
Abstract
DNA base damage is an important contributor to genome instability, but how the formation and repair of these lesions is affected by the genomic landscape and contributes to mutagenesis is unknown. Here, we describe genome-wide maps of DNA base damage, repair, and mutagenesis at single nucleotide resolution in yeast treated with the alkylating agent methyl methanesulfonate (MMS). Analysis of these maps revealed that base excision repair (BER) of alkylation damage is significantly modulated by chromatin, with faster repair in nucleosome-depleted regions, and slower repair and higher mutation density within strongly positioned nucleosomes. Both the translational and rotational settings of lesions within nucleosomes significantly influence BER efficiency; moreover, this effect is asymmetric relative to the nucleosome dyad axis and is regulated by histone modifications. Our data also indicate that MMS-induced mutations at adenine nucleotides are significantly enriched on the nontranscribed strand (NTS) of yeast genes, particularly in BER-deficient strains, due to higher damage formation on the NTS and transcription-coupled repair of the transcribed strand (TS). These findings reveal the influence of chromatin on repair and mutagenesis of base lesions on a genome-wide scale and suggest a novel mechanism for transcription-associated mutation asymmetry, which is frequently observed in human cancers.
DNA base lesions are the most frequent class of DNA damage, arising from reactive oxygen species, spontaneous base hydrolysis, deamination, or endogenous and exogenous alkylating agents. Base lesions must be efficiently repaired by the base excision repair (BER) pathway to avoid mutagenesis or cell death, outcomes which can contribute to carcinogenesis, neurodegenerative diseases, and aging (Wallace et al. 2012; Akbari et al. 2015). BER is initiated by lesion-specific DNA glycosylases that cleave the N-glycosidic bond of damaged bases to generate an abasic site. For example, alkyladenine glycosylase (AAG) and 3-methyladenine DNA glycosylase (Mag1) are responsible for recognizing and cleaving the major type of alkylation damage, N-methylpurine (NMP) lesions (e.g., 3-methyladenine [3meA] and 7-methylguanine [7meG]), in human and yeast cells, respectively (Bauer et al. 2015). The resulting abasic site is cleaved by apurinic/apyrimidinic endonuclease (APE1 in humans), followed by repair synthesis and DNA ligation (Bauer et al. 2015).
While it is well established that nucleotide excision repair (NER), which repairs helix-distorting DNA lesions such as UV photoproducts, is significantly modulated by cellular chromatin (Nag and Smerdon 2009), the effects of chromatin on BER in vivo are poorly understood. In vitro studies using purified BER enzymes have shown that each step in the BER pathway is significantly inhibited within a strongly positioned nucleosome, particularly when the lesion has an inward rotational setting or translational position near the nucleosome dyad (i.e., the position of intersection of DNA with the central dyad axis of the nucleosome) (Rodriguez et al. 2015). However, it is not clear to what extent nucleosomes affect BER in vivo, since nucleosome positioning in eukaryotic genomes is generally weaker than the positioning sequences used for in vitro studies (Mao et al. 2017) and the presence of cellular chromatin-remodeling enzymes can facilitate BER in nucleosomes (Hinz and Czaja 2015; Rodriguez et al. 2015). Moreover, nucleosomes in vivo are marked by different histone post-translational modifications, which could potentially alter repair efficiency (Rodriguez et al. 2016).
Our understanding of how chromatin and other genomic features influence the formation and repair of DNA base damage, such as DNA alkylation, has been limited by the lack of genome-wide methods for mapping this class of DNA lesions. Unlike NER, which has been extensively studied using novel genome-wide methods (Hu et al. 2015; Mao et al. 2016; Yu et al. 2016), there are currently no published studies measuring BER of DNA base lesions on a genomic scale. One recent study mapped the replicative incorporation of uracil (a form of base damage) across the yeast genome using a method called Excision-seq (Bryan et al. 2014). However, this technique required very high, nonphysiological levels of uracil incorporation (Wyrick and Roberts 2015) and did not measure subsequent repair of uracil lesions. Alternative methods have been proposed for mapping DNA base damage (Li et al. 2015; Riedl et al. 2016), but these have yet to be applied on a genome-wide scale. Moreover, little is known about how chromatin or other genomic features influence mutagenesis associated with DNA base damage across the genome, due to a lack of relevant genome-wide mutation data.
In this study, we developed a novel method known as N-methylpurine-sequencing (NMP-seq) to map methyl methanesulfonate (MMS)-induced alkylation damage across the yeast genome at single nucleotide resolution. We focused on DNA alkylation damage, as it is an important class of base lesions in eukaryotic cells that arises from endogenous (e.g., S-adenosyl methionine) and exogenous damaging agents, including chemotherapeutics such as temozolomide (Fu et al. 2012), and because it is commonly used as a model lesion for BER studies (e.g., Li and Smerdon 2002a; Li et al. 2015). We also analyzed the genome-wide distribution of mutations arising from MMS treatment in wild-type and repair-deficient strains and compared the patterns of mutagenesis with the observed variations in lesion formation and repair across the genome.
Results
A map of alkylation lesions at single nucleotide resolution across the yeast genome
To precisely map alkylation NMP lesions throughout the genome, we developed the NMP-seq method (Fig. 1A). NMP-seq is adapted from methods previously used for mapping ribonucleotide lesions (Ding et al. 2015) and UV-induced cyclobutane pyrimidine dimers (Mao et al. 2016). The major difference between these methods is that NMP-seq employs the AAG and APE1 BER enzymes to generate single-strand breaks containing free 3′OH immediately upstream of the NMP lesions 3-methyladenine (3meA) and 7-methylguanine (7meG) (see Fig. 1A), two DNA lesions which comprise >90% of all alkylation DNA damage induced by the model alkylating agent MMS (Friedberg et al. 2006). Free 3′OH ends generated by AAG and APE1 cleavage are then ligated to a biotinylated A adaptor, which is used to purify, PCR-amplify, and sequence the resulting DNA fragments. By mapping the sequencing reads back to the genome sequence, the location of the NMP lesion can be precisely determined.
Genome-wide map of DNA alkylation damage and repair. (A) Experimental strategy for mapping N-methylpurine (NMP) lesions using NMP-seq. Isolated genomic DNA fragments are ligated to the trP1 adaptor (green), cleaved at NMP lesions with human alkyladenine glycosylase (AAG) and AP endonuclease (APE1), ligated to the biotinylated A adaptor (purple), purified, and sequenced. “dd” indicates dideoxy (i.e., 3′H). (B) Nucleotide distribution of lesion sites associated with NMP-seq sequencing reads. In MMS-treated samples, there is a significant enrichment of sequencing reads at “G”, and to a lesser extent, “A” nucleotides. (C) Nucleosome organization surrounding the transcription start sites (TSSs) of 5762 yeast genes modulates repair of 7meG lesions. Top panel depicts the average number of 7meG lesions per G nucleotide following 2-h repair in WT yeast strain. Middle panel depicts the fraction of 7meG lesions remaining following 1- or 2-h repair in WT relative to a matched 0-h mag1Δ control. Lower panel depicts the average nucleosome center positioning score (Brogaard et al. 2012). (D,E) Snapshot of the distribution of 7meG lesions at 0 h (mag1Δ) and following 2-h repair (WT) for the ACS1, FLC2, RQC2, and CHL1 genes. Dark brown bars indicate positioned nucleosomes, and the associated number indicates the nucleosome dyad score. Only well-positioned nucleosomes (NCP score ≥ 1) are depicted.
