103 kb to 91 Mb and are randomly distributed, with the highest number of events on Chromosome 7 and no inversions found on Chromosomes 21 and 22 (Fig. 3). We successfully resolved the evolutionary history of 60 inversions and found that macaque has the highest number of lineage-specific inversions (n = 17), followed by chimpanzee (n = 7), human (n = 6), gorilla (n = 6), and orangutan (n = 3). A high number (n = 12) of inversions seems to have occurred in the African great ape ancestor. The remaining inversions occurred in the human–chimpanzee ancestor (n = 6) and in the great ape ancestor (n = 3). Our sequence analyses suggest that NAHR, mediated by segmental duplications, is the predominant mechanism underlying these events (Supplemental Table S3) in humans and great apes (82%). Only 41% of macaque inversion breakpoints contain segmental duplications, while LINEs and SINEs were found at the remaining breakpoints (47%) (Supplemental Tables S3, S8). This is expected, since most (80%) high-identity segmental duplications arose after the divergence of the Old World and hominoid lineages (Marques-Bonet et al. 2009).
We identified a very low number of inversion calls generated by the comparison of the net alignments of human (GRCh38/hg38) and gorilla (gorGor4.1/gorGor4) genomes. Experimental validations show that only previously reported large events are real (Ventura et al. 2011). The remaining smaller inversions are false calls or have not been verified because they are too small for FISH validation and BES mapping profiling results are unclear (Supplemental Table S3). The published gorilla genome assembly (gorGor4.1/gorGor4) used in this study was generated by a mix of capillary sequence and whole-genome shotgun Illumina sequencing, resulting in an assembly containing more than 400,000 gaps (Scally et al. 2012). The human genome was used to help guide the gorilla assembly, therefore generating an artificially low number of inversions. Unfortunately, the absence of the chromosome information for the latest gorilla assembly, GSMRT3/gorGor5, made it impossible to use it for our analysis.
In searching for genes that could be altered by the presence of the inversions, we found that 36 out of 67 loci are inverted in one or more primates and have breakpoints overlapping 81 human genes (Supplemental Table S10), prioritizing them as candidates for biological and evolutionary studies. These genes include members of several functional groups, including those related to drug metabolism, receptors, DNA-binding proteins, and transport proteins. Future experimental studies are required to demonstrate the functional significance of these genes in contributing to phenotypic differences among humans, great apes, and macaque.
Many previously identified inversion variants have been linked to susceptibility to further genomic rearrangements in offspring. We therefore investigated all inversion regions under 10 Mb in size in which humans carry the inverted orientation compared to the ancestral state and found seven inversions corresponding to sites of recurrent rearrangements associated with human disease, with four being polymorphic in humans (Table 1; Giglio et al. 2001, 2002; Osborne et al. 2001; Feuk et al. 2005; Antonacci et al. 2009; Schubert 2009; Cooper et al. 2011; Sanders et al. 2016).
This included four previously described events: a 5.1-Mb inversion on 4p16.3-p16.1 (Chr4_inv5) (Supplemental Fig. S3) and a 3.9-Mb inversion on 8p23.1 (Chr8_inv2), both known to be polymorphic in the human population and predisposing to recurrent t(4;8)(p16;p23) translocations and inv dup(8p) rearrangements in the offspring (Giglio et al. 2001, 2002; Antonacci et al. 2009; Sanders et al. 2016); a 2-Mb polymorphic inversion on 7q11.23 (Chr7_inv8) predisposing to Williams-Beuren syndrome (Osborne et al. 2001; Schubert 2009; Sanders et al. 2016); and a 569-kb inversion (Chr16_inv8) mapping to the 16p11.2 disease-associated region (Cooper et al. 2011; Nuttle et al. 2016). We also detected three novel smaller inversions, including a 1.5-Mb inversion on 15q13.1-q13.2 (Chr15_inv1), a 846-kb inversion on 1q21.1-q21.2 (Chr1_inv3), and a 580-kb inversion on 10q11.22 (Chr10_inv6) (Table 2; Supplemental Fig. S3). These three regions correspond to sites of recurrent deletions and duplications associated with intellectual disability and developmental delay (Cooper et al. 2011), providing further support for a link between human inversions and genomic disorders. The 10q11.22 region was recently resequenced and assembled using single-molecule, real-time (SMRT) sequencing of BAC clones (Chaisson et al. 2015) since the previous build of the human reference genome (GRCh37/hg19) contained seven gaps. The region is particularly enriched in segmental duplications in all the primate genomes analyzed (Marques-Bonet et al. 2009; Sudmant et al. 2013) and is polymorphic in human with a frequency of the minor allele in inverted orientation of 37%. All the other nonhuman primates tested are in opposite orientation compared to most humans, suggesting that the inversion occurred in the human lineage and is still polymorphic (Supplemental Fig. S3; Supplemental Table S4). It is possible that the inverted haplotype carries a genomic architecture that now predisposes this region to deletions and duplications associated with neuropsychiatric disease.
