
metilene: fast and sensitive calling of differentially methylated regions from bisulfite sequencing data
- Frank Jühling1,2,7,
- Helene Kretzmer1,2,7,
- Stephan H. Bernhart1,2,
- Christian Otto1,2,
- Peter F. Stadler2,3,4,5,6 and
- Steve Hoffmann1,2
- 1Transcriptome Bioinformatics Group, LIFE - Leipzig Research Center for Civilization Diseases, University of Leipzig, 04107 Leipzig, Germany;
- 2Interdisciplinary Center for Bioinformatics and Bioinformatics Group, Faculty of Computer Science, University of Leipzig, 04107 Leipzig, Germany;
- 3RNomics Group, Fraunhofer Institute for Cell Therapy and Immunology - IZI, 04103 Leipzig, Germany;
- 4Santa Fe Institute, Santa Fe, New Mexico 87501, USA;
- 5Department of Theoretical Chemistry, University of Vienna, 1090 Vienna, Austria;
- 6Max Planck Institute for Mathematics in Sciences, 04103 Leipzig, Germany
- Corresponding author: steve{at}bioinf.uni-leipzig.de
-
↵7 These authors contributed equally to this work.
Abstract
The detection of differentially methylated regions (DMRs) is a necessary prerequisite for characterizing different epigenetic states. We present a novel program, metilene, to identify DMRs within whole-genome and targeted data with unrivaled specificity and sensitivity. A binary segmentation algorithm combined with a two-dimensional statistical test allows the detection of DMRs in large methylation experiments with multiple groups of samples in minutes rather than days using off-the-shelf hardware. metilene outperforms other state-of-the-art tools for low coverage data and can estimate missing data. Hence, metilene is a versatile tool to study the effect of epigenetic modifications in differentiation/development, tumorigenesis, and systems biology on a global, genome-wide level. Whether in the framework of international consortia with dozens of samples per group, or even without biological replicates, it produces highly significant and reliable results.
As one of the most important mechanisms of epigenetic control, localized differential methylation has been associated with a wide variety of phenotypes and conditions. These include cell differentiation (Cortese et al. 2011; Sheaffer et al. 2014), tissue type and age (Day et al. 2013), and pain sensitivity (Bell et al. 2014). Differences in DNA methylation levels have also been connected to many different diseases like diabetes (Nilsson et al. 2014) or Alzheimer's disease (De Jager et al. 2014). Furthermore, differential DNA methylation plays a role in many cancers, such as medulloblastoma (Hovestadt et al. 2014) and B-cell lymphoma (Kretzmer et al. 2015), and connects risk factors like age (Teschendorff et al. 2010) and smoking (Teschendorff et al. 2015) with cancer.
Whole-genome bisulfite sequencing experiments (WGBS) and targeted protocols, e.g., reduced representation bisulfite sequencing (RRBS), made it possible to study cytosine methylation landscapes at single CpG resolution. Decreasing costs and a high availability of next-generation sequencing (NGS) facilities have made it feasible for a rapidly growing research community to study this important epigenetic layer. The identification of differentially methylated regions (DMRs) between different conditions in large groups of samples requires accurate and efficient algorithms. At present, this is a serious bottleneck in methylome analysis.
The objective problem of finding DMRs has two dimensions: The first dimension is to find a genomic region such that, in the second dimension, the individuals of two groups are significantly distinct in their methylation levels. Current solutions typically use pooled data and employ beta-binomial distributions or regression models fitted to single CpG methylation rates. After testing single CpGs for differential methylation (DMC), significant DMCs are merged to DMRs using various approaches (Akalin et al. 2012; Hebestreit et al. 2013; Park et al. 2014; Stockwell et al. 2014; Sun et al. 2014).