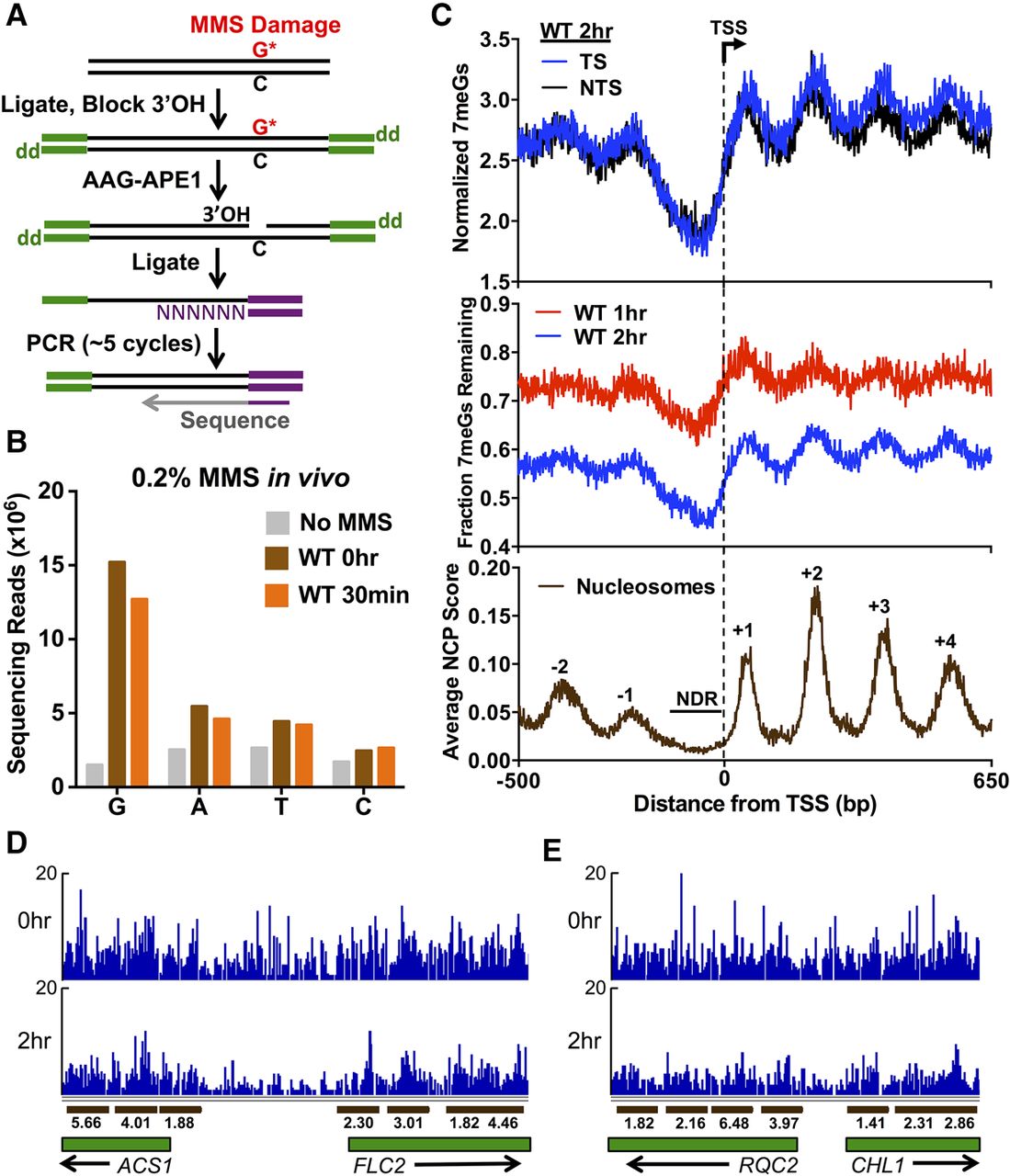
We used the NMP-seq method to map NMP lesions throughout the yeast genome following treatment with MMS. Wild-type yeast cells were treated with 0.2% MMS for 10 min, and then genomic DNA was isolated from yeast immediately after MMS treatment (0 h) or after an additional 30-min repair incubation in the absence of MMS. As a control, genomic DNA was also isolated from untreated yeast (No MMS). NMP lesions were mapped for each sample using the NMP-seq method. Analysis of the resulting NMP-seq data indicated a significant enrichment of sequencing reads associated with putative lesions at “G” nucleotides for the MMS-treated samples (10-fold higher) compared to the No MMS sample (Fig. 1B). There was also an apparent enrichment of sequencing reads associated with “A” nucleotides, although the magnitude of the enrichment was smaller (Fig. 1B). These results are in accordance with previous studies that have shown that ∼80% of MMS-induced DNA lesions are 7meG, and ∼10% are 3meA (Friedberg et al. 2006); hence, we attribute G-associated NMP-seq reads primarily with 7meG lesions and A-associated reads primarily with 3meA lesions. The enrichment of NMP-seq reads associated with 3meA lesions (relative to T-associated reads) was more apparent when yeast cells were treated with a higher dose of MMS (0.4% for 10 min) (Supplemental Fig. S1). Consequently, we used this dose (0.4% MMS) for all subsequent NMP-seq experiments. NMP-seq reads associated with other nucleotides (i.e., C or T) could represent rare alkylation lesions on pyrimidine bases (Friedberg et al. 2006), background due to low levels of AAG cleavage at undamaged DNA, or incomplete blocking of 3′OH groups by terminal transferase (Fig. 1A). The number of 7meG and 3meA NMP-seq reads decreased in the WT 30-min time point (Fig. 1B), likely reflecting ongoing BER of these NMP lesions.
Repair of NMP lesions is significantly modulated by the stereotypic organization of nucleosomes within yeast genes
To characterize the effects of genomic and chromatin contexts on repair of NMP lesions, we used NMP-seq to map NMP lesions following 1 or 2 h of repair in WT cells treated with 0.4% MMS. We focused on repair of 7meG lesions because this is the predominant class of lesion induced by MMS and thus could be more readily analyzed during the NMP-seq repair time course. We analyzed the average distribution of 7meG lesions adjacent to the transcription start sites (TSSs) of 5762 yeast genes. To account for potential sequence biases across yeast genes, the frequency of 7meG reads was normalized by the number of G nucleotides at each position relative to the TSS (i.e., from −500 bp upstream of, to +650 bp downstream from the TSS) on both DNA strands. The distribution of normalized 7meG reads adjacent to the TSS showed a striking distribution following 2-h repair, with low levels of 7meG lesions immediately upstream of the TSS, and periodic peaks of 7meG lesions downstream from the TSS (Fig. 1C, top panel). The distribution of 7meG lesions after 2-h repair was very similar on the transcribed strand (TS) and nontranscribed strand (NTS) (Fig. 1C, top panel; Supplemental Fig. S2), indicating that, unlike UV-induced DNA photoproducts (Hanawalt and Spivak 2008; Hu et al. 2015; Mao et al. 2016), MMS-induced 7meG lesions in WT yeast are not more rapidly repaired on the TS.
To determine whether the pattern of 7meG lesions represents differences in repair efficiency, as opposed to differences in initial damage levels, we calculated the fraction of 7meG lesions remaining (i.e., unrepaired) following 2-h repair relative to a 0-h control. Because repair is ongoing even during the 10-min MMS exposure in the WT strain, we used a matched mag1 deletion strain treated with the same MMS dose for the 0-h time point. Since Mag1 is the sole DNA glycosylase that recognizes NMP lesions in yeast (Bauer et al. 2015), the mag1Δ mutant should eliminate BER during MMS exposure and thus represent the initial distribution of NMP lesions. The fraction of unrepaired 7meG lesions showed the same trend as before, with low levels of unrepaired 7meG lesions immediately upstream of the TSS and periodic peaks of unrepaired 7meG lesions downstream from the TSS (Fig. 1C, middle panel). A similar, although less pronounced, trend of unrepaired 7meGs was apparent following 1-h repair (Fig. 1C, middle panel).
Downstream from the TSS, peaks of unrepaired 7meG lesions had a periodicity of ∼164 bp (1-h repair) and ∼160 bp (2-h repair), which matches the estimated 160- to 165-bp nucleosome repeat length in yeast (Cui et al. 2012). This suggests that the heterogeneous distribution of unrepaired 7meG reads following 2-h repair may reflect the inhibitory effect of nucleosomes on BER. To test this hypothesis, we analyzed a published map of nucleosome dyad positions in yeast (Brogaard et al. 2012). This analysis revealed that the peaks of unrepaired 7meG lesions downstream from the TSS coincided with the principal dyad locations of the +1, +2, +3, and +4 nucleosomes (cf. bottom panel with top and middle panels of Fig. 1C), while low levels of unrepaired 7meG upstream of the TSS largely coincided with nucleosome-depleted regions (NDR) present in many yeast promoter regions (Yuan et al. 2005). We also analyzed a published yeast DNase-seq data set (Zhong et al. 2016), which is an alternative measure of chromatin structure and accessibility. Comparison with the DNase-seq data revealed that peaks of unrepaired 7meG lesions corresponded to regions with a low density of DNase-seq reads (i.e., low chromatin accessibility due to positioned nucleosomes), while low levels of unrepaired 7meG in the NDR upstream of the TSS coincided with high levels of DNase-seq reads (Supplemental Fig. S3). This analysis indicates that differences in chromatin accessibility due to the stereotypic organization of nucleosomes across yeast genes significantly modulate the repair of 7meG lesions. Similar patterns of repair were also detected at individual genes (Fig. 1D,E).
To confirm that the observed patterns of unrepaired 7meG lesions were due to nucleosome-dependent variations in BER efficiency, we repeated the repair time course in a mag1Δ strain. Analysis of the mag1Δ NMP-seq data revealed that the levels of 7meG lesions showed only a marginal decrease following 1- or 2-h incubation (Supplemental Fig. S4A). Presumably, this reflects ongoing DNA replication and trans-lesion synthesis in the mag1Δ strain (Johnson et al. 2007), which would cause an apparent decrease in the levels of NMP lesions. Importantly, analysis of the 1- and 2-h mag1Δ data indicated that there was no correlation between the fraction of 7meG lesions remaining and nucleosome positions in the mag1Δ mutant (Supplemental Fig. S4B,C), indicating that Mag1 is required for the patterns of 7meG repair observed in WT cells. In summary, this analysis indicates that the stereotypic nucleosome organization within genes (e.g., −1 nucleosome, NDR, +1 nucleosome, etc. [Jiang and Pugh 2009; Rando and Winston 2012]) significantly modulates the efficiency of BER of 7meG lesions, with slower repair associated with the central dyad locations of nucleosomes and faster repair in NDRs.
