
Deep experimental profiling of microRNA diversity, deployment, and evolution across the Drosophila genus
- Jaaved Mohammed1,2,3,4,6,
- Alex S. Flynt3,5,6,
- Alexandra M. Panzarino3,
- Md Mosharrof Hossain Mondal5,
- Matthew DeCruz5,
- Adam Siepel4 and
- Eric C. Lai2,3
- 1Department of Biological Statistics and Computational Biology, Cornell University, Ithaca, New York 14853, USA;
- 2Tri-Institutional Training Program in Computational Biology and Medicine, New York, New York 10021, USA;
- 3Department of Developmental Biology, Sloan-Kettering Institute, New York, New York 10065, USA;
- 4Simons Center for Quantitative Biology, Cold Spring Harbor Laboratory, Cold Spring Harbor, New York 11724, USA;
- 5Department of Biological Sciences, University of Southern Mississippi, Hattiesburg, Mississippi 39406, USA
-
↵6 These authors contributed equally to this work.
Abstract
To assess miRNA evolution across the Drosophila genus, we analyzed several billion small RNA reads across 12 fruit fly species. These data permit comprehensive curation of species- and clade-specific variation in miRNA identity, abundance, and processing. Among well-conserved miRNAs, we observed unexpected cases of clade-specific variation in 5′ end precision, occasional antisense loci, and putatively noncanonical loci. We also used strict criteria to identify a large set (649) of novel, evolutionarily restricted miRNAs. Within the bulk collection of species-restricted miRNAs, two notable subpopulations are splicing-derived mirtrons and testes-restricted, recently evolved, clustered (TRC) canonical miRNAs. We quantified miRNA birth and death using our annotation and a phylogenetic model for estimating rates of miRNA turnover. We observed striking differences in birth and death rates across miRNA classes defined by biogenesis pathway, genomic clustering, and tissue restriction, and even identified flux heterogeneity among Drosophila clades. In particular, distinct molecular rationales underlie the distinct evolutionary behavior of different miRNA classes. Mirtrons are associated with high rates of 3′ untemplated addition, a mechanism that impedes their biogenesis, whereas TRC miRNAs appear to evolve under positive selection. Altogether, these data reveal miRNA diversity among Drosophila species and principles underlying their emergence and evolution.
MicroRNAs (miRNAs) are an extensive class of ∼22-nt RNAs that play important regulatory roles in diverse eukaryotic species (Flynt and Lai 2008; Axtell et al. 2011). In the canonical metazoan pathway, primary miRNA (pri-miRNA) transcripts are first cleaved by the nuclear RNase III enzyme Drosha to yield pre-miRNA hairpins. Upon export to the cytoplasm, these are further cleaved into miRNA duplexes by another RNase III enzyme, Dicer. One duplex strand is preferentially retained in an Argonaute (AGO) complex and guides it to complementary mRNA targets, whereas its partner miRNA* (star) strand is preferentially degraded. Beyond the canonical pathway, diverse noncanonical biogenesis pathways involving RNases that function in other processes have been uncovered (Yang and Lai 2011). Chief among these is the “mirtron” pathway, in which Drosha cleavage is substituted by the spliceosome, to define either or both pre-miRNA hairpin termini (Okamura et al. 2007; Ruby et al. 2007; Flynt et al. 2010).
Comparative genomics has provided key insights into miRNA functionality. For example, there is distinctive constraint on the miRNA “seed” sequence (positions 2–8 of the miRNA strand that predominantly mediate target recognition) and overall higher constraint on mature miRNA and star sequences relative to other partitions of pre-miRNA hairpins (Lai et al. 2003; Lim et al. 2003; Mohammed et al. 2013). Population data reveal miRNA evolution on a more recent timescale. For example, D. melanogaster polymorphism data uncover adaptive evolution of miRNAs within clusters with testes-restricted expression (Lu et al. 2008a; Lyu et al. 2014; Mohammed et al. 2014a). More generally, high-throughput sequencing has permitted diverse surveys of miRNA content across taxa, and it has been suggested that miRNA expansion may correlate with organismal complexity and body-plan innovation (Grimson et al. 2008; Christodoulou et al. 2010). MiRNA catalogs have been compared across broad phylogenetic distances, but mostly at the level of presence/absence of miRNA loci (Grimson et al. 2008; Christodoulou et al. 2010; Mohammed et al. 2014b). Much remains to be explored about miRNA evolutionary features across sets of related species, such as patterns and rates of gene emergence, decay and expansion, and consistency in processing across orthologs. Some relevant studies include analyses of four Caenorhabditis species (de Wit et al. 2009; Shi et al. 2013), up to six mammalian species (Berezikov et al. 2006; Meunier et al. 2013), and three Drosophila species (Lu et al. 2008b; Berezikov et al. 2010). A net gain rate of 12 miRNA genes/million years (Myr) was first estimated in Drosophila (Lu et al. 2008b). However, this estimate was later revised to 0.82–1.6 genes/Myr using a refined collection of miRNA loci (Berezikov et al. 2010), which proved relatively concordant with a subsequent estimate of 0.83 genes/Myr in mammals (Meunier et al. 2013). Since these studies, the annotation of Drosophila and mammalian miRNAs has expanded by many folds (Kozomara and Griffiths-Jones 2014). Nevertheless, despite thousands of miRNAs collected within the miRBase repository for these species, numbers of pan-mammalian (94) (Meunier et al. 2013) or pan-Drosophilid (123) miRNAs (Mohammed et al. 2013) have not changed much over the past decade. Thus, there is perhaps an order of magnitude more recently evolved miRNAs than well-conserved loci in some metazoans.
Most previous studies of miRNA evolutionary flux considered them as a unitary class. However, our recent studies show that miRNA subclasses exhibit distinct evolutionary parameters (Mohammed et al. 2013, 2014a,b). For example, mirtrons evolve more quickly than canonical miRNAs in both Drosophila (Berezikov et al. 2010) and in mammals (Wen et al. 2015), and studies in D. melanogaster identified a uridyltransferase with specificity for mirtrons (Bortolamiol-Becet et al. 2015; Reimão-Pinto et al. 2015). We speculate there should be diverse mechanisms that drive characteristic evolutionary behaviors of various miRNA classes, and a foundation to study these would be a deep empirical analysis of species-specific miRNAs across a phylogeny.
In this study, we characterized class-specific properties of miRNAs across 12 species of the Drosophila genus, which diverged from the common Dipteran ancestor ∼60 Myr. Building on previous deep analysis of D. melanogaster miRNAs (for review, see Berezikov et al. 2011; Wen et al. 2014), we sequenced an additional ∼1.5 billion sRNAs from embryos, heads, male bodies, and female bodies of the other 11 species. Using these comprehensive data, we elaborate numerous features of miRNA annotation and evolution, and show how these differ with respect to miRNA biogenesis types, tissues within an animal, and between different branches of the fruit fly phylogeny.
Results
Compendia of sRNA data across 12 Drosophila species
We previously annotated D. melanogaster miRNAs from approximately 1.9 billion small RNA reads, spanning more than 100 different developmental stages, tissue types, cell lines, and genetic and environmental manipulations (Berezikov et al. 2011; Wen et al. 2014). Although this scale is not currently practical to achieve across all other sequenced Drosophilids, we sought parameters of data collection that would permit deep annotations in other species.