Results
The segmentation algorithm
We present a segmentation algorithm to detect DMRs between single samples as well as in groups of samples (Fig. 1A). As a distinguishing feature, it does not make assumptions on underlying distributions or background models and is applicable to WGBS as well as RRBS data without further parameter adjustments. In contrast to other approaches, we propose a scoring model to find maximum intergroup methylation differences within a genomic interval of minimum length in combination with a nonparametric test. Our approach, based on a circular binary segmentation (CBS) (Siegmund 1986; Olshen et al. 2004), scans for pairs of change points within the mean difference signal (MDS), i.e., difference of CpG-wise mean methylation level in the groups, delimiting a region with homogeneous methylation difference. Subsequently, intervals are tested for similarity using a two-dimensional Kolmogorov–Smirnov test (2D-KS test) (Fasano and Franceschini 1987). Initially, the genome is presegmented to avoid calling DMRs containing long stretches without methylation information. These regions are recursively segmented until (1) a region contains less than a user-defined number of CpGs, or (2) no P-value improvement is achieved. Briefly, within a region [s, t], a window [a, b] is sought using the scoring function Zs,t(a, b), such that the MDS attains a maximal change. The algorithm checks for the existence of short methylation valleys embedded into longer differentially methylated regions and takes care of situations in which regions of differential up- and down-methylation are spatially adjacent.
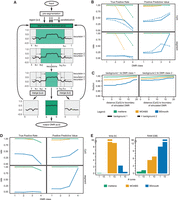
(A) Workflow of metilene. After a presegmentation step to exclude noninformative regions, the circular binary segmentation is used to identify regions with significant differential methylation. The segmentation algorithm is applied recursively trying to identify a window (a,b) with the maximum difference of the cumulative sum of the mean methylation difference, indicating a potential DMR. (B) The performances of metilene, MOABS, and BSmooth were assessed in terms of true positive rates and positive predictive values (PPVs) for four different classes of DMRs, starting with highly different DMRs in class 1 and ending up with a set containing more indifferent DMRs in class 4. The DMRs were simulated within two background settings: the homogeneous background 1 and the more heterogeneous background 2. The evaluation was performed in terms of the fraction of correctly predicted CpGs within simulated DMRs (top) as well as in terms of simulated and predicted DMR segments with an overlap of at least 50% (bottom). (C) Boundary detection analysis for strong (left) and weak (right) differences in the background methylation level. (D) Results for metilene, MOABS, and BSmooth on the low-coverage data sets. (E) Runtime and memory consumption on a single core and 10 cores.
A DMR caller should detect significant differences between groups of samples independently of the background and with the exact genomic boundaries. metilene was compared to three of the frequently used DMR detection tools, i.e., MOABS (v1.2.9) (Sun et al. 2014), BSmooth (v.1.0.0) (Hansen et al. 2012), and BiSeq (Hebestreit et al. 2013) (v1.2.5), using artificial and real-life data.
Performance tests on artificial data
We simulated DMRs for the human Chromosome 10 with different backgrounds and degrees of methylation difference, resulting in eight data sets with different levels of complexity (Supplemental Fig. S1; Methods). Two groups with 10 samples each were simulated. A performance evaluation in terms of the true positive rate (TPR) and positive predictive value (PPV) was carried out for CpGs within simulated and predicted DMRs as well as for predicted DMRs overlapping with ≥50% of simulated DMRs.
Almost all DMRs with larger methylation differences (DMR classes 1 and 2) were correctly predicted by metilene and MOABS on both the CpG (Supplemental Table S1) and the DMR level (Supplemental Table S2). With a TPR below 0.5, BSmooth had difficulties identifying simulated DMRs (Fig. 1B). For DMRs with smaller methylation differences (DMR class 3) in regions with difficult background methylation (background 2), the advantage of metilene was more apparent: While the TPR of MOABS dropped below 40%, metilene still reported 99.8% of the DMRs. A similar TPR was achieved by metilene for the most challenging DMR class 4 in background 1. A drop in sensitivity of metilene (TPR ≈ 0.5) was only observed for the complicated background. MOABS did not predict any class 4 DMRs. metilene showed a PPV ≥ 0.989 in all scenarios.
metilene predicted the starts and ends of DMRs (Fig. 1C; Supplemental Fig. S2) within a very small margin of error, independent of the background type and the DMR class. MOABS performed less accurately for the simple data. Likely due to the efficient smoothing step, BSmooth improved with more complicated background methylation. To verify the robustness of predictions, we simulated low coverage data. Here, metilene came up top in both PPV and TPR (Fig. 1D). In addition, different levels of noise were introduced (Methods). Except for very high noise, MOABS and metilene performed comparably (Supplemental Fig. S3).