BER efficiency is regulated by the translational and rotational settings of the lesions within strongly positioned nucleosomes in vivo
To better understand how the positioning of an NMP lesion within a nucleosome affects its repair in vivo, we analyzed the repair of 7meG lesions among a set of ∼10,000 strongly positioned nucleosomes (nucleosome score > 5) in the yeast genome (Brogaard et al. 2012; Mao et al. 2016). Analysis of 7meG lesions remaining (i.e., unrepaired) following 2-h repair revealed a greater fraction of unrepaired lesions near the nucleosome dyad relative to more distal regions of nucleosomal DNA (Fig. 2A) or the adjacent linker DNA (Supplemental Fig. S5A), indicating that the translational positioning of DNA lesions relative to the nucleosome dyad affects repair efficiency. A similar trend was observed following 1-h repair (Supplemental Fig. S5B), although the effect was less pronounced. Following 2-h repair, the fraction of unrepaired 7meG lesions also correlated with the rotational setting in strongly positioned nucleosomes. There was a smaller fraction of unrepaired lesions at outward rotational settings (dotted vertical lines in Fig. 2A) and a correspondingly higher fraction of unrepaired lesions at inward rotational settings. In contrast, there was no effect of translational or rotational setting on the fraction of 7meG lesions remaining in mag1Δ cells (Fig. 2B; Supplemental Fig. S5C), indicating that the patterns present in WT cells represent differences in BER efficiency.
Repair of 7meG lesions is regulated by their rotational and translational setting within strongly positioned nucleosomes. (A) Fraction of 7meG lesions remaining following 2-h repair in WT relative to 0-h mag1Δ control among ∼10,000 strongly positioned nucleosomes (nucleosome score > 5) across the yeast genome is plotted relative to the distance from the central dyad axis of the nucleosome (“dyad”). Nucleosomes overlapping with the MAG1 gene, which is deleted in the mag1Δ control strain, were excluded from this analysis. Vertical dotted lines indicate “Out” rotational settings. (B) Translational and rotational setting among strongly positioned nucleosomes does not affect the fraction of 7meG lesions remaining following 2-h repair in a mag1Δ mutant relative to a 0-h control. (C) Same as in part A, except the fraction of 7meG lesions remaining is plotted among ∼7500 weakly positioned nucleosomes (nucleosome score < 1). (D) Effect of rotational setting on 7meG repair occurs primarily 5′ of the nucleosome dyad. Data are plotted similarly to part A, except the plus and minus strands are shown individually and aligned in a 5′ to 3′ orientation. Bottom panel: weighted strand average of fraction of 7meG lesions remaining for the aligned plus and minus strands. (E) The effect of rotational setting on DNase-seq read density, a measure of chromatin accessibility, also is more prominent 5′ of the nucleosome dyad. Bottom panel: average DNA-seq reads for the aligned plus and minus strands. (F) At “Out” rotational settings (indicated with arrows), the 5′ half of the nucleosomal DNA (pink/red) faces the solvent, while the 3′ half of the nucleosomal DNA (green) faces the other DNA gyre. Image was created using PyMOL (http://www.pymol.org/) to visualize PDB ID 1kx5.
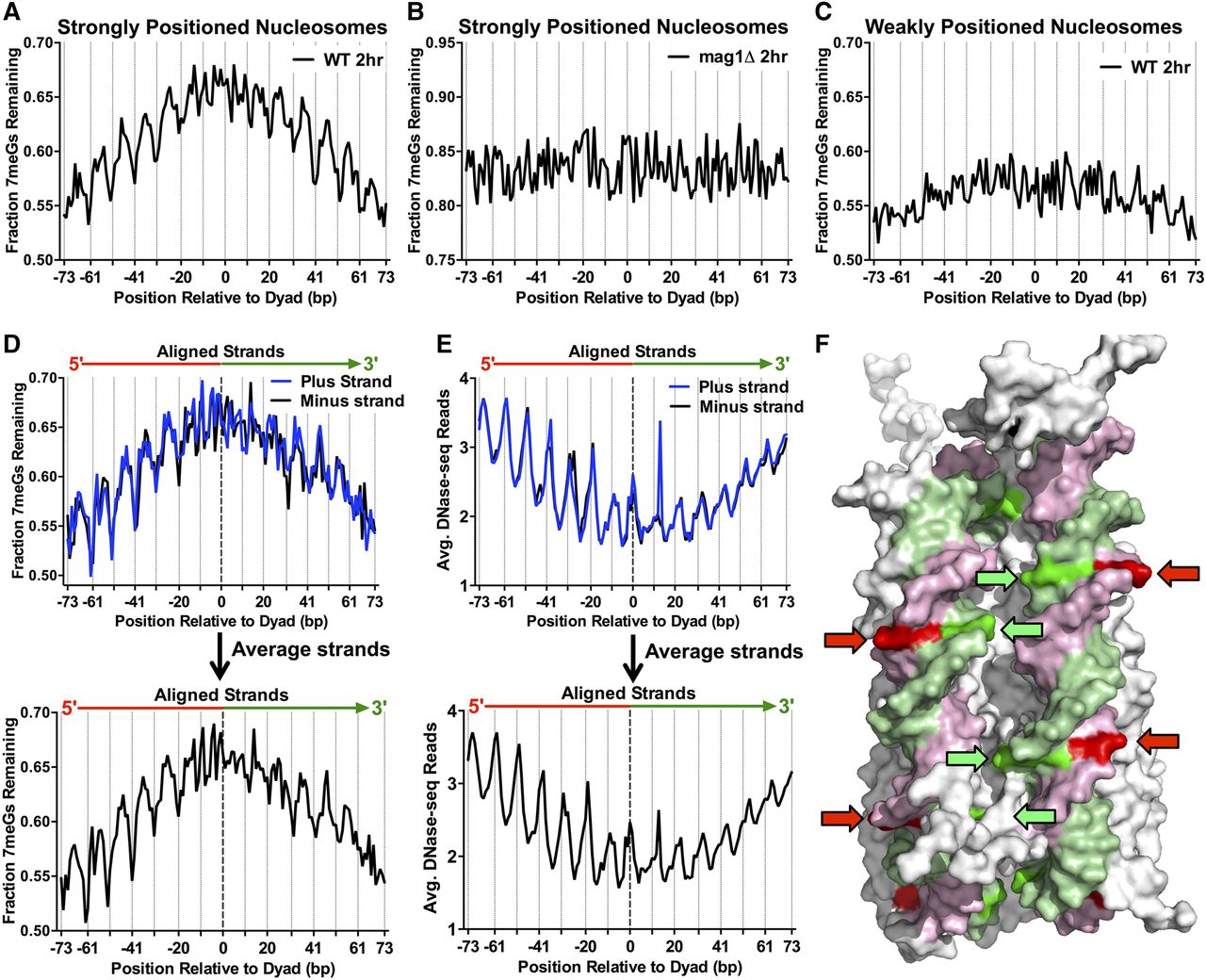
Analysis of ∼7500 weakly positioned nucleosomes in the yeast genome (nucleosome positioning score < 1) revealed a much less pronounced effect of translational positioning on repair, as there was only a slightly higher fraction of lesions remaining near the nucleosome dyad (Fig. 2C). Similarly, there was only a relatively small difference in 7meG lesions remaining at outward versus inward rotational settings within weakly positioned nucleosomes. These data indicate that the impact of nucleosomes on BER efficiency in vivo is dependent upon nucleosome positioning strength.
Analysis of 7meG removal for each individual DNA strand within the high-resolution Brogaard et al. nucleosome map (Brogaard et al. 2012) also revealed a striking asymmetry relative to the nucleosomal dyad (Fig. 2D). The fraction of unrepaired 7meG lesions following 2-h repair among strongly positioned nucleosomes in the yeast genome (Brogaard et al. 2012) was analyzed for each individual DNA strand aligned in a 5′ to 3′ orientation. For both the plus and minus strands, positions upstream (or 5′) of the nucleosome dyad showed a significant effect of rotational setting on the fraction of unrepaired 7meG lesions, while this effect was less pronounced at positions downstream (or 3′) from the nucleosome dyad (Fig. 2D). Quantification of the data confirmed that there was a higher relative difference in unrepaired 7meG lesions at “In” relative to “Out” rotational settings for positions 5′ of the nucleosome dyad relative to positions 3′ from the dyad (P < 0.05) (Supplemental Table S1). In parallel, we analyzed chromatin accessibility along the DNA strands within strongly positioned nucleosomes using a high-resolution yeast DNase-seq map (Zhong et al. 2016). There was a more pronounced effect of rotational setting on DNase I accessibility at positions 5′ of the nucleosome dyad relative to positions 3′ from the dyad (P < 0.05) (Fig. 2E; Supplemental Table S1), consistent with our 7meG repair data (and a recent study [Zhong et al. 2016]).
Closer examination of the data indicates that the rotational asymmetry across the dyad for both the repair and DNase I data sets was largely driven by differences at “Out” rotational settings. There was a significant difference in the fraction of unrepaired 7meG lesions for “Out” rotational settings 5′ of the dyad relative to the equivalent settings 3′ from the dyad (P < 0.05); in contrast, there was no significant difference in the fraction of 7meG lesions remaining at “In” rotational settings across the dyad (P > 0.05). A similar trend was observed for the DNase-seq data, as “Out” rotational settings 5′ of the nucleosome dyad were more accessible to DNase I cleavage than “Out” rotational settings 3′ from the dyad (P < 0.05), whereas there was no significant difference in accessibility at “In” rotational settings (P > 0.05). It has previously been suggested that this asymmetry in DNase I chromatin accessibility may be due to the orientation of the DNA strands relative to the other DNA gyre (Zhong et al. 2016). Inspection of the nucleosome structure supports this model, as DNA strands 5′ of the dyad consistently orient toward the solvent at “Out” rotational settings, while at the same locations DNA strands 3′ from the dyad consistently orient toward the other DNA gyre (Fig. 2F) and thus are less accessible to DNase I and BER factors such as Mag1.
