
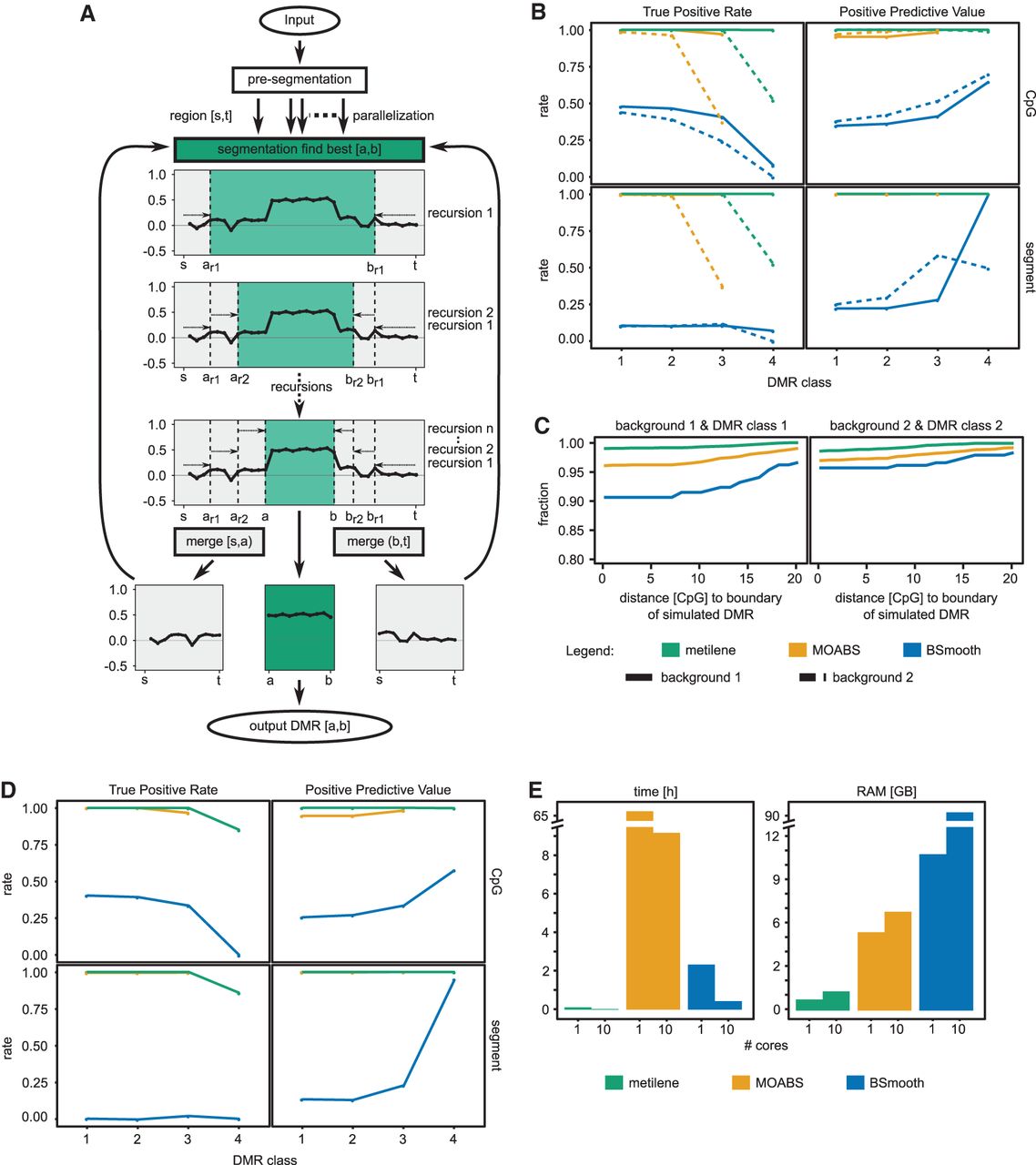
(A) Workflow of metilene. After a presegmentation step to exclude noninformative regions, the circular binary segmentation is used to identify regions with significant differential methylation. The segmentation algorithm is applied recursively trying to identify a window (a,b) with the maximum difference of the cumulative sum of the mean methylation difference, indicating a potential DMR. (B) The performances of metilene, MOABS, and BSmooth were assessed in terms of true positive rates and positive predictive values (PPVs) for four different classes of DMRs, starting with highly different DMRs in class 1 and ending up with a set containing more indifferent DMRs in class 4. The DMRs were simulated within two background settings: the homogeneous background 1 and the more heterogeneous background 2. The evaluation was performed in terms of the fraction of correctly predicted CpGs within simulated DMRs (top) as well as in terms of simulated and predicted DMR segments with an overlap of at least 50% (bottom). (C) Boundary detection analysis for strong (left) and weak (right) differences in the background methylation level. (D) Results for metilene, MOABS, and BSmooth on the low-coverage data sets. (E) Runtime and memory consumption on a single core and 10 cores.