On a single core, metilene used ∼4 min to analyze the simulated data set (Chromosome 10) with 2 × 10 samples. The runner-up with respect to the results, MOABS, needed >65 h to perform the same task, whereas BSmooth took 2.3 h. The memory consumption of metilene was <1 GB, whereas MOABS (5.4 GB) and BSmooth (10.7 GB) used substantially more RAM. On 10 cores, metilene used ∼1 min, while MOABS took ∼9 h. BSmooth used 23 min. The memory consumption of metilene and MOABS increased only slightly, while BSmooth used >90 GB in the multithreaded mode (Fig. 1E).
We explored the performance of metilene on RRBS data and found similar results without further parameter adjustments. We added another specialized RRBS tool, i.e., BiSeq, and showed that metilene still performs favorably regarding the prediction power (Supplemental Tables S3, S4; Supplemental Fig. S4), boundary detection (Supplemental Fig. S5), and runtime and memory requirements (Supplemental Tables S5, S6). In some test scenarios, BiSeq and metilene showed comparable results regarding TPRs. However, BiSeq did not reach metilene's PPV. The results of MOABS and BSmooth on RRBS data are comparable to the results on WGBS data.
DMR calling on biological data
A human WGBS (Hovestadt et al. 2014) data set comprising 12 samples from human medulloblastomas and eight controls was retrieved to compare the three tools, i.e., BSmooth, MOABS, and metilene, on real data. For the genome-wide analysis of the WGBS data set, our tool used 10 min on 10 cores, and the peak memory usage was 88.3 MB. Because of the memory and runtime requirements of MOABS and BSmooth (Supplemental Table S7), the qualitative comparison of the results had to be restricted to Chromosome 10.
Among the tools tested, metilene found the highest number of DMRs (n = 4602), followed by MOABS (n = 2108) and BSmooth (n = 1935) (Fig. 2A). This observation is in line with the observed advantages in sensitivity in the simulations. The distribution of additional DMRs detected by metilene peaks around mean methylation differences between 0.2 and 0.3 (Fig. 2B). MOABS has difficulties reporting DMRs with smaller differences, and BSmooth reports only a rather small number of DMRs with higher differences. Using an independent nonparametric test (Wilcoxon) confirms that the DMRs exclusively reported by metilene are significantly differential. In general, metilene's unique DMRs tend to have lower P-values than MOABS or BSmooth (Fig. 2C). We found that some of metilene's unique DMRs do not only show a high absolute methylation difference but are also long (Fig. 2D). To test whether the DMRs exclusively reported by metilene can be confirmed with respect to the estimated mean methylation difference, and thus the homogeneity inside the predicted boundaries, we compared the WGBS results to matched 450k methylation arrays. The high correlation coefficient (r = 0.96) indicates the good reproducibility of metilene predictions (Fig. 2E). An example of a DMR exclusively found by metilene is shown on Figure 2F. It is located in an intragenic region and shows a strong difference level.
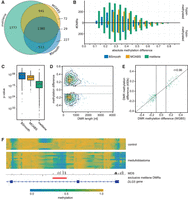
(A) Venn diagram of DMRs found by metilene, BSmooth, and MOABS in the WGBS medulloblastoma data on human Chromosome 10. (B) Count of DMRs exclusively found by metilene, BSmooth, and MOABS binned into methylation difference classes. (C) Box plots of P-values of exclusive DMRs using an independent Wilcoxon test. (D) Scatter plot of length and mean methylation differences of DMRs exclusively reported by metilene. Isoclines indicate their distribution, while labels denote the fraction of DMRs found inside the respective area. Note the minimum methylation cutoff at 0.1 (gray line). (E) Correlation of mean difference of exclusive metilene DMRs and 450k methylation beta values. The plot shows all DMRs covered by at least two probes on the array. (F) Figure of the DLG5 gene containing a DMR (red line) exclusively found by metilene. Methylation rates of control (top) and medulloblastoma (below) are heatmap color-coded, indicating low methylation rates in blue and high methylation rates in yellow. The MDS is shown above the DMR annotation and the gene annotation (bottom).