The effect of translational positioning on BER efficiency is regulated by histone modifications
Histone post-translational modifications (PTMs) such as histone acetylation alter nucleosome dynamics (Neumann et al. 2009), and particular histone acetylation sites are associated with efficient NER (Yu et al. 2016); however, it is not known how histone acetylation or other histone PTMs affect BER. To address this question, we analyzed our NMP-seq data using a nucleosome-resolution map of steady-state histone modifications in yeast (Weiner et al. 2015). This study showed that yeast histone modifications can be clustered based on their genome-wide distribution into two major groups: (1) histone PTMs associated with the 5′ coding regions of yeast genes (e.g., H3K4me3, H3K9ac, H3K14ac, etc.), and (2) histone PTMs associated with the 3′ coding regions of yeast genes (H3K36me3, H3K79me3, etc.). Analysis of representative histone PTMs from each of these groups (i.e., H3K14ac and H3K36me3) revealed significant differences in BER of 7meG lesions among nucleosomes with high or low levels of these histone PTMs. There was a smaller fraction of unrepaired 7meG lesions following 2-h repair at distal translational positions in nucleosomes with high levels of H3K14ac relative to nucleosomes with low levels of H3K14ac, while there was a relatively higher fraction of unrepaired 7meG lesions near the nucleosome dyad (Fig. 3A). A similar trend was observed for a number of other histone PTMs associated with group 1 (e.g., H3K4me3, H3K23ac, H4K5ac) (Supplemental Fig. S6A–C). These results suggest that high levels of pre-existing histone acetylation are associated with more rapid BER at distal translational locations within nucleosomes and paradoxically slower repair at translational positions near the nucleosome dyad.
Histone post-translational modifications (PTMs) alter 7meG repair within nucleosomes. (A) Fraction of 7meG lesions remaining following 2-h repair in WT relative to 0-h mag1Δ control among top 10,000 nucleosomes with highest levels of H3K14 acetylation (High H3K14ac) and 10,000 nucleosomes with the lowest levels of H3K14 acetylation (Low H3K14ac), based on data from Weiner et al. (2015), is plotted relative to the distance from the central dyad axis of the nucleosome (“dyad”). Vertical dotted lines indicate “Out” rotational settings. Nucleosomes overlapping with the MAG1 gene, which is deleted in the mag1Δ control strain, were excluded from this analysis. (B) Same as in part A, except for H3K36 trimethylation (H3K36me3). (C) Same as in part A, except for H3K79 monomethylation (H3K79me1).
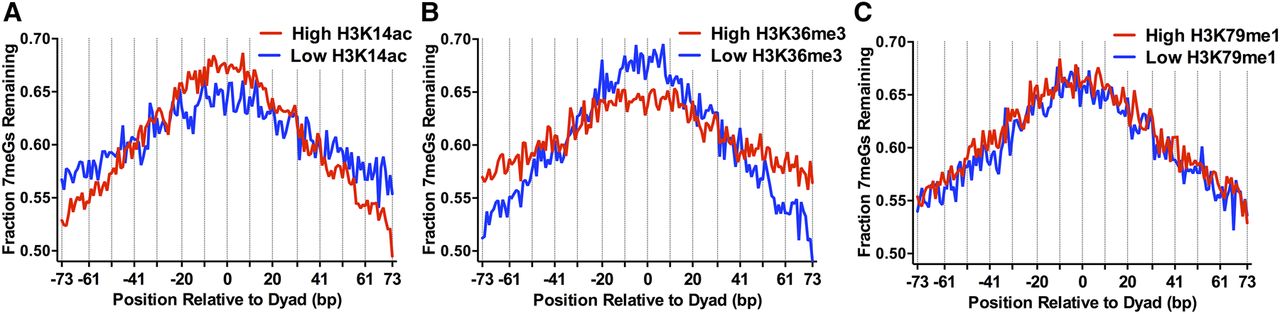
The opposite trend was observed for nucleosomes with high levels of group 2 histone PTMs (i.e., H3K36me3). Nucleosomes with high levels of H3K36me3 had a higher fraction of 7meG lesions remaining at distal translational locations relative to the low H3K36me3 controls following 2-h repair but a lower fraction of unrepaired 7meG lesions near the nucleosome dyad (Fig. 3B). Roughly similar trends were observed for a number of other group 2 histone PTMs (e.g., H3K36me2, H3K79me3) (Supplemental Fig. S6D,E). These results may in part be due to the negative correlation between H3K36me3 and histone acetylation (Weiner et al. 2015), since H3K36 methylation functions to recruit histone deacetylase complexes to the 3′ coding region of yeast genes (Lee and Shilatifard 2007).
However, not all histone modifications were associated with distinct patterns of repair. For example, there was no apparent difference in the pattern of unrepaired 7meG lesions between nucleosomes with high and low levels of H3K79 monomethylation (H3K79me1) (Fig. 3C). While pre-existing histone PTMs were associated with distinct patterns of repair associated with translational positioning in nucleosomes, the effect of rotational setting on repair was not apparent in this nucleosome map. This is likely due to the relatively low precision of the MNase-ChIP-seq method in defining nucleosome dyad positions (Weiner et al. 2015).
MMS-induced mutations are enriched near the dyad of strongly positioned nucleosomes in yeast
Since nucleosome structure causes such striking effects on the repair of NMPs in vivo, we next sought to determine whether the decreased repair efficiency within strongly positioned nucleosomes ultimately resulted in an increase in MMS-induced mutation density in these regions. We therefore subjected WT and mag1Δ yeast to 10 repeated 2-d exposures of 0.016% MMS to accumulate MMS-induced mutations and subsequently sequenced the genomes of 24 independent clonal isolates of each genotype (Fig. 4A) to determine the location and spectra of the acquired mutations. Treatment of WT and mag1Δ yeast with a single 2-d exposure to 0.016% MMS increased CanR mutation frequencies ∼71- and ∼450-fold over untreated yeast, respectively (P = 0.0022 for both by Mann-Whitney U test) (Fig. 4B), indicating that greater than 98.6% and 99.8%, respectively, of accumulated mutations in the MMS-treated strains are the result of MMS-induced DNA lesions. Whole-genome sequencing of MMS-treated WT yeast revealed that, in accordance with previously published MMS-induced mutation spectra (Roberts et al. 2012), nearly equal numbers of mutations occurred at “A” nucleotides compared to “G” nucleotides (∼59% and ∼41%, respectively) (see Fig. 4C), despite 7meG lesions being formed approximately eightfold more frequently than 3meA. This result is consistent with prior studies (Roberts et al. 2012) and highlights the greater mutagenicity of 3meA lesion relative to 7meG (Shrivastav et al. 2010). Compared to WT yeast, mag1Δ yeast displayed a much higher overall number of mutations per genome (Fig. 4D) and a greater number of A-mutations relative to G-mutations (∼86% and ∼14%, respectively, P < 0.0001) (Fig. 4C), indicating that MMS-treatment is inducing mutagenic NMP lesions that are normally repaired by Mag1 and highlighting the greater efficiency of Mag1 in removing 3meA lesions (Bjoras et al. 1995).
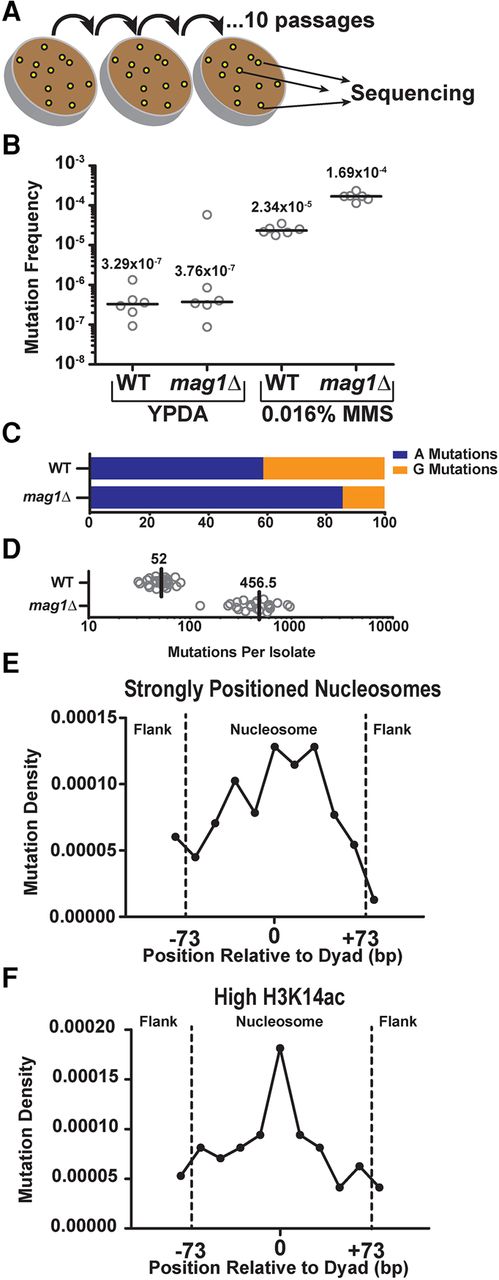
Analysis of MMS-induced mutations across the genome of WT and mag1Δ yeast. (A) Individual isolates (n = 24) of WT and mag1Δ yeast strains were grown on 0.016% MMS-supplemented yeast extract peptone dextrose adenine (YPDA) media at 30°C for 48 h. Separate lines were then streaked for singles (not explicitly shown) on additional MMS plates, for a total of 10 passages. At the end of the passaging, unique isolates were picked for whole-genome sequencing. (B) CanR frequencies were measured for six independent isolates of WT and mag1Δ strains of yeast (open circles), which were either untreated (grown on YPDA media) or treated for 2 d with 0.016% MMS. Black horizontal bars and numbers indicate the median frequency of CanR for each genotype and treatment. Data were analyzed using a two-tailed Mann-Whitney U test. (C) Percentage of A- and G-mutations in whole-genome sequencing of MMS-treated mag1Δ and WT strains. (D) Number of mutations per isolate from whole-genome sequencing of MMS-treated WT and mag1Δ strains. (E) Strongly positioned nucleosomes have elevated mutation density relative to flanking DNA regions in WT yeast. Mutations from MMS-treated WT isolates were mapped to strongly positioned nucleosomes (score > 5) and normalized by sequence context. An area of ∼180 bp was examined, which consisted of the main nucleosome (147 bp) and an additional flank on either side (∼30 bp total). Each DNA sequence was then divided into 11 bins, each of ∼16 bp. Comparisons of the number of mutations observed between nucleosome-bound and flanking DNA was performed using χ2 analysis. (F) Same as in part E, except mutation density in WT yeast was analyzed for 10,000 nucleosomes with the highest levels of H3K14 acetylation (H3K14ac) (Weiner et al. 2015).