Our prior experience indicated that mixed embryos, adult heads, male bodies, and female bodies are efficacious for broad capture of miRNA diversity. To test this, we performed recovery analyses of D. melanogaster miRNAs by subsampling data from these four tissue types (Methods). At an aggregate depth of 100 million reads, we recovered 94%–98% of conserved (128) miRNAs with at least 30 mature miRNA reads and three miRNA* reads from 100 simulation experiments (Fig. 1A). Of the 135 miRNAs that emerged recently in the melanogaster group, we recovered 21%–27% of miRNAs using these miRNA/miRNA* thresholds. Because increasing depths provided minor returns (Fig. 1B; Supplemental Fig. S1), we considered 100 million reads from these tissue types as a strong empirical foundation to assess miRNA evolution across the Drosophilid phylogeny.
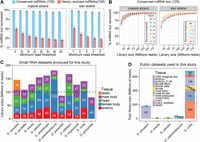
Summary of Drosophila species small RNA sequencing data and analysis of sequencing depth sufficiency. (A) The recovery rate of known D. melanogaster miRNAs using sets of 100 million total reads sampled randomly from D. melanogaster head, mixed embryo, male-body, and female-body public libraries. These libraries mimic those we sought to create for the 11 other Drosophila genomes. Bars represent the fraction of conserved or newly evolved D. melanogaster miRNAs recovered at various miRNA and miRNA* minimum read thresholds, and error bars represent the standard error of the recovery rate across 100 independent samples of 100 million reads. (B) Saturation curve of miRNA (mature and star) strand recovery at varying minimum read depth cutoffs. Based upon these results, we sought to acquire approximately 100 million reads per species. (C) Summary statistics of the actual Drosophila species small RNA libraries across tissues generated for this study. (D) Summary of publicly available Drosophila species small RNA libraries utilized in this study.
We sequenced 52 small RNA libraries (about 1.5 billion total reads) from these four tissue types across 11 species (some samples were resequenced), exceeding 100 million reads for nearly all species (Fig. 1C). Read lengths of most libraries peaked at 21–22 nt, representing miRNAs, and most body libraries showed an additional 24–28 nt peak representing piRNAs (Supplemental Table S1; Supplemental Fig. S2). These data broadly extend the limited collection of publicly available sRNA data from other fly species, primarily D. simulans (about 200 million reads) and D. virilis (about 700 million reads), which we aggregated with our libraries (Fig. 1D; Supplemental Table S2).
A genus-wide catalog of Drosophila miRNA annotations
Several approaches to annotate miRNAs have been developed, which collectively have distinct merits. However, no single strategy suffices to discover the full range of confident miRNAs, especially ones with atypical structures and/or noncanonical biogenesis. Therefore, we deployed a multipronged framework including (1) miRDeep2 (to cast a wide net of candidate hairpins with evidence of cloned small RNA duplexes); (2) an independent set of predicted hairpin structures (useful for identifying miRNAs from extended hairpins disallowed by miRDeep2); (3) intron annotations (to identify mirtrons and tailed mirtrons, which are systematically overlooked by canonical miRNA finders such as miRDeep2); and (4) whole-genome alignments to identify putative miRNA orthologs across multiple Drosophila species (to “rescue” miRNA loci from the candidate pool that have confidently cloned orthologs). Since all initial computational scans include substantial false positives, we subsequently utilized stringent criteria (e.g., abundance and patterns of sRNA read pileups indicative of RNase III cleavage) and systematic visual inspection of all loci before assigning final annotations (Supplemental Fig. S3).
We first queried miRBase (v21) loci for Drosophilid orthologs whose cloned small RNAs had not previously been explicitly identified. This served as an initial check on the overall quality of the data sets, since genomic conservation of a miRNA is usually taken to imply processing into mature small RNAs. Our data support the first cloning evidence for 592 unannotated homologs of conserved Drosophilid miRNA loci (Fig. 2A). Of these loci, 512 were cloned at thresholds of at least 30 miR reads and three miR* reads, which would have supported high-confidence de novo annotation. The remainder were cloned at lower thresholds, and would initially have been segregated as “candidates,” but could be recovered based on their orthology to loci well-cloned in other species (80 candidate-rescued, and 42 candidate miRNAs). This supported a rationale to “rescue” certain candidate miRNA loci that fall below high stringency thresholds, but could be reasonably considered as genuine based on sequence homology to a cloned miRNA locus in another species. On the other hand, our deep data sets supported our decision to demote 47 annotations from Drosophilid miRBase loci. For example, we previously demoted miR-280/287/288/289 due to lack of read support in deep D. melanogaster data (Berezikov et al. 2011), and we do not see experimental support for these loci in other species. As these loci are conserved across Drosophilid genomes, they may have other regulatory functions. Most of the other downgraded miRNA annotations are from D. pseudoobscura (Supplemental Fig. S4; Supplemental Table S3). Our reassessment of these miRBase loci emphasizes the rigor of our miRNA scoring criteria.
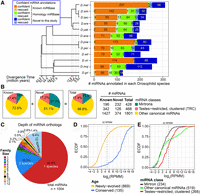
Summary of all known and novel miRNAs recovered within 12 Drosophila genomes. (A) Counts of known and novel miRNAs recovered or identified, respectively, at our two highest confidence classes—“confident” and “candidate-rescued.” MiRNAs from a third confidence class—“candidate” miRNAs—are shown in Supplemental Figure S5. (B) The proportion of miRNAs recovered within three classes defined by biogenesis pathway, and testes-restricted, clustered status. Pie charts are provided for all novel or known annotations and for the merged collection. (C) The distribution of alignment sizes upon assignment of all miRNAs into 1004 alignments. Paralogous miRNAs were assigned to single-species alignment. The majority of miRNAs identified are singletons (species-specific) or doubletons (clade-specific). (D,E) Empirical cumulative distribution function (ECDF) of alignment expression. Alignments are segregated based on age (D) and miRNA class (E). Empirical CDFs are plotted using the maximum expression values computed across all constitutive members of each alignment. (RPMM) Reads per Million mapped miRNA reads.
For the remainder of our analysis, we grouped newly cloned homologs of miRBase loci with extant miRBase miRNAs, so as to distinguish the collection of truly novel miRNAs lacking homology to previously annotated loci. In particular, our data support the annotation of 649 novel, confident miRNAs across 12 Drosophilid species (Fig. 2A). Consistent with the phylogeny, many of the highest expressed novel miRNAs are from the virilis subclade, which is most distant from D. melanogaster. The example of Dvir_264 was cloned at more than 100,000 reads (Fig. 3A). At this depth, we observe not only precision of 5p and 3p reads, but also recover loop reads, which provide evidence of a dominant diced product and indicate 3′ trimming of the predominantly cloned 20-nt species (Fig. 3A). However, perhaps unexpected was that even in species relatively close to D. melanogaster, we still recovered scores of novel confident miRNAs, although we sampled their small RNAs at less than one-tenth the read depth and tissue/cell diversity assayed in D. melanogaster. For example, Dere_50 illustrates a melanogaster subgroup miRNA expressed only in D. erecta and recovered at more than 1000 reads, but absent from D. melanogaster (Fig. 3A). Such observations provide indications of evolutionary flux that we explore later in this study.

