
Abstract
Enhancers and promoters commonly occur in accessible chromatin characterized by depleted nucleosome contact; however, it is unclear how chromatin accessibility is governed. We show that log-additive cis-acting DNA sequence features can predict chromatin accessibility at high spatial resolution. We develop a new type of high-dimensional machine learning model, the Synergistic Chromatin Model (SCM), which when trained with DNase-seq data for a cell type is capable of predicting expected read counts of genome-wide chromatin accessibility at every base from DNA sequence alone, with the highest accuracy at hypersensitive sites shared across cell types. We confirm that a SCM accurately predicts chromatin accessibility for thousands of synthetic DNA sequences using a novel CRISPR-based method of highly efficient site-specific DNA library integration. SCMs are directly interpretable and reveal that a logic based on local, nonspecific synergistic effects, largely among pioneer TFs, is sufficient to predict a large fraction of cellular chromatin accessibility in a wide variety of cell types.
Genomic DNA comprises multiple overlapping codes that contain information specifying cellular function. Although the “genetic code” that governs how DNA encodes protein sequence through triplet codons was cracked more than 40 years ago, the codes governing how genes are regulated remain largely unsolved.
Chromatin accessibility, which we define to be a measure of the relative depletion of local nucleosome contact with genomic DNA (see Supplemental Information for in-depth definition), is a critical component of transcription factor (TF) binding, gene regulation, and cellular identity (Weintraub and Groudine 1976; Wu 1980; Soufi et al. 2012; Sherwood et al. 2014). Several measurement techniques reveal a common set of regions with accessible chromatin (Giresi et al. 2007; Boyle et al. 2008; Gaulton et al. 2010; Song et al. 2011; Buenrostro et al. 2013), and in this work, we primarily measure chromatin accessibility genome-wide using DNase-seq (Boyle et al. 2008), a method for identifying DNase I hypersensitive sites (DHS) (Weintraub and Groudine 1976; Wu 1980). DNase I hypersensitivity is a common feature of most gene regulatory elements, including enhancers and promoters (Thurman et al. 2012), and thus systematic understanding of what governs chromatin accessibility would be an enormous advance in understanding the genomic regulatory code.
Yet, is there a DNA logic underlying chromatin accessibility? There is evidence that the accessibility of specific genomic regions is governed by binding of “pioneer” TFs, which are capable of binding to inaccessible, nucleosome-bound DNA and inducing accessibility (Gualdi et al. 1996; Zaret and Carroll 2011; Soufi et al. 2012). However, pioneer TFs do not bind to every instance of their binding motif in the genome as might be expected by their imperviousness to prior chromatin state (Sherwood et al. 2014), and thus there must be additional components determining whether a pioneer TF will induce accessibility at a genomic motif instance. Additionally, a causal role for pioneer TF binding in determining accessibility has thus far only been confirmed at a small number of genomic loci, and so it is unknown whether pioneer TF binding is sufficient to explain the chromatin accessibility of all genomic loci.
These observations suggest that chromatin accessibility is regulated by interactions among chromatin-regulating DNA sequences that are more complex than the absence or presence of a single bound pioneer factor. We consider a specific type of interaction between such regulatory sequences in which every short DNA sequence (k-mer) is given a fixed, spatial effect that multiplicatively combine to form overall chromatin accessibility. This stands in contrast to the single-factor hypothesis in which overall chromatin accessibility is formed by the existence of a single pioneer factor.
We reasoned that if a DNA logic for chromatin accessibility exists, then we could discover a general model that predicts chromatin accessibility directly from DNA sequence. Although prior work has focused upon prediction of regulatory sequences using bags of k-mers (Lee et al. 2011; Ghandi et al. 2014) or identification of motifs enriched in regulatory regions (Stergachis et al. 2013), our goal is to construct a generative model of the DNase-seq assay directly linking DNA sequence to DNase-seq read count. Such a model should predict the expected number of reads observed at any base in a DNase-seq assay and the locations of all accessible chromatin regions in the genome, including promoters and enhancers. If the computational model is sufficiently accurate at predicting sequence-dependent chromatin accessibility on novel sequences, we can expect this framework to yield testable hypotheses of how pioneers and other regulatory mechanisms control chromatin accessibility. To minimize bias from incomplete biological understanding, we utilized a computational framework that ignores any preconceptions of what factors or motifs might be involved in this regulatory process and that makes predictions genome-wide rather than over some curated functional subset.