Detected and validated novel inversion events
Notably, FISH results for the 1q21.1-q21.1 region (Chr1_ inv3), inverted in the chimpanzee and orangutan genome references (calls hg38_panTro5_22 and hg38_ponAbe2_22), showed that 20 of 20 human chromosomes tested were inverted relative to GRCh38/hg38, suggesting a potential error in the orientation of the reference genome assembly involving 10 genes (Supplemental Table S3). Alternatively, the human reference at this complex region of the genome may represent a rare haplotype. Therefore, this disease-associated region of 846 kb represents a real evolutionary inversion event that is misassembled in chimpanzee, gorilla, and orangutan reference genomes.
Of note, our analysis yielded a total of 74 regions that are not properly assembled in the published versions of one or more primate genomes, containing over 300 Mb of DNA and 1978 human (RefSeq curated) genes. Using BES pair mapping analyses and Illumina sequencing of BAC inserts, we identified species-specific clones for future further characterization through long-read single-molecule sequencing (Chaisson et al. 2015) in order to refine the sequence and orientation at these complex genomic regions. Further efforts should be devoted toward producing primate genome sequences with greater accuracy and completeness. This is critical to detect subtle differences in genes or their regulatory regions that could lead to hominid physiological and behavioral differences and, importantly, to know that such differences are due to biology rather than poor sequence quality. In addition, many primate species are significant biomedical research models, and high-quality sequencing of primate genomes is needed to provide comparative sequence information that has implications for the understanding of the genetic basis for human disease.
Methods
Sequence alignments between human and primate genome assemblies
Tables of net alignments between a human (GRCh38/hg38) and a primate genome, i.e., chimpanzee (Pan_tro 3.0/panTro5), gorilla (gorGor4.1/gorGor4), orangutan (WUGSC 2.0.2/ponAbe2), and macaque (BCM Mmul_8.0.1/rheMac8), were downloaded from the University of California, Santa Cruz “goldenPath” website (http://hgdownload-test.cse.ucsc.edu/goldenPath/). For every primate genome, the net-alignment table shows the best human/primate genome chain alignment, with a gap scoring system allowing longer gaps than traditional affine gap scoring systems. This analysis is useful for finding orthologous regions and for studying genomic rearrangements (Chiaromonte et al. 2002; Schwartz et al. 2003). All alignments labeled as inversions were filtered for size (longer than 100 kb) and percentage of repeats or segmental duplications (<90% of the inversion size). To reduce the number of false positives, we filtered out the nested inversions, considering only alignments better than level four of the net-alignment output, as previously described by Feuk et al. (2005). We repeated the analyses using the complementary net alignments (primate species versus human genome) and manually inspected inversion calls that were missed in the first analysis. This step allowed us to add three additional calls (rheMac8_hg38_98; gorGor4_hg38_5; panTro5_hg38_31). Alignments on sex chromosomes were excluded from the analysis.
Inversion maps of large known genomic inversions
All previously reported inversions larger than 5 Mb were used to draw ideograms for each chromosome in all the species analyzed (Fig. 1; Supplemental Fig. S1; Ventura et al. 2007, 2011; Cardone et al. 2008). Each colored block represents a synteny block—a region previously described as inverted in at least one of the species analyzed—but the order of the markers within the block is conserved in all the different species.
BES pair mapping
During the sequencing of the chimpanzee, gorilla, orangutan, and macaque genomes, BAC libraries (CHORI-251, CHORI-277, CHORI-276, CHORI-250, and CHORI-259) were constructed from the same individuals used to generate the sequence assemblies. We obtained the sequence for all traces from the NIH trace repository (http://www.ncbi.nlm.nih.gov/Traces/trace.cgi?). We aligned BES against the human reference genome sequence (GRCh38/hg38) as part of a three-step process (recruitment, quality rescoring, and pairing) optimized and published by Tuzun et al. (2005). Since the original protocol was applied using fosmid paired ends, we adapted it based on longer expected BAC insert sizes (range: 160–180 kb). BACs spanning inversions are discordant since they have end pairs that map abnormally far apart and have ends that are incorrectly oriented when mapped to the human reference genome sequence. Clones spanning regions in the same orientation as in human are concordant in size and for orientation of the ends.