Examples of novel miRNAs identified in this study. (A) Dvir_264 and Dere_50 are examples of confident, canonical miRNAs. Small RNAs were cloned from both arms of their pre-miRNA, and an alignment of these sequences reveal patterns of precise 5′ end cleavage and 1- to 2-nt 3′ blunt end overhang in the hairpin structure. These are all signatures of Drosha- and Dicer-mediated cleavage. (B) Examples of orthologous confident (Dsim_14614) and candidate-rescued (Dsec_989) mirtrons within the two sister species, D. simulans and D. sechellia. Total cloned reads for Dsec_989 fell below our threshold for “confident” classification, and this mirtron was effectively placed into the “candidate” confidence group. We “rescued” this locus due to its synteny with Dsim_14614, a confident mirtron. (C) Two novel, conserved, canonical miRNAs identified within the host gene cwo. Dmel_164 sequence alignment indicates higher conservation of the miR and miR* arms in comparison to the loop and flanking basal stem regions, a classic signature of pre-miRNA sequence conservation. However, it possesses conserved A-rich regions that flank the duplex, which are seemingly incompatible with a lower stem needed for Drosha processing. Dmel_373 exhibits a large terminal loop atypical of most canonical miRNAs. The full alignments of Dmel_373 loops across Drosophila species can be accessed in the Supplemental Material. (RPM) Reads per Million mapped reads.
Figure 3B illustrates Dsim_14614 as a novel splicing-derived mirtron; note that nearly all Dsim_14614-3p reads are uridylated (Fig. 3B). Our comparative sRNA data allowed us to recover “candidate-rescued” miRNAs that are orthologous to confident ones; i.e., that we consider as genuine miRNA products. For example, Dsec_989 was initially classified as a candidate mirtron due to the lack of miR* reads (Fig. 3B); however, its synteny to Dsim_ 14614 allowed us to elevate the confidence of this mirtron. From an initial collection of 765 “candidate” loci bearing small RNA evidence reminiscent of miRNAs, we reclassified 82 as candidate-rescued miRNAs or mirtrons that were clearly orthologous to confident loci (Fig. 2A; Supplemental Fig. S5).
The remaining 683 “candidate” loci have small RNA evidence reminiscent of miRNAs, but do not meet minimum criteria (Supplemental Fig. S5). We set them aside for now and do not presently consider these genuine miRNAs, although the veracity of some may emerge from deeper or specialized sequencing. Nevertheless, our comprehensive small RNA data allows us to expand the collection of 1965 known and unannotated miRBase loci of confident and rescued status with 732 novel miRNAs and mirtrons to arrive at a final collection of 2697 total miRNAs and mirtrons present within the Drosophila genus. These annotations can be explored in Supplemental Table S4 and the supplemental website, which provides extensive information regarding read pileups, secondary structures, aligned sequences in other Drosophilid species, and so forth (http://compgen.cshl.edu/mirna/12flies/12flies_alignments.html; Supplemental Material).
miRNA classes, alignments, and expression
We segregated our annotations into loci of distinct biogenesis and genomic clustering classes. We separated mirtrons from canonical miRNAs and used expression profiles to define Testes-restricted, Recently evolved, Clustered canonical miRNAs (TRC miRNAs) (Mohammed et al. 2014a). More than half (374 loci; 51.1%) of the novel miRNAs identified in our study and 72.6% (1427) of known and unannotated miRBase loci were solo (i.e., nonclustered) canonical miRNAs (Fig. 2B). On the other hand, TRC miRNAs comprised 31.7% (126) of our novel collection, and mirtrons accounted for 17.2% (232). Therefore, specific pools of hairpins, namely noncanonical and testes-restricted loci, contribute disproportionately to the aggregate catalog of miRNA substrates (Supplemental Fig. S6).
We grouped miRNA orthologs into alignments by building upon previous, manual alignments for D. melanogaster miRNAs (Mohammed et al. 2013) and assigning new miRNAs into groups via genome-wide homology identification and multispecies whole-genome alignments (Methods). Altogether, we grouped 2697 miRNAs into 1004 miRNA alignments. Species-specific miRNAs (comprising the majority of loci newly annotated in this study) comprise the dominant class (65.4%), and miRNAs with one cloned ortholog comprise the next largest class (15.2%). On the other end, 115 alignments (11.4% of loci) contained 10 or more members, and thus were present at the base of the Drosophilid phylogeny (Fig. 2C).
We next assessed the range and variation of miRNA expression. We computed the log2 (reads per million mapped miRNA reads [RPMM]) score for all loci and recorded the maximum expression level per miRNA in any individual library. We evaluated this metric, instead of the “average” expression of miRNAs, to account for the tissue-specific deployment of many miRNAs. We find that 92.3% of conserved miRNA loci had a maximum expression greater than 32 RPMM [i.e., log2(5)], whereas 24.1% of newly evolved miRNAs achieved this level (Fig. 2D). At a lower threshold, 80.2% of novel miRNAs reached more than 1 RPMM in at least one library. When comparing miRNA classes, we observed that TRC miRNA alignments outperformed other classes at the higher (greater than 32 RPMM) cutoff (e.g., 45% for TRC versus 19.7% for mirtrons and 34.1% for other canonical miRNAs) (Fig. 2E).
Novel, deeply conserved miRNAs
Catalogs of well-conserved miRNAs are considered largely complete, as it is generally believed the set of “clonable” hairpins with miRNA-like evolutionary signatures were exhausted years ago. However, some conserved miRNAs continue to be found, many of which derive from unusual genomic locations or noncanonical pathways, perhaps explaining why they were overlooked earlier. For example, we recently reported that deeply conserved, noncanonical, dme-mir-10404 is processed from the internal spacer regions of highly repetitive rRNA loci (Chak et al. 2015). Indeed, we find this miRNA is well-cloned from across the Drosophilid phylogeny (Supplemental Fig. S7).
Among our novel miRNA annotations, a handful of loci appeared to be cloned from a broad range of Drosophilid species (Supplemental Fig. S8A). The behavior of pasha 5′ UTR hairpins was instructive. A feedback loop in which Drosha cleaves 5′ UTR foldbacks in pasha/DGCR8 is conserved from mammals to fruit flies (Han et al. 2009; Smibert et al. 2011). Although mammalian DGCR8 5′ UTR hairpin products are nuclearly retained (Han et al. 2009), sufficient mature reads of DGCR8 hairpins exist to justify annotation of mir-3618 and mir-1306. We identified small RNA duplexes from the corresponding species ranges for both the deeply conserved (Dmel_422) and melanogaster group–restricted (Dmel_474) pasha 5′ UTR hairpins (Supplemental Fig. S8A). Although their resultant small RNAs are not very abundant, the coupling with our prior evidence of in vivo cleavage of these hairpins by Drosha (Smibert et al. 2011) indicates the depth of our small RNA profiling.