Analysis of the trinucleotide sequence context in which MMS-induced mutations and lesions occurred (Supplemental Figs. S7, S8) indicated that the relative abundances of G- and A-mutations, as well as 7meG and 3meA lesions, strongly correlated with the trinucleotide composition of the yeast genome (P ≤ 0.002; Pearson coefficient > 0.75 for independent comparisons of G-mutation to G bases in WT yeast and A-mutations to A bases in mag1Δ by Pearson correlation tests; P = 0.042; Pearson coefficient = 0.51 for A-mutations compared to A bases in WT yeast), indicating that MMS-induced lesions and mutations occur largely independent of sequence specificity. Additionally, both 7meG- and 3meA-induced mutations produce biased base substitutions (favoring G to A or T and A to G) and occur with little to no sequence specificities (Supplemental Fig. S8).
We next determined the frequency of mutations occurring within ∼10,000 strongly positioned nucleosomes and their surrounding linker DNA, normalizing the number of mutations observed across the nucleosome by the relevant sequence content of A and G bases. We found that, similar to the patterns observed with NMP-seq data (Supplemental Fig. S5A), strongly positioned nucleosomes had significantly higher mutation densities compared to the flanking linker DNA (P = 0.0149), with mutation density peaking near the dyad axis (Fig. 4E; Supplemental Fig. S9B). In contrast, mutation densities in weakly positioned nucleosomes (∼7500 nucleosomes) displayed no significant difference (P = 0.2935) between positions near the nucleosome dyad and those in the flanking linker DNA (Supplemental Fig. S9A,B). These findings indicate that reduced BER efficiency in strongly positioned nucleosomes, particularly near the dyad axis, is a key determinant of MMS-induced mutation density in vivo.
We also examined mutation density among nucleosomes with differing levels of histone PTMs, since our NMP-seq data indicated that histone PTMs such as H3K14ac and H3K36me3 significantly altered the pattern of repair of NMP lesions within nucleosomes. Consistent with the repair data (Fig. 3B), nucleosomes with low levels of H3K36me3 showed a peak of mutation density near the dyad axis (Supplemental Fig. S9C). In contrast, the effect of translational positioning on mutation density was less apparent for nucleosomes with high levels of H3K36me3 (Supplemental Fig. S9D). A similar trend was observed for nucleosomes with high levels of H3K14ac (Fig. 4F), again consistent with our repair data (Fig. 3A), although the difference between nucleosomes with high and low H3K14ac was less clear (cf. Fig. 4F and Supplemental Fig. S9E). These findings suggest that variations in repair efficiency in nucleosomes due to histone PTMs can influence mutation rates.
MMS-induced A-mutations are enriched on the NTS of yeast genes and are associated with higher levels of 3meA lesions
Analysis of MMS-induced mutations at adenine nucleotides (i.e., A to G, A to C, or A to T mutations) revealed a novel asymmetry in mutation density between the TS and NTS of yeast genes. This effect was particularly apparent in the MMS-treated mag1Δ strain, as there were fivefold more A-mutations on the NTS relative to the TS in yeast coding sequences, even after normalizing for the frequency of “A” nucleotides in the TS and NTS (P < 0.0001 by χ2 test; Fig. 5A). In MMS-treated WT yeast, we also observed a significant enrichment of A-mutations occurring on the NTS relative to the TS (P = 0.0023) (Fig. 5B), although the magnitude of the enrichment was smaller than in the mag1Δ strain. In contrast, flanking promoter and downstream DNA lacked a significant strand asymmetry in the mag1Δ (P = 0.9014) (Fig. 5A) and wild-type strains (P = 0.5473). Surprisingly, the densities of A-mutations in both the TS and NTS in the mag1Δ strain significantly differed from that of the promoter and downstream flanking regions, with A-mutations being elevated in the NTS and reduced in the TS relative to flanking DNA (Fig. 5A). This suggests that two independent processes occurring on the NTS and TS may contribute to the transcriptional asymmetry of MMS-induced mutations.
MMS-induced A-mutations are enriched on the nontranscribed strand (NTS) and depleted from the transcribed strand (TS) of yeast genes, particularly in a BER-deficient (mag1Δ) strain. (A,B) The density of mutations of “A” nucleotides in the MMS-treated (A) mag1Δ and (B) WT strains was plotted for 5762 yeast genes. The number of A-mutations in the transcribed strand (TS; orange) and nontranscribed strand (NTS; blue) was determined for bins spanning 501 bp upstream of the transcription start site (TSS), the transcribed region of the gene, and 501 bp downstream from the transcription termination site (TTS), and normalized by the number of A nucleotides in each bin. (C,D) 3-methyladenine (3meA) lesions are elevated on the NTS immediately following MMS-treatment (i.e., 0 h) in (C) WT and (D) mag1Δ strains. The average number of 3meA lesions per A nucleotide was plotted for bins spanning 5762 yeast genes and flanking promoter and downstream DNA sequences, as described in part A. (E,F) Levels of 3meA lesions are preferentially depleted from the TS in a mag1Δ strain during a repair time course. The average number of 3meA lesions per A nucleotide in the mag1Δ (E) 1-h and (F) 2-h time points was plotted as described in part C.
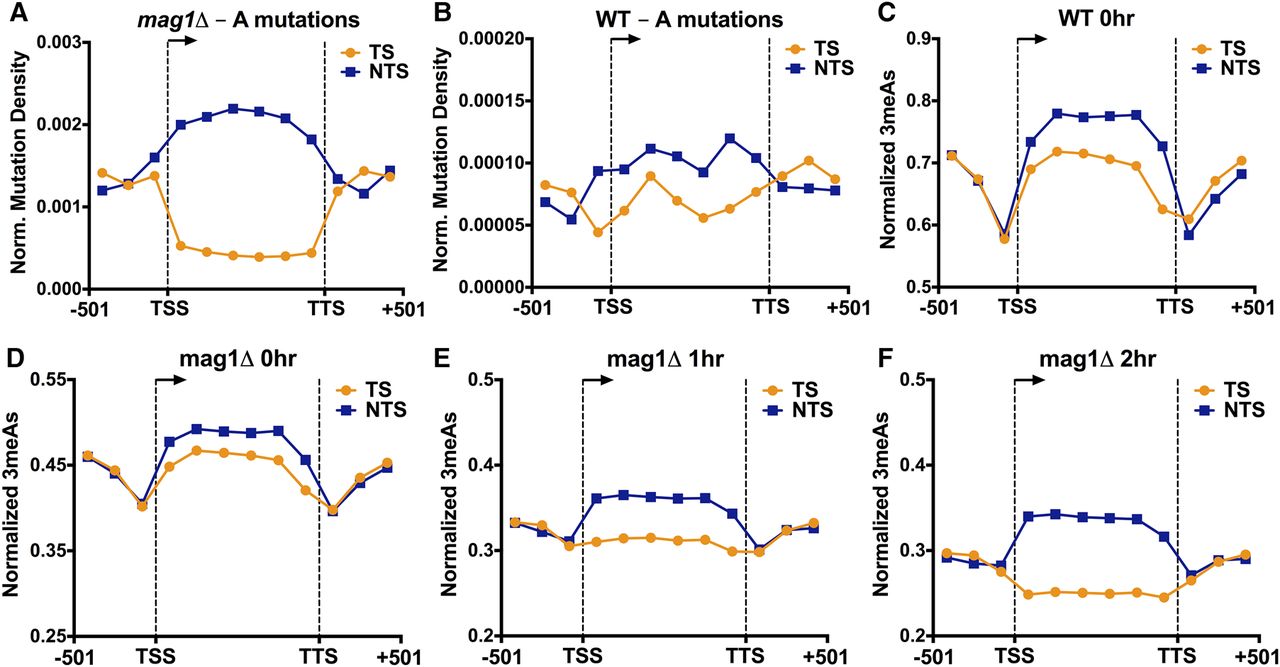
To investigate the molecular mechanisms underlying the strand asymmetry in MMS-induced A-mutations, we first analyzed the initial formation of 3meA lesions in our NMP-seq data sets. While the levels of 3meA lesions were much lower than 7meG lesions overall, we consistently observed higher levels of 3meA lesions on the NTS of yeast genes relative to the TS immediately following MMS treatment (i.e., WT 0-h and mag1Δ 0-h samples) (see Fig. 5C,D). The magnitude of this difference was relatively small, ranging from 6%–11% more 3meA lesions on the NTS relative to the TS after normalizing for the frequency of A nucleotides on the NTS and TS, but was consistent across multiple experiments (Supplemental Fig. S10A–C). There were also consistently higher 3meA lesions on the NTS relative to flanking DNA (∼11%–18% more 3meA lesions) (see Fig. 5C,D). These differences were magnified in a mag1Δ strain treated with a chronic dose of 0.02% MMS for 3 h (∼24% more 3meA lesions on the NTS relative to the TS and ∼17% more 3meA lesions on the NTS relative to flanking DNA) (see Supplemental Fig. S10D), experimental conditions that more closely mimic the mag1Δ MMS mutation experiments. These results indicate that higher 3meA lesion formation on the NTS of yeast genes likely contributes to transcriptional asymmetry in MMS-induced A-mutations.