FISH analysis
Metaphase spreads and interphase nuclei were obtained from lymphoblast and fibroblast cell lines from 10 human HapMap individuals (Coriell Cell Repository), four chimpanzees, three gorillas, two orangutans, two macaques, and one marmoset (Supplemental Table S4). FISH experiments were performed using human fosmid (n = 46) or BAC (n = 41) clones (Supplemental Table S5; Supplemental Fig. S2) directly labeled by nick-translation with Cy3-dUTP (PerkinElmer), Cy5-dUTP (PerkinElmer), and fluorescein-dUTP (Enzo) as described by Lichter et al. (1990), with minor modifications. Briefly, 300 ng of labeled probe were used for the FISH experiments; hybridization was performed at 37°C in 2×SSC, 50% (v/v) formamide, 10% (w/v) dextran sulphate, and 3 mg sonicated salmon sperm DNA, in a volume of 10 mL. Post-hybridization washing was at 60°C in 0.1×SSC (three times, high stringency, for hybridizations on human, chimpanzee, gorilla, and orangutan), or at 37°C in 2×SSC and 42°C in 2×SSC, 50% formamide (three times each, low stringency, for hybridizations on macaque and marmoset). Nuclei were simultaneously DAPI-stained. Digital images were obtained using a Leica DMRXA2 epifluorescence microscope equipped with a cooled CCD camera (Princeton Instruments). DAPI, Cy3, Cy5, and fluorescein fluorescence signals, detected with specific filters, were recorded separately as grayscale images. Pseudocoloring and merging of images were performed using Adobe Photoshop software. Interphase three-color FISH was used to validate inversions between 500 kb and 2 Mb in size, and metaphase two-color FISH was used for inversions larger than 2 Mb (Supplemental Table S5). In the case of interphase three-color FISH, each region was interrogated using two probes within the putative inversion region and a reference probe outside. A change in the order of the probes mapping within the inversion was indicative of the presence of the inversion. Examining inversions in interphase, distance, position, and order between FISH dots of different colors has to be measured in order to reveal their spatial pattern. Therefore, a minimum of 50 interphase cells were scored for each region in order to determine if the pattern observed was casual or due to a real inversion. For this reason, only patterns where the probes were positioned on a straight line and whose distance was consistent with their mapping position were considered. Probes overlapping or whose pattern resembled a triangle were excluded. A region was considered homozygously inverted if scoring of the probes in inverted orientation exceeded 80% of the total count and heterozygously inverted if probes in direct and inverted orientation were equally scored. A minimum of 10 cells were scored instead for FISH on metaphase chromosomes. In this case, the orientation of the region was more easily assessed by simply visualizing a switch in the order of the probes mapping within the inversion.
Illumina sequencing of BAC clones
DNA from 11 CH251, nine CH277, eight CH276, 26 CH250, and five CH259 BAC clones (Supplemental Table S6) was isolated, prepped into genomic libraries, and sequenced (PE250) on an Illumina MiSeq using a Nextera protocol (Antonacci et al. 2010). DNA from 37 clones was barcoded before library preparation, while DNA from 22 clones mapping to different chromosomes and free of segmental duplications was pooled two at a time before library preparation, and then barcoded and sequenced. Sequencing data were mapped with mrsFAST (Alkan et al. 2009) to the human reference genome and singly unique nucleotide (SUN) identifiers were used to discriminate between highly identical segmental duplications (Sudmant et al. 2010).
Gene Ontology analysis
Genes at the inversions breakpoints were analyzed using InterPro (http://www.ebi.ac.uk/interpro/), which is a database that integrates diverse information about protein families, domains, and functional sites and makes it freely available to the public via web-based interfaces and services (Hunter et al. 2012). Gene Ontology codes were extracted from the InterPro output and manually clusterized.
Data access
Raw sequencing data from Illumina sequencing experiments from this study have been submitted to the Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra/) under BioProject PRJNA429373.
Acknowledgments
We thank T. Brown for the critical review of the manuscript. This work was supported by the “Fondazione Cassa di Risparmio di Puglia” grant to F.A. and, in part, by a US National Institutes of Health (NIH) grant HG002385 to E.E.E. C.R.C. is supported by the “Fondo di Sviluppo e Coesione 2007–2013 – APQ Ricerca Regione Puglia Programma regionale a sostegno della specializzazione intelligente e della sostenibilità sociale ed ambientale – Future In Research” grant. E.E.E. is an investigator of the Howard Hughes Medical Institute.
Author contributions: F.A., C.R.C., and F.A.M.M. designed the study. C.R.C., F.A.M.M., M.B., O.C., M.L.S., and M.M. performed FISH experiments. C.R.C. and F.A.M.M. performed library construction for Illumina sequencing. P.D. performed sequencing data analysis. N.A., M.V., and E.E.E. contributed to data collection. F.A., C.R.C., and F.A.M.M. contributed to data interpretation. F.A., C.R.C., and F.A.M.M. wrote the manuscript. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.234831.118.
- Received January 19, 2018.
- Accepted April 26, 2018.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