Among novel conserved miRNAs, a notable pair is Dmel_373 and Dmel_164, which are clustered in the first intron of clockwork orange (cwo). Both loci are highly conserved, exhibit greater loop divergence relative to the hairpin arms that is diagnostic of conserved miRNAs, and are processed into small RNA duplexes across the Drosophilids (Fig. 3C). However, both loci exhibit atypical features: Dmel_164 harbors a conserved, A-rich lower stem that likely precludes Drosha access, whereas Dmel_373 contains an unusually large (∼50 nt) loop (mean loop size of 209 D. melanogaster miRNAs is 22 nt). Although corresponding reads were detected throughout the Drosophilid phylogeny, their accumulation was modest (Supplemental Fig. S9), and efforts to detect them by Northern blotting were negative (Supplemental Fig. S8B). Thus, although the deep conservation of these hairpins implies functional utility, it remains to be seen whether cwo miRNAs are matured via noncanonical mechanisms, or if they serve another regulatory role but happen to be sampled in deep small RNA sequencing. Additional examples of conserved miRNAs are shown in Supplemental Figure S8A with read details in Supplemental Figure S9.
Evolutionary shifted processing of some conserved miRNA loci
It is generally assumed that genomic conservation of miRNA loci goes hand-in-hand with conserved processing of mature small RNAs, which in turn are locked into conserved regulatory networks. Since even a 1-nt shift in miRNA 5′ identity can redirect its target network, conserved miRNAs are inferred to maintain precise processing. However, in the absence of systematic small RNA sequencing analysis across a genus, the tenets of this assumption have not systematically been challenged by empirical data.
We investigated the consistency in 5′ end processing for 129 well-conserved Drosophilid miRNAs using our comparative data (Fig. 4). As expected, the strong majority of conserved miRNAs exhibit 5′ identities (Supplemental Fig. S10). Of note, some miRNA loci are documented to generate substantial iso-miR species bearing distinct 5′ ends. In general, we observed concordance in iso-miR abundance across these Drosophila genomes. For example, two iso-miRs from the 3′ arm of pre-mir-79 accumulated at a consistent abundance of approximately 3:1 ratio in all species (Fig. 4A). Such conservation in iso-miR 5′ end processing suggests that multiple species from a single locus are incorporated into conserved regulatory networks.
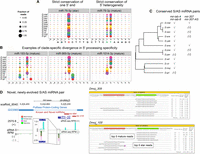
Shifted processing and alternate biogenesis pathways of Drosophila miRNAs. (A,B) Consistency in 5′ end processing for conserved miRNAs. (A) sRNA read alignment for mir-79, represented compactly in these bubble plots, show two miR sequences with unique 5′ ends. These represent two seed-distinct iso-miRs that are both produced in several Drosophila species. Position 0 represents the proportion of reads that begin with the base of the most abundant 5′ arm sequence at either the 5′ strand (miR*) and 3′ strand (miR) for all 12 Drosophila genomes. Proportions shown at positions less than or greater than 0 represent proportion of reads with shifted processing. For mir-79, two iso-miRs are produced in similar proportions. (B) Panels of bubble plots depict the heterogeneity of 5′ end processing for the miR sequence of other conserved miRNAs and mirtrons. Greater than 4 alternate iso-miR-193 sequences in D. melanogaster were noted previously. This heterogeneity is preserved in the genomes of the other Drosophilids, and conserved, dominant iso-miRs is not apparent. We identified clade-specific iso-miRs for one canonical miRNA (mir-969) and one mirtron (mir-1014). Specifically, two unique iso-miR-969 sequences are each preferentially abundant in the Sophophora group and Drosophila group species, respectively, and for mir-1014, the melanogaster group species produces one iso-miR-1014 sequence that is distinct from the dominant iso-miR of other Sophophorans. (C) mir-iab-4/8 and mir-307/mir-307-as are the only two reasonably conserved miRNAs with sense and antisense transcription and processing based upon our genus-wide data. (D) We identified dozens of recently evolved antisense miRNAs; shown is the example of dmo_105/dmo_309 that arose adjacent to the conserved mir-994/318 cluster.
In contrast, the heterogeneous processing of D. melanogaster mir-193 (Berezikov et al. 2011) is not consistent across the Drosophilid phylogeny (Fig. 4B). In particular, its 5′ end shifts by 2 nt in the virilis subgroup relative to the other species. As well, all Drosophila group species consistently processed mir-969 into a particular species, but a different iso-miR appears substantially in the Dana/Dpse/Dper/Dwil ancestor, whereas Sophophora group species dominantly accumulate a completely distinct, third iso-miR-969 (Fig. 4B). We also observe clade-specific 5′ shifts for noncanonical miRNAs, illustrated by mirtronic miR-1014-3p (Fig. 4B). Such alterations in miRNA processing and targeting capacity of certain conserved miRNAs were hidden until the availability of deep, evolutionary profiling of small RNAs.
Antisense miRNAs
Certain miRNA loci are transcribed and processed on both strands (Tyler et al. 2008). A marquee example in Drosophila is mir-iab-4/mir-iab-8, for which sense (S) and antisense (AS) miRNAs have distinct and genetically overt neural functions (Garaulet et al. 2014; Picao-Osorio et al. 2015). Since S/AS transcription of this miRNA locus has been observed in beetles (Hui et al. 2013), one might assume this is a conserved feature throughout the Drosophilids. Indeed, we confidently annotated mir-iab-4 and mir-iab-8 in all Drosophila species, except for D. persimilis and D. grimshawi, in which the lower-expressed mir-iab-8 locus had reads but had to be recovered through the “candidate-rescue” pipeline (Fig. 4C).
We observed 18 other confident S/AS miRNA pairs, as well as several dozen candidate antisense miRNAs (Supplemental Table S5). A few of these involve conserved miRNAs. Of these, the one whose antisense processing was most broadly detected mir-307-AS, which was confidently or candidately recorded in seven related Drosophilid species (Fig. 4C). The modest, but clear, cross-species accumulation of mir-307-AS might simply reflect low expression, but alternatively it may have spatially or temporally restricted deployment. However, most AS miRNAs were poorly conserved; in fact, a substantial fraction of them were present in species other than D. melanogaster (Supplemental Fig. S11). For example, D. mojavensis generated a novel sense/antisense locus Dmoj_105/Dmoj_309 adjacent to the deeply conserved intronic miRNAs mir-994/mir-318 (Fig. 4D). We present additional examples of S/AS miRNA pairs in Supplemental Figure S11, including loci that originated de novo in individual species. Overall, although S/AS miRNA pairs have originated across all branches of the Drosophilid phylogeny, few instances have been retained over evolution.
Vast evolutionary flux of testes-restricted miRNA clusters
The second largest class of novel miRNAs we annotated classified as testes-restricted, recently evolved, clustered (TRC) miRNAs, based on their residence in genomic clusters and preferred or exclusive accumulation in male-body/testis libraries relative to other tissue libraries. We identified 126 novel TRC miRNAs, all of which were recently evolved, which represented 17% of all new miRNA annotations (Fig. 5). Collectively, this abundance of novel miRNAs clustered into nine novel genomic regions within the genomes of different Drosophila species (Supplemental Figs. S12–S15).