Hovestadt et al. (2014) reported 39 promoter downstream correlating regions (pdCRs) on Chromosome 10, i.e., differential methylated regions correlating with gene expression. Of these 39 pdCRs, 29 were retrieved by metilene, whereas MOABS reported only 21 and BSmooth reported 12. Most pdCRs (seven of 10) not reported by metilene contain large stretches of >300 nt between CpG sites, whereas the remaining three show highly heterogeneous intra-group methylation levels. With the exception of a single BSmooth prediction, all pdCRs reported by BSmooth and MOABS were also detected by metilene.
Discussion
In summary, metilene does not rely on any assumptions on underlying distributions or background models, nor on joining DMCs. Thus it is able, in contrast to MOABS and BSmooth, to analyze sample pairs without replicates (Supplemental Fig. S6). Furthermore, it can estimate missing methylation data (Supplemental Fig. S7) to include CpG positions not fully covered in all samples. Additionally, it features modes for testing differential methylation in user-defined regions and finding DMCs.
Despite its superior performance in terms of sensitivity, specificity, and accuracy on simulated data, metilene achieved a speed-up of nearly 1000× on a single core of a cluster machine compared to MOABS, the only remaining competitor after analyzing the DMR prediction results of all tools (Supplemental Table S5). On a single core of a desktop machine, metilene was able to process the human WGBS set with eight versus 12 human samples in ∼15 min. Speed and memory requirements are an important issue concerning future studies. We compared all tools and found only metilene to be prepared for larger sample sizes (Supplemental Table S8). Although metilene still performed as well for small group sizes, MOABS and BSmooth took 4.5 and 18.75 d, respectively, to finish the eight versus eight run. Furthermore, although metilene's memory consumption was constantly low (<150 MB) and independent of the number of samples, MOABS and BSmooth consumed between 17 and 300 GB RAM for the same tasks. Both the 16 versus 16 and the 50 versus 50 run could be performed only for metilene due to time/memory issues of both other tools. The results impressively demonstrated the future-proof design of metilene that is capable of dealing with large sample sizes without a large loss of performance. Although we have demonstrated that metilene works well on lowly covered data (sevenfold), we still recommend to provide our tool with methylation rates calculated from 15 reads or more (cf. Ziller et al. 2015).
The nonparametric test at the heart of the strategy integrates two dimensions, i.e., for each given genomic interval (first dimension) the methylation signal of all samples (second dimension). Our simulations have shown that this method outperforms alternative tools in particular in situations in which group-wise methylation differences are subtle and/or the methylation background is variant. This capability comes in handy when dealing with contaminated or heterogeneous samples. Therefore, metilene seems to be especially suitable for DMR prediction in cancer samples or other samples with a superposition of different methylation signals.
Methods
Data acquisition
The biological data that were used within this study are publicly available and were published in the context of the Pediatric Brain Cancer project (PBCA-DE) of the International Cancer Genome Consortium (ICGC) through the following submitter IDs: ICGC_MB2, ICGC_MB6, ICGC_MB7, ICGC_MB15, ICGC_MB19, ICGC_MB24, ICGC_MB26, ICGC_MB32, ICGC_MB38, ICGC_MB40, ICGC_MB49, ICGC_MB51, ICGC_A1, ICGC_A2, ICGC_A3, ICGC_A4, ICGC_F1, ICGC_F2, ICGC_F3, and ICGC_F4. We used ENCODE RRBS data (ENCODE: ENCSR000DFT) and an ENCODE chromatin state segmentation, both of GM12878, for data simulations.
Foundations
The segmentation is based on a circular binary segmentation (Olshen et al. 2004) in conjunction with a scoring function similar to Siegmund (1986) and Zhang and Siegmund (2012). In a first step, the mean difference signal for each CpG (MDS),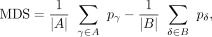 (1)is calculated. Here, A, B are the sample groups to be compared and pγ(pδ) is the methylation level of the given CpG position in sample γ(δ)
(1)is calculated. Here, A, B are the sample groups to be compared and pγ(pδ) is the methylation level of the given CpG position in sample γ(δ)
We use a slightly modified scoring function to account for decreasing values in the MDS: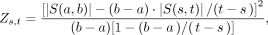 (2)
where Zs,t(a, b) denotes the score of window [a, b] in segment [s, t] and S(i, j) the sum of the MDS for the interval [i, j].