3meA lesions are preferentially repaired on the TS in a BER-deficient yeast strain
While strand-specific differences in 3meA lesion formation can explain the elevated levels of A-mutations on the NTS, this mechanism cannot explain the significantly lower levels of A-mutations on the TS relative to flanking DNA, since initial levels of 3meA lesions on the TS were generally higher than flanking DNA (Fig. 5C,D). However, analysis of 3meA lesions in subsequent repair time points (i.e., 1- and 2-h) in the mag1Δ strain revealed a progressive loss of 3meA lesions on the TS, both relative to the NTS and flanking DNA (Fig. 5E,F). To compare repair efficiency between TS and NTS, we analyzed the fraction of 3meA lesions remaining at the 1- and 2-h repair time points in the mag1Δ strain relative to initial damage levels (i.e., mag1Δ 0-h). While overall 3meA lesions decreased in the mag1Δ strain following a 1- or 2-h repair incubation, presumably reflecting ongoing DNA replication and trans-lesion synthesis in the mag1Δ strain (Johnson et al. 2007), the levels of 3meA lesions decreased more rapidly on the TS relative to the NTS or flanking DNA (Fig. 6A,B). While the transcribed region showed a clear strand-specific difference in 3meA repair, there was no difference in 3meA removal in the flanking promoter and downstream sequences (Fig. 6A,B). Moreover, there was no strand-specific difference in 7meG repair in the mag1Δ strain at the 1- and 2-h repair time points (Fig. 6C; Supplemental Fig. S11), indicating the observed repair activity is specific for 3meA lesions. These results indicate that in a BER-deficient strain there is preferential repair of 3meA lesions on the TS of yeast genes, providing a potential explanation for the significantly lower levels of A-mutations on the TS in the MMS-treated mag1Δ strain.
Transcription-coupled repair of 3meA lesions in a BER-deficient (mag1Δ) strain. (A,B) Fraction of 3meA lesions remaining in a mag1Δ 1- and 2-h sample relative to mag1Δ 0-h control was plotted for bins spanning 501 bp upstream of the transcription start site (TSS), the transcribed region of the gene, and 501 bp downstream from the transcription termination site (TTS) for 5762 yeast genes. (C) No transcription-coupled repair of 7meG lesions in a mag1Δ strain. Same as in part A, except the fraction of 7meG lesions remaining following 2-h repair in a mag1Δ strain was plotted. (D) MMS sensitivity of yeast strains containing single or double mutants in MAG1 or the indicated NER genes (i.e., RAD16, RAD26, and RAD14) when spotted on plates containing the indicated concentrations of MMS.
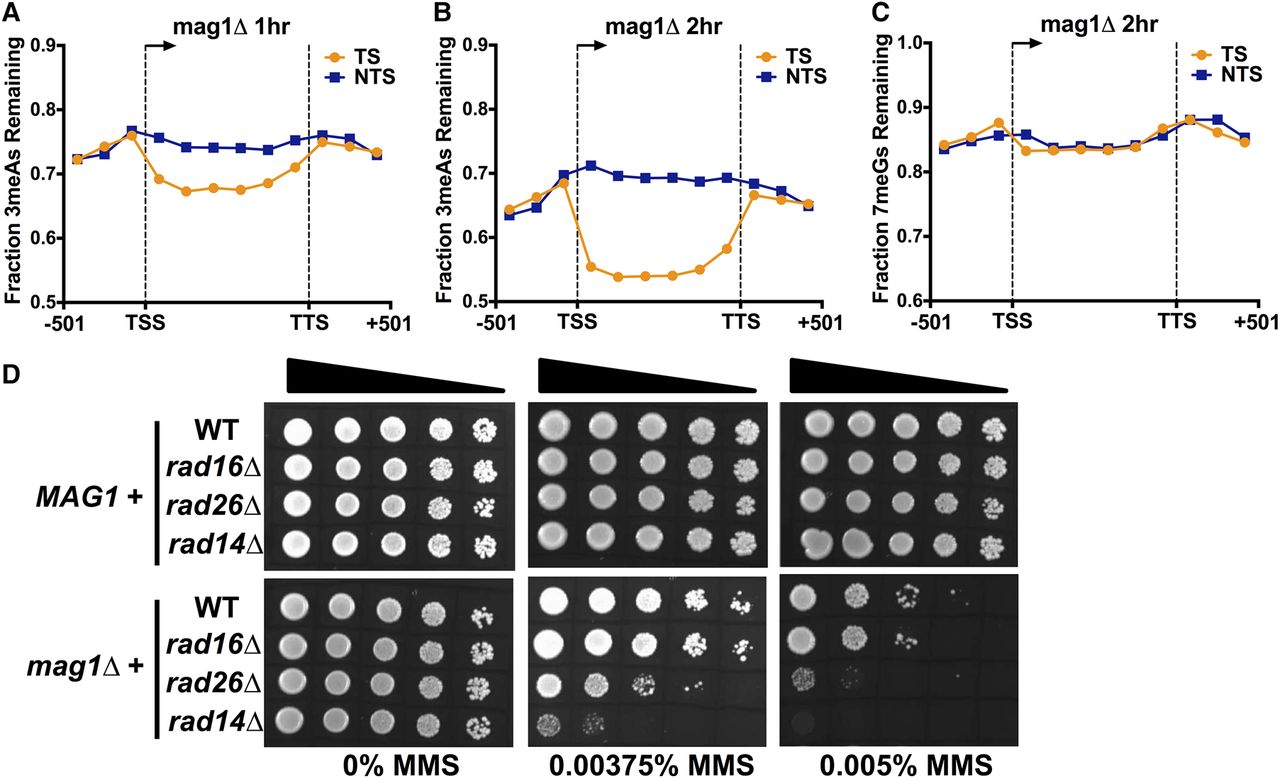
Based on these results, we hypothesized that transcription-coupled nucleotide excision repair (TC-NER) functions as a backup repair pathway to preferentially remove 3meA lesions from the TS of yeast genes to prevent MMS-induced mutagenesis and cytoxicity in BER-deficient cells. To test this hypothesis, we analyzed the MMS sensitivity of yeast mutants lacking both BER (mag1Δ) and TC-NER (rad26Δ) pathways. While the mag1Δ mutant was sensitive to MMS, as expected, none of the NER-deficient mutants (i.e., rad14Δ, rad16Δ, and rad26Δ) were sensitive to low doses of MMS when BER was functional (Fig. 6D). However, the mag1Δrad26Δ double mutant showed enhanced MMS sensitivity (Fig. 6D), consistent with a previous study (Lee et al. 2002), indicating that TC-NER may function in NMP removal in a BER-deficient strain. In contrast, deletion of RAD16, which causes a specific defect in the global genome nucleotide excision repair (GG-NER) pathway and is highly sensitive to UV irradiation (Supplemental Fig. S12) but does not affect TC-NER, did not show elevated MMS sensitivity in a mag1Δ background (Fig. 6D). Deletion of RAD14, which eliminates both TC-NER and GG-NER, also significantly enhanced the MMS sensitivity of a mag1Δ strain to an even greater extent than the mag1Δrad26Δ double mutant (Fig. 6D). Previous studies have shown that deletion of RAD26 does not completely eliminate TC-NER in yeast (Li and Smerdon 2002b), which may explain the higher MMS sensitivity in the mag1Δrad14Δ double mutant. These data are consistent with the model that the Rad26-mediated TC-NER pathway specifically repairs cytotoxic and mutagenic 3meA lesions on the TS of yeast genes.
Discussion
While efficient repair of DNA base lesions is critical to maintaining genome integrity and preventing mutations, the impact of chromatin and other genomic features on BER and mutagenesis in vivo is not well understood. Here, we utilized a novel method known as NMP-seq to map the initial formation and repair of DNA alkylation damage across the yeast genome at single nucleotide resolution. Our results indicate that BER of MMS-induced alkylation damage is significantly modulated by cellular chromatin, with slower repair of lesions at translational positions near the nucleosome dyad and at inward rotational settings. Furthermore, we show that the effect of translational and rotational positioning on repair is asymmetric relative to the nucleosome dyad and significantly modulated by histone PTMs. Importantly, slower repair within strongly positioned nucleosomes correlated with an increased frequency of MMS-induced mutations in yeast, indicating that chromatin-associated variations in BER efficiency can impact mutation rates in vivo. Finally, we find that MMS-induced mutations at adenine nucleotides show a striking DNA strand asymmetry among yeast genes, particularly in the absence of BER, with a significantly higher mutation frequency on the NTS relative to the TS. We show that this is due to higher initial 3meA formation on the NTS of yeast genes and preferential removal of 3meA lesions from the TS. In summary, our data reveal that chromatin structure, backup DNA repair activities, and transcription are prominent sculptors of mutation distributions associated with alkylation DNA base damage.