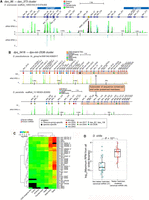
Testes-restricted, Recently evolved, Clustered (TRC) canonical miRNAs in Drosophila. (A) An example of a novel TRC miRNA cluster (dan_86 → dan_373) in D. ananassae. The majority of miRNAs show high expression in the male-body libraries. (B) An example of a TRC miRNA cluster (dps_3416 → dps-mir-2536) in the obscura subgroup species. The D. pseudoobscura cluster contains 36 miRNAs, whereas its sister species, D. persimilis, contains 26 miRNAs. MiRNAs within the 3′ end region of these orthologous clusters (orange highlight) have preserved their order, whereas miRNAs within the 5′ region show high gene duplication. Colored circles and numbers represent miRNAs of the same family. (C) Expression heatmap for all D. pseudoobscura copies reveals a predominant testes-restricted profile. (D) Comparison of expression difference between TRC and solo canonical miRNAs present in D. virilis alone or within the virilis/mojavensis clade alone. TRC miRNAs of the virilis-subgroup show significantly higher expression than their age-matched solo canonical cohorts (Mann-Whitney U test, P < 10−8). All Drosophilid subclades have their own distinct TRC loci, and details of all the novel TRC loci cloned in this study are provided in Supplemental Figs. S12–S15.
Strikingly, we find one or more novel TRC clusters specific to each major Drosophilid branch. Beyond the previously described miRNA clusters composed of conserved and recently emerged testis-expressed miRNAs (Mohammed et al. 2014a), we discovered many new TRC specific to D. ananassae (2 TRC, containing 34 miRNAs) (Fig. 5A) or D. willistoni (1 TRC, containing 19 miRNAs) and orthologous clusters that were only traceable between closely related sister species, such as between D. virilis and D. mojavensis (2 TRC shared by these species, containing 31–34 miRNAs, with two additional virilis-specific clusters containing 18 miRNAs), or between D. pseudoobscura and D. persimilis (2 TRC shared by these species, containing 47–50 miRNAs) (Supplemental Figs. S12–S15). Some of these clusters dwarf the largest previously known fly miRNA clusters. For example, we expanded the membership of the D. pseudoobscura Dpse_3416 → Dpse-mir-2536 TRC to 36 miRNAs, and identified an orthologous 26-member D. persimilis cluster (Fig. 5B). Small RNA expression showed significantly higher expression in male-body and testis libraries than other tissues (Fig. 5B,C). Interestingly, although miRNAs in the 3′ region of this cluster preserve their order between the two species, miRNAs near the 5′ end of the cluster evolved rapidly via both local gene duplication and de novo miRNA emergence, as evident from precursor and miRNA sequence alignments (Fig. 5B, family assignments; Supplemental Fig. S14).
The wealth of male-body sRNA libraries for all 12 genomes and testis data for D. pseudoobscura and D. virilis permitted the evaluation of the relative expression of TRC miRNAs to that of age-matched, solo canonical miRNA cohorts. In D. virilis, the species with the greatest number of TRC miRNAs (66 in total, including known and novel TRC miRNAs), we observed significantly higher expression for TRC miRNAs than for solo canonical miRNAs (Mann-Whitney U test, P < 10−8) (Fig. 5D). Although we observed a similar shift for substantially increased average expression of TRC miRNAs in D. pseudoobscura, this did not quite achieve significance over age-matched, non-TRC canonical miRNAs due to a small number of highly expressed loci in the latter category (Supplemental Fig. S16).
Altogether, the remarkable flux of clustered testis miRNA loci across the Drosophilid phylogeny generalizes their distinct evolutionary features that we had established from studies based on a D. melanogaster-centric viewpoint. These data provide strong evidence that TRC miRNAs are unlikely to be evolving along a purifying selection route, but instead may be utilized for adaptive regulatory purposes.
Distinct rates of gain and losses among miRNA classes
Using our updated Drosophila miRNA collection, we characterized rates of gain and loss across our three miRNA classes. To do so, we developed a phylogenetic probabilistic graphical model with the intention of estimating miRNA gene birth and death rates by maximum likelihood (Fig. 6A). This method allows us (1) to infer universal, clade-, or branch-specific birth (λ) and death (µ) rate parameters; (2) to predict node-wise ancestral miRNA presence or absence; and (3) to estimate expected counts of edge-wise gain and loss events. Although data were sampled more deeply in some species (e.g., D. melanogaster), we chose not to subsample the data because of the “rescue” approach used in the annotation process (Methods).
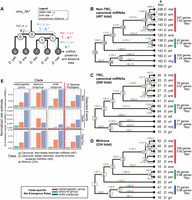
Estimation of miRNA birth and death rates in Drosophila. (A) A phylogenetic probabilistic graphical model for estimating rates of gene birth and death. The model takes binary data representing miRNA presence or absence at the leaves of the tree and uses numerical optimization methods to estimate model parameter (µ, λ) values by maximum likelihood. Branch lengths (τ) are fixed. Maximum-likelihood parameter estimates are then used to reconstruct node-wise miRNA presence/absence and edge-wise birth and death events. (B–D) Summary of estimated ancestral miRNA content and edge-wise birth and death events for three classes of miRNAs. miRNA classes are canonical miRNAs (B), testes-restricted, recently evolved, clustered canonical miRNAs (TRC) (C), and mirtrons (D). Estimates of edge-wise birth and death events are shown in green and red, respectively. Net emergence rate (i.e., total birth − death events/Myr) are shown in each class for the melanogaster group, obscura subgroup, and virilis subgroup species. (E) Rate of birth and death and net miRNA gain rate for three clades—melanogaster group, obscura subgroup, and virilis subgroup—are shown. Note that mirtrons and TRC miRNAs exhibit much higher rates of flux than do canonical non-testes-restricted miRNAs.
We applied our method to the pooled collection of miRNA families from our three classes and estimated model parameters (i.e., λ, µ). Using these rate estimates, we then reconstructed node-wise presence and absence of ancestral miRNAs, and subsequently branch-wise miRNA birth and death events, for each miRNA alignment family and across all miRNA classes (for summary, see Fig. 6B–D; for examples of all possible tree configurations, see Supplemental Fig. S17; for trees per miRNA alignments across three miRNA classes, see Supplemental Figs. S18–S20). Consistent with previous studies, the canonical miRNA class contained the largest number of ancient miRNAs, that is, those present at the root of the Drosophila phylogeny. Of 236 D. melanogaster canonical miRNAs, 106 were clearly present in the Drosophilid ancestor (Fig. 6B). As mentioned, there are more canonical miRNAs annotated in D. melanogaster than any other fly species owing to its depth of sequencing; otherwise, the majority of canonical miRNAs that are not testes-restricted are conserved (Fig. 6B).