(2)
where Zs,t(a, b) denotes the score of window [a, b] in segment [s, t] and S(i, j) the sum of the MDS for the interval [i, j].
The window [amax, bmax] with the maximal increase or decrease in the MDS within segment [s,t] is given by (3)
(3)
Since we consider all samples of both groups, we have slightly modified the originally proposed formula. By maximizing, we find the window in [s,t] that shows the maximal possible change in the MDS. This window is marked as a potential DMR.
The statistical significance of potential DMRs is assessed by a two-dimensional version of the Kolmogorov–Smirnov test (Fasano and Franceschini 1987) to calculate P-values during segmentation and to use it as a termination criterion during recursive segmentations. The output of metilene provides the adjusted and unadjusted P-value for the 2D-KS test and additionally the results of an independent Mann-Whitney U test.
Algorithm
Initially, the genome is presegmented into regions that do not have subintervals longer than tdist (default: 300 nt) without methylation information. The presegmented regions are recursively segmented. First, the window [a, b] with the maximal absolute change in the MDS in a region [s, t] is identified via Zmax (Equation 3). This results in three subregions: [s, a − 1] [a, b] and [b + 1, t]. The effect of different tdist settings is shown in Supplemental Table S9. The parameter affects the performance of metilene only slightly as long as it is kept in the range ≤500 nt. For the sake of biological interpretation, we do not recommend to include longer stretches of the genome without any methylation information.
Second, termination criteria are checked for all three subregions. Every subregion that fulfills none of the termination criteria is further segmented, starting with the first step. We use two distinct termination criteria: (1) number of CpGs < tmin (default: 10); and (2) a P-value-based termination criterion, i.e., the P-value of a subregion must be larger than the P-value of the parental region. To save computational time, the P-value of a region is not calculated if (1) the segment contains a window of tmin consecutive CpGs with a much weaker MDS than the total segment; or if (2) the signs of MDS have no major trend, i.e., we find a balanced number of both positive and negative values. In such regions, segmentation is directly continued.
Please note that except for the tmin criteria, there are no additional constraints or parameters for the minimum length of an interval [a, b] or [s, t]. The effect of different tmin settings is shown in Supplemental Table S10. The parameter does affect the performance of metilene only in WGBS data when it is set to large window sizes.
Third, after the termination of the recursion, the subregion associated with the lowest P-value is flagged as a potential DMR. All surrounding subregions are merged and recursively segmented again (see Supplemental Fig. S8 for the pseudocode).
Missing data estimation
Methylation rates of missing data (pmis) are estimated from a beta distribution pmis ∼ Beta (α, β). The parameters α and β are calculated from the mean (μr) and variance 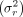 of the remaining methylation rates at the corresponding CpG position:
of the remaining methylation rates at the corresponding CpG position:
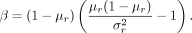
Positions with more than a user-defined number of missing values are omitted.
Data simulation
All DNA methylation simulations were performed on the human Chromosome 10 using a Beta-Binomial approach implemented in R (R Core Team 2013) to simulate both biological methylation and the sequencing step. The simulated data that were used for the benchmarks are available at http://www.bioinf.uni-leipzig.de/Software/metilene/. The scripts and parameters to simulate these data from scratch can also be found in the Supplemental Material.
We simulated two different backgrounds to account for promoter and nonpromoter differences. Since the overall sequence composition in promoters is expected to be different from nonpromoter regions, and to do the simulation in a more realistic fashion, we used the ENCODE chromatin state segmentation of a lymphoblastoid cell line (GM12878) (Ernst and Kellis 2010; Ernst et al. 2011; Raney et al. 2011) to obtain a set of real promoter and nonpromoter regions on Chromosome 10. The GM12878 segmentation has only been used in the simulation and for this specific purpose.
For the 20 simulated samples, methylation rates p were drawn from beta distributions (Beta) with parameters α and β to obtain p ∼ Beta(α, β). The distributions of the two simulated methylation backgrounds (Supplemental Table S11) are visualized in Supplemental Figure S1A.
Junctions between the nonpromoter and the promoter backgrounds were blurred using a weighted, local polynomial regression fitting (loess; span value of 0.2). Specifically, the methylation rate of 25–50 CpGs (uniform sample) around each junction was smoothed by setting it to the average of the raw and the fitted value.