We find that repair of 7meG lesions in cellular chromatin is significantly modulated by the translational and rotational setting of the lesion, with faster repair of lesions at “Out” rotational settings and translational positions distal from the nucleosome dyad, including linker DNA, and slower repair at “In” rotational settings and translational positions near the dyad. While these findings are generally consistent with in vitro studies of BER in “designed” nucleosomes (Hinz and Czaja 2015; Rodriguez et al. 2015), the effects of translational and rotational setting on BER efficiency have not been previously demonstrated in vivo (Li and Smerdon 2002a; Li et al. 2015). These nucleosome-dependent differences in BER efficiency translate to a striking pattern of BER surrounding yeast genes. Peaks of unrepaired 7meG lesions correlate with the stereotypic positioning of nucleosomes downstream from the TSS (e.g., the +1 nucleosome, +2 nucleosome, etc.), while repair of 7meG lesions occurs much more rapidly in NDRs upstream of the TSS. Furthermore, we show that the effects of rotational setting on BER efficiency are primarily confined to the 5′ half of each nucleosomal DNA strand due to strand-specific differences in chromatin accessibility at “Out” rotational settings. This asymmetry has not previously been detected in biochemical studies of BER in nucleosomes but could be tested using defined nucleosome substrates in vitro.
Histone PTMs have been shown to regulate other repair pathways (e.g., NER and DNA double-strand break repair), but their role in BER is unclear (Mao and Wyrick 2016). Our analysis indicates that pre-existing histone PTMs cause subtle but significant differences in BER efficiency among nucleosomes in vivo. For example, high levels of histone acetylation, such as H3K14ac, are associated with faster repair at distal translational positions near the DNA exit/entry sites but paradoxically slower repair near the nucleosome dyad. Histone acetylation is often associated with increased nucleosomal DNA unwrapping, particularly at distal translational positions (Neumann et al. 2009), which could explain the faster repair of 7meG lesions at these positions in highly acetylated nucleosomes. While slower repair near the nucleosome dyad among highly acetylated nucleosomes seems counterintuitive, a recent report suggests that H3K14ac and other histone acetylations (i.e., H3K56ac) inhibit DNA polymerase β activity during repair of lesions located near the dyad of nucleosome substrates in vitro (Rodriguez et al. 2016). The opposite trend was observed for histone PTMs associated with the 3′ coding regions of yeast genes (e.g., H3K36me3), presumably because these histone PTMs are associated with histone deacetylation (Lee and Shilatifard 2007; Weiner et al. 2015). While our data indicate that MMS-induced mutations in wild-type yeast are significantly elevated near the nucleosome dyad relative to flanking regions among strongly positioned nucleosomes, this effect appears to be magnified among nucleosomes with high levels of H3K14ac or correspondingly low levels of H3K36me3. Notably, in wild-type yeast, MMS-induced mutagenesis mirrors the effect of translational positioning on BER efficiency within these nucleosomes, suggesting that differences in repair due to histone PTMs may be an important contributor to mutation rates. It would be interesting to determine to what extent this mechanism regulates mutation rate in human cancers, since it is known that certain histone PTMs correlate with mutation density in sequenced cancer genomes (Schuster-Bockler and Lehner 2012).
In the course of analyzing MMS-induced mutations, we discovered a significantly higher frequency of mutations at adenine nucleotides on the NTS relative to the TS of yeast genes. This DNA strand asymmetry was magnified in the BER-deficient mag1Δ strain, suggesting that rapid removal of 3meA lesions by Mag1 suppresses this effect. Our data suggest that two distinct mechanisms contribute to this novel transcriptional asymmetry.
First, there is higher initial 3meA formation on the NTS of yeast genes, even after normalizing for differences in DNA sequence context. Previous studies have suggested that ongoing transcription can render the NTS more susceptible to chemical modifications, as the NTS becomes transiently single-stranded during transcription-associated R-loop formation (Kim and Jinks-Robertson 2012); it is possible that a similar mechanism contributes to the higher frequency of 3meA formation and A-mutations on the NTS of yeast genes. However, single-stranded DNA also transiently accumulates on the lagging strand during DNA replication and A-mutations were not enriched on the lagging strand in mag1Δ yeast, instead showing a slight (but significant) enrichment on the leading strand (Supplemental Fig. S13). Thus, either mutagenesis associated with increased formation of 3meA on single-stranded DNA occurs in a transcription-specific context or other mutagenic processes occurring during leading strand synthesis (possibly strand bias in the usage of error-free polymerases) obscure this mutation signature at the replication fork.
Second, 3meA lesions are preferentially repaired from the TS of yeast genes. This effect was apparent in a mag1Δ mutant strain, indicating that TC-NER may serve as a backup strategy to remove mutagenic and cytotoxic 3meA lesions from the TS of yeast genes. Previous studies have suggested that NER can serve as a potential backup repair pathway for DNA alkylation damage in the absence of functional BER in a variety of organisms, including yeast (Fu et al. 2012); however, the mechanism underlying this effect was unclear. Our data are consistent with the model that 3meA lesions are specifically removed from the TS by TC-NER, which, coupled with higher initial 3meA formation on the NTS, comprise a novel mechanism for transcription-associated mutation asymmetry due to DNA base damage. The TC-NER factor Rad26, which is a homolog of the human CSB gene (Friedberg et al. 2006), appears to play an important role, although Rad26 has also been reported to have TC-NER-independent functions in cell survival following acute alkylation damage (Lee et al. 2002). 3meA lesions are known to stall replicative DNA polymerases (Plosky et al. 2008) and have been reported to inhibit T7 phage RNA polymerase (Racine et al. 1993); however, it is not known to what extent this occurs with eukaryotic RNA polymerases. We hypothesize that persistent 3meA lesions may trigger TC-NER in yeast by stalling RNA polymerase II, implying that TC-NER may play a broader role in repairing nonbulky DNA lesions than previously appreciated. While our data suggest that TC-NER repairs certain classes of alkylation damage in yeast, it is not clear if TC-NER plays a similar role in mammalian cells. A previous study reported that there was no strand bias in the repair of 3meA lesions at the DHFR gene in a BER-deficient murine cell line lacking AAG (Plosky et al. 2002), even though NER contributes to NMP lesion removal at this locus. It will be important to analyze the genome-wide repair of alkylation damage in human cells, since these findings may have important implications for understanding the molecular basis of transcriptional asymmetries in mutations found in many human cancers (Alexandrov et al. 2013; Roberts and Gordenin 2014).
Methods
MMS treatment, NMP-seq library preparation, and sequencing
Yeast cells (BY4741) were grown in YPD (yeast extract, peptone, dextrose) medium to OD600 ≈ 0.8. MMS (Sigma-Aldrich) was added to the yeast culture to a final concentration of 0.2% (v/v) or 0.4%, and incubated at 30°C for 10 min with shaking to induce alkylation damage. Cell pellets were spun down and washed twice with sterile deionized water to remove remaining MMS. Aliquots were taken before adding MMS and immediately after MMS incubation for “No MMS” and “MMS 0-h” samples, respectively. The remainder of the cells was resuspended in prewarmed YPD medium and incubated in a 30°C shaker, and samples were collected at different repair times as indicated. For chronic MMS treatment, yeast cells (OD600 ≈ 0.6) were grown in YPD containing 0.02% MMS for 3 h. MMS was then removed, and cells were collected for DNA isolation.
Yeast genomic DNA was isolated as previously described (Mao et al. 2016). The procedure of NMP-seq library preparation is similar to our published CPD-seq method (Mao et al. 2016), except that damaged DNA was incubated with recombinant human AAG glycosylase (AAG 1-79Δ, a gift from Dr. Leona Samson at MIT) to cleave NMPs. The resulting abasic sites were further processed by incubating DNA with recombinant AP endonuclease APE1 (NEB) to generate new 3′OHs at NMP sites. The resulting library was amplified for approximately five cycles by PCR, using primers complementary to trP1 and A, size-selected with AMpure XP beads, and sequenced on an Ion Torrent Proton platform (Life Technologies). Validation of the NMP-seq library, including trP1 ligation, free 3′OH blocking, and A adaptor ligation, was conducted as previously described for CPD-seq damage mapping (Mao et al. 2016). DNA adaptor and primer sequences are provided in the Supplemental Methods.
NMP-seq data analysis
NMP-seq data analysis was performed using a modified version of the published protocol for analyzing CPD-seq data (Mao et al. 2016). A detailed description of the data analysis methods is provided in the Supplemental Methods.