The tables are turned when examining the fraction of conserved loci in the other categories of miRNAs. The strong majority of testes-restricted canonical miRNAs across the Drosophilid phylogeny, most of which are arranged in genomic clusters, are not conserved. Indeed, only 13 such miRNAs are deeply conserved and include specific members of the mir-972 → 979 cluster and the mir-959 → 964 cluster. Otherwise, most fly species harbor dozens of lineage-restricted TRC miRNAs (Fig. 6C). Similarly, the mirtron class also contains very few conserved loci. This notion was suggested earlier (Berezikov et al. 2010), but we now broadly extend this principle using empirical annotation of mirtrons across 12 Drosophila species. Although the genomes of many individual species bear more than 45 mirtrons (e.g., D. melanogaster with 52, D. pseudoobscura with 50, and D. virilis with 47 mirtrons), only seven mirtrons were present at the root of the Drosophilid phylogeny (Fig. 6D).
Next, we computed rates of miRNA birth and death by first aggregating birth and death events across important clades (melanogaster group, obscura subgroup, and virilis subgroup) or the entire phylogeny for each miRNA class, and normalizing them by both the total branch length (Myr) and by conserved members of each class (i.e., present at the root of the tree). These clade-specific and tree-wide rate estimates permitted additional intra-miRNA-class comparisons of total miRNA flux (i.e., birth plus death) in each of these representative clades or across the entire phylogeny.
Interestingly, we saw striking rate variation across the three classes of miRNAs (Fig. 6E). In general, canonical non-testes-restricted miRNAs exhibited the lowest rates of birth, death, and total miRNA flux in each clade and across the Drosophila phylogeny when compared to the two other classes. At the other end of the spectrum, mirtrons exhibited the highest rate estimates. This is due not only to the large collection of single-species mirtron loci (Fig. 6D), but also to certain atypical patterns of mirtron presence within species that do not group along clade boundaries (Supplemental Fig. S21). Testes-restricted canonical miRNAs exhibited birth, death, and total flux rates in between those of canonical non-testes-restricted miRNA and mirtrons. Notably, we also observed branch-specific behavior, since TRC miRNAs showed significantly elevated death rate within the obscura subgroup compared to the other miRNA classes. Altogether, these findings indicate substantial heterogeneity among evolutionary rates of multiple classes of canonical and noncanonical miRNAs and also highlight differential behavior along individual lineages.
Multiple mechanisms underlie distinct flux behaviors of different miRNA classes
We explored several molecular strategies that could underlie the distinct evolutionary behaviors of different miRNA classes using these comprehensive novel miRNA annotations.
cis-Mutations affecting processing
The impact of nucleotide changes themselves, especially those that are sparse among orthologous pre-miRNAs sequences, are little known on miRNA expression, genesis, or decay. We identified compelling sets of miRNA orthologs for experimental tests, including ones exhibiting large variations in apparent biogenesis despite sometimes full genomic identity in the mature miRNA species.
For example, the melanogaster subgroup–specific locus mir-4984 is highly similar across five genomes, yet exhibits large expression differences between orthologs; only D. melanogaster, D. simulans, and D. sechellia orthologs are processed (Supplemental Fig. S22A). We tested a panel of species mir-4984 expression constructs to validate preferential processing and activity of the D. mel-mir-4984 versus other orthologs (Supplemental Fig. S22B–D). Another example is the alignment of Dpse_41 and Dper_ 2484, which are specific to the obscura subgroup. These miRNAs contain a few hairpin divergences, including a single seed change, but Dpse_41 is much more highly expressed than Dper_2484 in sRNA data (Fig. 7A). Expression constructs recapitulated stronger maturation of Dpse_41 by Northern blotting (Fig. 7B). Thus, cis-changes and not transcriptional changes account for processing differences. This was corroborated by functional assay of a luciferase reporter bearing two seed matches for each ortholog; Dpse_41 but not Dper_2484 conferred strong repression of this sensor (Fig. 7C).
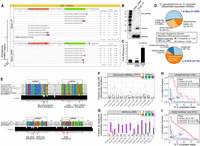
Multiple distinct cis-molecular signatures associated with miRNA flux. (A–C) Duplex alterations affect miRNA maturation and function. (A) The Dpse_41/Dper_2484 ortholog pair, with only a few duplex divergences, exhibits divergent expression between very closely related species. Functional assays confirm differential biogenesis of Dpse_41/Dper_2484 expression constructs by Northern blotting (B) and differential activity by luciferase sensor assay (C). (D) Transcriptome comparison of miRNAs differentially expressed between sister species Dpse and Dper. In general, significantly more duplex divergent miRNAs are differentially expressed miRNAs for both newly evolved and conserved miRNAs. (E) Evolution of seed regions of testes-restricted, clustered (TRC) miRNAs. Shown are examples of one-to-one orthologs of TRC miRNAs between Dpse and Dper, including available Dpse population data. Highlighted are examples of seed divergence between expressed TRC miRNA orthologs between these closely related species, consistent with adaptive evolutionary behavior. (F–I) Impact of terminal uridylation system on evolutionary suppression of mirtrons and behavior of canonical miRNAs. (F,G) Compared to canonical miRNAs (F), mirtrons (G) in every Drosophilid species acquire much higher rates of terminal untemplated uridylation (purple) on the 3′ ends of their 3p species, compared to any other nucleotide modifications. (H,I) 3′ uridylation of canonical miRNAs is sensitive to terminal hairpin nucleotide. In these graphs, miRNA loci are divided by biogenesis type (canonical versus splicing-derived), by terminal nucleotide (3′-G versus 3′-A/U/C, i.e., “3′-H”), and by evolutionary age. Analysis of deeply conserved miRNA loci (H) and recently evolved loci (I) shows that canonical miRNA hairpins that end in G acquire higher levels of 3′ uridylation than do other canonical miRNA hairpins. P-values computed from a two-tailed Wilcoxon rank-sum test.
We broadened our analysis of obscura species, as the D. pseudoobscura/persimilis sister pair was closely related (0.93 Myr) and harbored numerous novel miRNA annotations that might include other expression fluctuations. We labeled miRNA alignments with greater than or equal to sixfold RPMM expression difference as differentially expressed and identified six conserved and nine newly evolved miRNAs as such (Supplemental Fig. S23). The conserved loci included several mir-309 cluster members and seemed to be a sampling artifact given they are expressed as a highly stage-specific operon (Bushati et al. 2008). Otherwise, there was high correlation (r2 = 0.938) between the remaining 205 D. pseudoobscura and D. persimilis ortholog pairs (Supplemental Fig. S23).
The 15 differentially expressed, obscura subgroup ortholog pairs shared high miR:miR* duplex sequence similarity. We asked if duplex substitutions were more prevalent between differentially expressed miRNAs than nondifferentially expressed ones, for conserved and newly evolved miRNAs. Indeed, within the obscura subgroup, we saw significantly more differentially expressed miRNAs with duplex substitutions within the newly evolved group (Fisher's exact test P < 0.003), and within the conserved group (Fisher's exact test P ≈ 0.03) (Fig. 7D).