Subsequently, an artificial read coverage n was assigned to the CpGs by sampling from a normal distribution (N) with mean μ = 30 and standard deviation σ = 5. The minimum required coverage was set to ɛ = 15 reads. For the simulation
of lowly methylated data, the parameters were set to μ = 7, σ = 2, and ɛ = 3: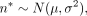
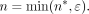
To determine the number of these reads with CpGs indicating methylation, m ∼ B(n, p) was drawn from a binomial distribution (B) with parameters n and P for coverage and methylation rate, respectively.
To simulate the Foreground, i.e., the DMRs, the 20 samples were split into two groups of 10 samples. A total of 1000 DMRs
were introduced into the simulated background of each group. We introduced an equal number of hypermethylated DMRs into promoter
(500) and hypomethylated DMRs into nonpromoter regions (500). To generate the DMRs, the parameters α and β of the background
where swapped, i.e., a DMR within a promoter region was simulated using the beta-distribution for the nonpromoter regions
and vice versa. To account for DMRs of different intensities, the values were mixed from both beta distributions and multiplied
with different mixing factors C,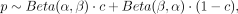 to generate a total of four different DMR sets. The mixture factors c are given in Supplemental Table S12, and all resulting distributions of DMR classes 1–4 are visualized in Supplemental Figure
S1B. Again, the number of reads without conversion was drawn from a binomial distribution.
to generate a total of four different DMR sets. The mixture factors c are given in Supplemental Table S12, and all resulting distributions of DMR classes 1–4 are visualized in Supplemental Figure
S1B. Again, the number of reads without conversion was drawn from a binomial distribution.
Reduced representation bisulfite sequencing data were simulated in a similar fashion as the WGBS data. To make our simulation more realistic, we used the regions covered by the publicly available RRBS data set of GM12878. In total, we placed 200 DMRs, 100 DMRs in promoters and 100 DMRs in nonpromoters, with a maximum length of 40 nt inside these regions using the same parameters as for the WGBS simulations.
One of the artificial data sets (background 1 and class 1 DMRs) was used to perform the noise analysis. All DMRs were gradually distorted by replacing 10%–90% of all methylation rates with uniformly distributed methylation rates pr ∈ [0, 1] for all samples.
The data set containing 10 versus 10 samples of class 1 DMRs on background 1 was simulated, and methylation values were removed at a probability of 10%–90% to resemble data sets with different degrees of missing values.
Performance evaluation
All segmentation tools were run in their default settings; and for MOABS, the maximal distance between two consecutive DMCs
to be considered in a DMR (maxDistConsDmcs option) was set to 300 nt to be comparable to metilene and BSmooth. Because BSmooth
terminated with a runtime error, local corrections were switched off for the RRBS data analysis (local.correct = F). Predicted
DMRs were filtered to a minimal absolute methylation difference (average MDS) larger than 0.1, which is in accordance with
the cutoff recommended also by BSmooth. By default, BSmooth reports only significant DMRs. The output of MOABS and metilene
was filtered for DMRs with an (adjusted) P-value ≤ 0.05. The performance was evaluated in terms of CpG-wise and DMR-wise true positive rates (TPR) and positive predictive
values (PPV). In the following we distinguish between sets of CpGs (C) and sets of CpGs within DMRs (D). A set of DMRs is denoted by (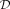 ). The sets of simulated and predicted CpGs inside simulated and predicted DMRs are defined by
). The sets of simulated and predicted CpGs inside simulated and predicted DMRs are defined by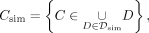
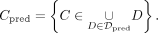
Furthermore, we define true positive (TP) CpGs (CTP) as those CpGs in simulated DMRs that were correctly predicted by the segmentation tool. This definition includes all those
CpGs that are in the overlap between simulated and predicted DMRs. Thus,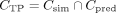 is the set of true positive CpGs. We calculate
is the set of true positive CpGs. We calculate
 to obtain the benchmarks on the CpG resolution level. For the region-wise evaluation,
to obtain the benchmarks on the CpG resolution level. For the region-wise evaluation, 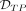 is the set of predicted DMRs in which >50% of the DMRs’ CpGs are simulated to be differential, i.e., elements of
is the set of predicted DMRs in which >50% of the DMRs’ CpGs are simulated to be differential, i.e., elements of 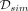 . On the other hand,
. On the other hand, 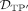 is the set of simulated DMRs in which >50% of the DMRs’ CpGs are in
is the set of simulated DMRs in which >50% of the DMRs’ CpGs are in 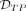 . Given
. Given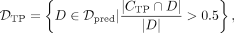

 we calculate
we calculate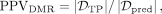
 to obtain the benchmarks for the region-wise comparison analogously. Note that the distinction between
to obtain the benchmarks for the region-wise comparison analogously. Note that the distinction between 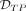 and
and 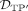 is necessary due to a possible m-to-n rather than a 1-to-1 association between simulated and predicted DMRs.