Accumulation of MMS-induced mutations in yeast
Diploid wild-type or mag1Δ yeast (BY4743) accumulated mutations by being repeatedly passaged on YPDA media containing 0.016% MMS. Briefly, individual colonies were streaked for singles on YPDA supplemented with 0.016% MMS, then allowed to grow for 48 h. Then, a single colony unique to each streak was picked and struck out onto another MMS-containing YPDA plate. This was repeated for 10 passages. Total genomic DNA was purified from 24 independent WT and mag1Δ isolates, sequenced, and mutations called similar to Sakofsky et al. (2014). A detailed description of these methods and the analysis of the mutation distribution are provided in the Supplemental Methods. All mutations identified are listed in Supplemental Table S2.
Data access
The NMP-seq data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE98031. The MMS-induced mutation sequencing data from this study have been submitted to the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession number SRP105152.
Acknowledgments
We thank Mark Wildung and Wei Wei Du for technical assistance with Ion Proton sequencing and Amelia Hodges for helpful comments and suggestions. This research was supported by grants from the National Institute of Environmental Health Sciences (NIEHS) (R01ES002614 to J.J.W. and M.J.S., R03ES027945 to P.M., and R00ES022633 to S.A.R.), Department of Defense Congressionally Directed Medical Research Programs (BC141727 to S.A.R.), and an internal grant from the Washington State University College of Veterinary Medicine (to J.J.W.).
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.225771.117.
References
- ↵Akbari M, Morevati M, Croteau D, Bohr VA. 2015. The role of DNA base excision repair in brain homeostasis and disease. DNA Repair 32: 172–179.
- ↵Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, 2013. Signatures of mutational processes in human cancer. Nature 500: 415–421.
- ↵Bauer NC, Corbett AH, Doetsch PW. 2015. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res 43: 10083–10101.
- ↵Bjoras M, Klungland A, Johansen RF, Seeberg E. 1995. Purification and properties of the alkylation repair DNA glycosylase encoded the MAG gene from Saccharomyces cerevisiae. Biochemistry 34: 4577–4582.
- ↵Brogaard K, Xi L, Wang JP, Widom J. 2012. A map of nucleosome positions in yeast at base-pair resolution. Nature 486: 496–501.
- ↵Bryan DS, Ransom M, Adane B, York K, Hesselberth JR. 2014. High resolution mapping of modified DNA nucleobases using excision repair enzymes. Genome Res 24: 1534–1542.
- ↵Cui F, Cole HA, Clark DJ, Zhurkin VB. 2012. Transcriptional activation of yeast genes disrupts intragenic nucleosome phasing. Nucleic Acids Res 40: 10753–10764.
- ↵Ding J, Taylor MS, Jackson AP, Reijns MA. 2015. Genome-wide mapping of embedded ribonucleotides and other noncanonical nucleotides using emRiboSeq and EndoSeq. Nat Protoc 10: 1433–1444.
- ↵Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. 2006. DNA repair and mutagenesis. ASM Press, Washington, DC.
- ↵Fu D, Calvo JA, Samson LD. 2012. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer 12: 104–120.
- ↵Hanawalt PC, Spivak G. 2008. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol 9: 958–970.
- ↵Hinz JM, Czaja W. 2015. Facilitation of base excision repair by chromatin remodeling. DNA Repair 36: 91–97.
- ↵Hu J, Adar S, Selby CP, Lieb JD, Sancar A. 2015. Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes Dev 29: 948–960.
- ↵Jiang C, Pugh BF. 2009. A compiled and systematic reference map of nucleosome positions across the Saccharomyces cerevisiae genome. Genome Biol 10: R109.
- ↵Johnson RE, Yu SL, Prakash S, Prakash L. 2007. A role for yeast and human translesion synthesis DNA polymerases in promoting replication through 3-methyl adenine. Mol Cell Biol 27: 7198–7205.
- ↵Kim N, Jinks-Robertson S. 2012. Transcription as a source of genome instability. Nat Rev Genet 13: 204–214.
- ↵Lee JS, Shilatifard A. 2007. A site to remember: H3K36 methylation a mark for histone deacetylation. Mutat Res 618: 130–134.
- ↵Lee SK, Yu SL, Prakash L, Prakash S. 2002. Yeast RAD26, a homolog of the human CSB gene, functions independently of nucleotide excision repair and base excision repair in promoting transcription through damaged bases. Mol Cell Biol 22: 4383–4389.
- ↵Li S, Smerdon MJ. 2002a. Nucleosome structure and repair of N-methylpurines in the GAL1-10 genes of Saccharomyces cerevisiae. J Biol Chem 277: 44651–44659.
- ↵Li S, Smerdon MJ. 2002b. Rpb4 and Rpb9 mediate subpathways of transcription-coupled DNA repair in Saccharomyces cerevisiae. EMBO J 21: 5921–5929.
- ↵Li M, Ko T, Li S. 2015. High-resolution digital mapping of N-methylpurines in human cells reveals modulation of their induction and repair by nearest-neighbor nucleotides. J Biol Chem 290: 23148–23161.
- ↵Mao P, Wyrick JJ. 2016. Emerging roles for histone modifications in DNA excision repair. FEMS Yeast Res 16: fow090.
- ↵Mao P, Smerdon MJ, Roberts SA, Wyrick JJ. 2016. Chromosomal landscape of UV damage formation and repair at single-nucleotide resolution. Proc Natl Acad Sci 113: 9057–9062.
- ↵Mao P, Wyrick JJ, Roberts SA, Smerdon MJ. 2017. UV-induced DNA damage and mutagenesis in chromatin. Photochem Photobiol 93: 216–228.
- ↵Nag R, Smerdon MJ. 2009. Altering the chromatin landscape for nucleotide excision repair. Mutat Res 682: 13–20.
- ↵Neumann H, Hancock SM, Buning R, Routh A, Chapman L, Somers J, Owen-Hughes T, van Noort J, Rhodes D, Chin JW. 2009. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol Cell 36: 153–163.
- ↵Plosky B, Samson L, Engelward BP, Gold B, Schlaen B, Millas T, Magnotti M, Schor J, Scicchitano DA. 2002. Base excision repair and nucleotide excision repair contribute to the removal of N-methylpurines from active genes. DNA Repair 1: 683–696.
- ↵Plosky BS, Frank EG, Berry DA, Vennall GP, McDonald JP, Woodgate R. 2008. Eukaryotic Y-family polymerases bypass a 3-methyl-2′-deoxyadenosine analog in vitro and methyl methanesulfonate-induced DNA damage in vivo. Nucleic Acids Res 36: 2152–2162.
- ↵Racine JF, Zhu Y, Mamet-Bratley MD. 1993. Mechanism of toxicity of 3-methyladenine for bacteriophage T7. Mutat Res 294: 285–298.
- ↵Rando OJ, Winston F. 2012. Chromatin and transcription in yeast. Genetics 190: 351–387.
- ↵Riedl J, Fleming AM, Burrows CJ. 2016. Sequencing of DNA lesions facilitated by site-specific excision via base excision repair DNA glycosylases yielding ligatable gaps. J Am Chem Soc 138: 491–494.
- ↵Roberts SA, Gordenin DA. 2014. Hypermutation in human cancer genomes: footprints and mechanisms. Nat Rev Cancer 14: 786–800.
- ↵Roberts SA, Sterling J, Thompson C, Harris S, Mav D, Shah R, Klimczak LJ, Kryukov GV, Malc E, Mieczkowski PA, 2012. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol Cell 46: 424–435.
- ↵Rodriguez Y, Hinz JM, Smerdon MJ. 2015. Accessing DNA damage in chromatin: preparing the chromatin landscape for base excision repair. DNA Repair 10.1016/j.dnarep.2015.04.021.
- ↵Rodriguez Y, Hinz JM, Laughery MF, Wyrick JJ, Smerdon MJ. 2016. Site-specific acetylation of histone H3 decreases polymerase β activity on nucleosome core particles in vitro. J Biol Chem 291: 11434–11445.
- ↵Sakofsky CJ, Roberts SA, Malc E, Mieczkowski PA, Resnick MA, Gordenin DA, Malkova A. 2014. Break-induced replication is a source of mutation clusters underlying kataegis. Cell Rep 7: 1640–1648.
- ↵Schuster-Bockler B, Lehner B. 2012. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 488: 504–507.
- ↵Shrivastav N, Li D, Essigmann JM. 2010. Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation. Carcinogenesis 31: 59–70.
- ↵Wallace SS, Murphy DL, Sweasy JB. 2012. Base excision repair and cancer. Cancer Lett 327: 73–89.
- ↵Weiner A, Hsieh TH, Appleboim A, Chen HV, Rahat A, Amit I, Rando OJ, Friedman N. 2015. High-resolution chromatin dynamics during a yeast stress response. Mol Cell 58: 371–386.
- ↵Wyrick JJ, Roberts SA. 2015. Genomic approaches to DNA repair and mutagenesis. DNA Repair 36: 146–155.
- ↵Yu S, Evans KE, van Eijk P, Bennett M, Webster RM, Leadbitter M, Teng Y, Waters R, Jackson SP, Reed SH. 2016. Global genome nucleotide excision repair is organized into domains that promote efficient DNA repair in chromatin. Genome Res 26: 1376–1387.
- ↵Yuan GC, Liu YJ, Dion MF, Slack MD, Wu LF, Altschuler SJ, Rando OJ. 2005. Genome-scale identification of nucleosome positions in S. cerevisiae. Science 309: 626–630.
- ↵Zhong J, Luo K, Winter PS, Crawford GE, Iversen ES, Hartemink AJ. 2016. Mapping nucleosome positions using DNase-seq. Genome Res 26: 351–364.