Seed-targeting alterations of TRC miRNAs
Functional miRNAs are not expected to diverge between closely related species, especially within seed regions. However, we previously used D. melanogaster population data and melanogaster group species orthologs to provide evidence for adaptive evolution of TRC miRNAs in this clade, including within seeds (Mohammed et al. 2014a). There is limited population data in other Drosophilids, but analysis of TRC loci with clear one-to-one orthologs between obscura subgroup species revealed unambiguous cases of TRC miRNAs with intraspecies divergences within the seed and a reduction in interspecies polymorphism (Supplemental Methods). For example, the mature (3′) arm of D. pseudoobscura mir-2523 contained a G-to-T substitution at the eighth seed position relative to its D. persimilis ortholog Dper_106 (Fig. 7E). Analysis of D. pseudoobscura population data (McGaugh et al. 2012) indicated that all individuals were monomorphic for the “T” allele (i.e., a fixed difference). We also observed several other nonseed divergent and polymorphic bases within the star strand within the D. pseudoobscura population, likely indicative of relaxed constraint. As well, both mature and star arms of Dpse-mir-2542-1 exhibit multiple positions of seed divergence with its ortholog Dper_101 (Fig. 7E).
We further note that Dpse_41/Dper_2484 tested in Figure 7A–C are TRC miRNAs, which have a functionally validated seed change and altered activity between related species. Overall, multiple obscura TRC loci defy conventional behavior for purifying selection of seed regions and instead alter their seed regions between closely related species, consistent with the notion of adaptive targeting behavior.
Preferential 3′ untemplated uridylation of mirtrons
Approximately 54% (232 of 428) of mirtron and tailed-mirtron annotations in Drosophila are new to our study, and are recently emerged. This large set of novel spliced miRNAs prompted us to ask if they exhibit characteristic properties of 3′-untemplated uridylation, as we reported in D. melanogaster (Bortolamiol-Becet et al. 2015; Reimão-Pinto et al. 2015). In comparisons of 1777 canonical miRNAs and 289 mirtrons (Supplemental Table S6), we observed that mirtrons exhibited much greater 3′ untemplated uridylation than canonical miRNAs (Fig. 7F,G). This was also the case even after conditioning on canonical miRNAs, whose 3′ arm read ended in AG dinucleotide as with mirtrons (Supplemental Fig. S24A).
Next, we examined whether the elevated frequency of uridylation at canonical miRNAs and mirtrons whose 3′ read ended with G was consistent across conserved and newly evolved loci. For the conserved loci, we recapitulated previous signatures of uridylation (Fig. 7H). Namely, mirtrons exhibited a significantly higher frequency of uridylation than canonical miRNAs (Mann-Whitney U test, P < 10−21), and comparisons among canonical miRNAs revealed that loci whose 3′ arm read ended with G were more uridylated than loci ending in other bases (i.e., IUPAC “H”) (P < 10−12). Of note, newly evolved mirtrons and canonical miRNAs also exhibited the same signature as conserved loci (Fig. 7I), and comparisons of loci within individual species revealed similar significant results in many species (Supplemental Fig. S24B). Altogether, these findings from small RNA sequencing across the Drosophilid phylogeny broadly support the notion that adventitious access of splicing-derived hairpins to Dicer is suppressed via 3′ uridylation.
Discussion
A deep and broad empirical analysis of miRNA flux across the Drosophila genus
In this study, we extended our previous deep curation of D. melanogaster miRNAs (Berezikov et al. 2011) with largescale empirical analysis of sRNA data across the Drosophila genus, first yielding cloning of hundreds of orthologs of conserved miRNAs, and then identification of 649 completely novel miRNA loci. Overall, these data yield diverse insights into miRNA processing and evolution. These include the surprising existence of novel conserved miRNAs, unexpected clade-specific shifts in processing register, and post-transcriptional modifications of miRNAs. Beyond conserved loci, the trove of “young” miRNAs allows us to quantify distinct rates of miRNA flux according to biogenesis type, genomic locale, tissue restriction, and evolutionary clade. We identify patterns of structural change associated with flux in expression of evolutionarily nascent canonical miRNAs, providing a mechanistic basis for their instability. With this rich foundation of species-specific miRNA annotations in hand, a clear challenge for the future will be to discern whether these loci impart species-specific regulatory impacts.
Divergent rationales for rapid evolution of mirtrons and TRC miRNAs
We solidify the perspective that miRNAs do not comprise a unitary class, but encompass a diversity of functional loci with distinct evolutionary imperatives. In particular, among our extensive collection of recently emerged miRNAs, we discern two major subclasses of rapidly evolving loci: splicing-derived miRNAs (i.e., mirtrons) and testes-restricted clustered (i.e., TRC) miRNAs. We propose divergent functional explanations for their distinct evolutionary behavior, relative to the bulk collection of recently emerged miRNAs that either evolve under mild purifying selection or lack substantial utility and evolve neutrally (Mohammed et al. 2013).
Mirtrons mature via the dominant noncanonical mechanism that bypasses the Drosha/DGCR8 “Microprocessor,” which otherwise serves as a molecular gatekeeper for generation of specific and accurate Dicer substrate hairpins. Mirtrons occasionally yield regulatory species that incorporate into beneficial regulatory networks, but the vast majority are not retained during evolution. Indeed, molecular mechanisms involving uridylation have recently been shown to selectively suppress splicing-mediated miRNA biogenesis and promote their evolutionary flux (Bortolamiol-Becet et al. 2015; Reimão-Pinto et al. 2015).
Our current studies across the Drosophila genus broadly confirms that the accelerated evolutionary dynamics of mirtrons correlates well with their remarkably high rates of 3′ uridylation. Indeed, only six of the more than 400 mirtrons we annotated across the Drosophilid phylogeny were present in the fruit fly ancestor. We and others showed this is mediated by the uridyltransferase Tailor, which recognizes hairpins bearing 3′-(A)G, a characteristic for splicing-derived hairpins (Bortolamiol-Becet et al. 2015; Reimão-Pinto et al. 2015). We hypothesized this may carryover to suppress the evolutionary emergence of canonical miRNAs that happen to end in 3′-(A)G, and our experimental data support this notion. Therefore, a uridylation mechanism shapes the evolution of both noncanonical and canonical miRNA substrates.
On the other hand, the rapid dynamics of TRC miRNAs in all subclades of the Drosophila genus provides compelling evidence for their adaptive evolution. Not only do TRC miRNA sequences evolve more quickly than canonical miRNA substrates of matched age, the total flux in TRC miRNA numbers between Drosophila subclades outpaces that of canonical miRNA loci. For example, clear sequence orthologs of 106 of 497 canonical miRNAs not in the TRC class were present in the pan-Drosophilid ancestor, whereas this is only true of 13 of 265 TRC miRNA loci (Fig. 6C). An alternative possibility is that some TRC miRNAs, owing to positive selection, have evolved in primary sequence so quickly that their ancestral relationships are not possible to assess. In any case, it is clear that the wholesale appearance and disappearance of extensive TRC loci in different clades reflects a fundamentally different usage of these miRNAs than for maintenance of conserved seed-driven target networks as with typical canonical miRNAs.