is necessary due to a possible m-to-n rather than a 1-to-1 association between simulated and predicted DMRs.
Accuracy of DMR boundaries
Boundaries of predicted DMRs were compared to simulated DMRs. More precisely, the distance (in CpGs) between the boundaries of each simulated DMR and its closest correctly predicted (TP) DMR was calculated. To analyze and compare the accuracy regarding the boundary detection of the different segmentation tools, the empirical cumulative distribution function of the absolute values of these distances was used.
Whole-genome bisulfite data of medulloblastoma tumors
DNA methylation data of 12 human medulloblastoma tumors (subgroup 4) and eight human normal controls were taken from Hovestadt et al. (2014). The qualitative analysis of the WGBS data set with 12 versus eight samples (22,524,970 data points without missing values) was restricted to Chromosome 10 with a total of 1,111,583 methylation data points. All segmentation tools were run in their default settings; and for MOABS, the maximal distance between two consecutive DMCs to be considered in a DMR (maxDistConsDmcs option) was set to 300 nt to be comparable to metilene and BSmooth. DMRs were required to contain at least 10 informative CpGs, i.e., CpGs with an associated methylation rate, and an absolute methylation difference (average MDS) larger than 0.1. As described above, outputs of MOABS and metilene were filtered with the critical P-value 0.05. For the analysis of promoter downstream correlating regions (pdCRs), the full set of pdCRs published in Hovestadt et al. (2014) was downloaded and restricted to Chromosome 10. For each pdCR, the difference between the average methylation of the tumor samples and the average methylation of the control samples was calculated using BEDTools (Quinlan and Hall 2010).
Evaluation of runtime and memory requirements
The running time of all tools was measured using the Unix time command, while the maximal residual memory consumption (RAM) was tracked by the Unix ps command. The evaluation was done separately with 10 cores or one core of a cluster with the following specifications: Intel XEON E7540 CPU at 2.00 GHz × 24, 520 GB RAM. To measure memory and runtime requirements for the WGBS data set, we used 10 cores of the same cluster machine. To evaluate metilene's performance in an environment with restricted computational resources, the WGBS data set was additionally processed on a desktop machine with the following specifications: Intel Core i5-4570 CPU at 3.20 GHz × 4, 7.8 GB RAM.
Software availability
The software is published under the GNU GPL v2.0 license. The source code of metilene is available in the Supplemental Material and at http://www.bioinf.uni-leipzig.de/Software/metilene/. The implementation that was used for benchmarking (metilene version 0.2-4) as well as the scripts for simulating DMRs is also part of the Supplemental Material.
Acknowledgments
This publication is supported by the German Bundesministerium für Bildung und Forschung (BMBF; PTJ grant HNPCCSys 031 6065A and ICGC MMML-Seq 01KU1002J), the European Union in the framework of the BLUEPRINT (HEALTH-F5-2011-282510) project, and by LIFE: Leipzig Research Center for Civilization Diseases, University of Leipzig. LIFE is funded by means of the European Union, by the European Regional Development Fund (ERDF), the European Social Fund (ESF), and by means of the Free State of Saxony within the framework of the excellence initiative.
Author contributions: F.J., H.K., P.F.S., and S.H. conceived the project. F.J. and S.H. implemented metilene. H.K., F.J., C.O., and S.H.B. tested metilene on artificial and biological data. F.J., H.K., and S.H. wrote the manuscript with contributions from all authors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.196394.115.
- Received July 1, 2015.
- Accepted November 25, 2015.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