Moreover, the atypical dynamics of TRC miRNAs are substantially accelerated in both species examined in the obscura subclade. In fact, D. pseudoobscura and D. persimilis themselves exhibit substantial differences in their TRC repertoire, underlying nearly an order of magnitude greater birth estimate in the obscura branch than other branches of the phylogeny. The functional underpinnings of this remain to be tested, but they go hand-in-hand with the recent observation of proliferations of testes-restricted AGO2 paralogs specifically in the obscura subclade, and not in other Drosophila subclades (Lewis et al. 2016).
Overall, our study provides a wealth of small RNA data that can guide functional studies of miRNA biogenesis, regulation of miRNA processing, and will underlie discovery of novel small RNA types (such as siRNAs and piRNAs). In addition, our deep and broad sampling across an entire genus provides many insights into the distinct evolutionary trajectories of multiple miRNA subtypes, affirming that miRNAs cannot be considered a unitary class with respect to their functional impact and utilization.
Methods
Drosophila species small RNA libraries
To analyze miRNA evolution in Drosophila species, we obtained cultures of whole-genome sequenced D. simulans, D. sechellia, D. yakuba, D. erecta, D. ananassae, D. pseudoobscura, D. persimilis, D. willistoni, D. virilis, and D. mojavensis strains from the UCSD Drosophila Species Stock Center. Adult D. grimshawi samples were a gift of Dr. Kevin White (University of Chicago). Small RNAs (∼18–28 nt) were isolated from male bodies, female bodies, heads, and mixed embryos using polyacrylamide gel electrophoresis, and we prepared libraries as described (Berezikov et al. 2010, 2011). Libraries were sequenced on Illumina GaIIx or HiSeq 2000 instruments. Some libraries were resequenced to reach desired read depth (Supplemental Fig. S1).
Annotation of miRNA genes
To identify novel miRNAs and assess the expression levels of all miRNA loci, we first mapped reads from each of the 11 Drosophila species unto their reference genomes. All reference genomes, except for D. simulans, were obtained from FlyBase (Gramates et al. 2017). We utilized a revised D. simulans genome assembly created from an isogenic w501 female within our analysis (Hu et al. 2013). Reads were mapped using the Bowtie program (Langmead et al. 2009) by allowing for up to three mismatches (parameters: -v 3 -k 20 --best --strata). Perfectly mapped reads, and reads with 3′ end mismatches characteristic of untemplated additions were used for the identification of miRNAs.
We supplemented existing Drosophila miRNA annotations from miRBase v21 (Kozomara and Griffiths-Jones 2014) with novel miRNAs and mirtrons using a multistage pipeline. First, canonical miRNA and mirtrons were predicted using miRDeep2 using default software settings (Friedlander et al. 2012). To identify short mirtron and long pre-miRNA hairpins, two classes systematically missed by miRDeep2, we mapped sRNA data sets to introns from FlyBase annotations, and hairpins were predicted using einverted from the EMBOSS package (Rice et al. 2000) in a genome-wide manner per species. We used the invert_it.pl utility from ShortStack (Shahid and Axtell 2014) to filter einverted results. The parameters specified to this script were: -f 0.6 -p 30. Introns or hairpin structures with at least one mapped read were retained and ranked by P-values calculated from a Random Forest classifier. We trained this classifier with a balanced set of positive training examples comprised of known D. melanogaster and D. pseudoobscura miRNAs downloaded from miRBase (v21) and a negative training set composed on non-miRNA predictions identified manually in this study. We used 37 features per training case representing sequence, structure, and sRNA read alignment features (Supplemental Table S7). Minimum free energy, and suboptimal secondary structures were predicted using RNAfold and RNAsubopt in the ViennaRNA software (Lorenz et al. 2011). All hairpin candidates from the pipelines were vetted manually and bioinformatically, and additional considerations are described in the Supplemental Text.
Identification of miRNA orthologs and alignments
miRNA orthologs were identified using the LASTZ program with the following parameters: H = 2000, Y = 3400, L = 4000, K = 2200, and Q = HoxD55.q (Harris 2007). Hits were ranked by a score based on the consistency, continuity, and percent identity metrics from LASTZ. A 12-species sequence alignment was created for each miRNA prediction using best scoring orthologs and the Fast Statistical Aligner program (Bradley et al. 2009). Paralogs were a by-product of this procedure because they attained lower rank during orthology assignments. All orthologs and paralogs were automatically included in our annotation pipeline and were vetted by the same criteria.
Birth and death model
To assess birth and death rate variation across classes of miRNAs and across Drosophila clades of interest, we designed and implemented a phylogenetic probabilistic graphical model. This model permits estimation of parameters of gene birth (λ) and death (μ) (Fig. 6A) based on our assignments of miRNA presence and absence in each species per miRNA family alignment. Parameter estimation required two sets of precomputed data. The first datum needed was a binary encoding of miRNA presence (1) and absence (0) as leaf node labels of the phylogenetic model. The second datum needed was phylogenetic branch-length estimates for the 12 Drosophila species phylogeny (Clark et al. 2007). Given these two data sets, we used our model to infer maximum-likelihood parameter estimates (i.e., λ, μ) using the standard belief-propagation algorithm to compute likelihoods, and the Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm to obtain maximum-likelihood parameter estimates (Zhu et al. 1997). Parameter estimates were computed for the merged miRNA collection (final estimate: λ = 0.292, μ = 0.694), which we later used to compute (1) ancestral gene presence or absence states, and (2) probabilities of observable edge-wise birth and death events (Supplemental Figs. S17–S20). To assess cumulative counts of observable birth and death events per miRNA class or Drosophila clade, we computed edge-wise joint posterior probabilities (i.e., P[child, parent]) by belief propagation. For simplicity, we called birth [P(1,0)], death [P(0,1)], and “no change” events [P(0,0) or P(1,1)] if these probability estimates were ≥0.5 (Fig. 6B–D). Further details and considerations are provided in the Supplemental Text.
Northern blotting and luciferase assays
We used previously described methods (Okamura et al. 2007), with cloning strategies and detailed methods provided in the Supplemental Text.
Data access
The Drosophila sRNA sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE98013. This birth and death model is implemented as a Java software package and available at http://compgen.cshl.edu/mirna/12flies/software/MirnaTreeML.zip as well as a zip file in the Supplemental Materials. Read pileup, structure prediction, and 12-fly sequence alignments of all Drosophila miRNAs are provided as Supplemental Material via an online website (http://compgen.cshl.edu/mirna/12flies/12flies_alignments.html) as well as in a zip file in the Supplemental Materials.
Acknowledgments
We thank the UCSD Drosophila Species Stock Center for fly stocks. J.M. was supported in part by the Tri-Institutional Training Program in Computational Biology and Medicine (via National Institutes of Health [NIH] training grant 1T32-GM083937). A.S. was supported by the NIH (R01-GM102192). Work in A.S.F.'s group was supported by NIH (R15-GM120716) and Mississippi INBRE funded by the NIH (IDeA P20-GM103476). Work in E.C.L.'s group was supported by the NIH (R01-GM083300 and R01-NS083833) and MSK Core Grant P30-CA008748.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.226068.117.
- Received June 7, 2017.
- Accepted November 20, 2017.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