The guiding philosophy behind SCM is that the entire genome is one continuous regulatory sequence in which are imbedded “code words” that induce invariant spatial effects on proximal chromatin accessibility wherever they occur and that interact with each other in a predictable way. Based on evidence from our previous work (Sherwood et al. 2014), we utilized the following small set of biological assumptions to build the SCM: (1) The building blocks of chromatin accessibility are short stretches of DNA (k-mers) 8 bp or smaller; (2) k-mers have exclusively local effects on chromatin accessibility within ±1 kb of their occurrence; (3) a small number of k-mers play a role in determining chromatin accessibility; (4) a particular k-mer always produces the same effect on chromatin accessibility wherever it occurs in the genome; and (5) k-mer effects on chromatin accessibility nonspecifically synergize such that the chromatin accessibility at any DNA base is the multiplicative product of the effects of all nearby chromatin accessibility-affecting k-mers. This extends a line of work in transcriptional regulation in which a similar multiplicative model is used with a logistic link function to model the effect of transcription factors on gene expression (Veitia 2003; He et al. 2010). We define a synergistic model of chromatin accessibility as a model in which sequence effects are log-linear in the observed chromatin accessibility data. The rationale behind these assumptions is discussed in more detail in the Supplemental Information. It is notable that these simple assumptions do not allow for far-reaching spreading of chromatin accessibility state or effects of specific protein–protein interactions on chromatin accessibility unless such interactions occur at short, fixed distances from each other (Supplemental Fig. 1).
Our SCM approach to identifying regulatory sequences is distinct from traditional motif-finding and represents a conceptual advance in the identification of functional sequences. In traditional motif-finding and discriminative motif-finding approaches (Bailey 2011; Huggins et al. 2011; Lee et al. 2011; Ghandi et al. 2014), the practitioners must predetermine a class of interesting regions such as accessible chromatin regions, footprints, peaks, or enhancers, making arbitrary in versus out cutoffs in what is often continuous data. Our work removes the step of defining an “interesting region” and instead identifies any sequence that induces changes in the observed data. This distinction gives our approach two major advantages. First, SCM automatically yields information on the role (or lack thereof) of every DNA sequence, whereas traditional motif-finding approaches only return sequences that correlate with the desired function. This allows us to gauge the genome-wide accuracy of our method through comparing model output to actual data at high spatial resolution, as opposed to traditional methods whose accuracy can only be gauged after grouping data into classes. We believe that a method aimed at genomic prediction should be able to predict the status of every region in the genome without preconceptions, and SCM is the first approach capable of doing so. Second, our automated approach yields spatial information about how each DNA sequence contributes to local chromatin accessibility, which immediately suggests the function of sequences.
Results
We train a SCM model on DNase-seq data from a particular cell type and its underlying DNA sequence so the model can generate cellular state-specific predictions of the chromatin accessibility of any DNA sequence. DNase-seq data from a subset of the chromosomes are used to train the model, and we test the model on DNase-seq data from the held-out chromosomes. The SCM model automatically learns which “code words” in the genome have local cis-regulatory effects on chromatin accessibility. Each code word is a k-mer between 1 and 8 bases long and is associated with a profile of how it is predicted to effect chromatin accessibility at every base position ±1 kb at each site where it occurs (Fig. 1A). Because the model computes the synergistic effect of thousands of overlapping k-mers at any given site, the pattern of predicted chromatin accessibility at a given site is not always the straightforward effect of the strongest k-mers in the vicinity (Fig. 1A). Once the SCM has discovered the chromatin accessibility code words and their spatial patterns from cell-type–specific training data, it can output predicted chromatin accessibility patterns for any DNA sequence, be it genomic DNA or novel DNA sequence. Since our model is trained on a particular cell type, we will denote an instance of the model that has been trained on data from a specific cell type as “SCM (cell type),” such as SCM (K562). To further validate the model, we computed novel sequences with varying degrees of predicted accessibility, synthesized these sequences, and observed their DNase-seq accessibility in vivo in a matched cell type. Thus, the model is capable of predicting chromatin accessibility of variants of the original genome. In addition, it is interpretable, allowing us to learn and explore the precise sequences that direct chromatin accessibility.
Multiplicative effects of local k-mers accurately predict chromatin accessibility. (A) A SCM uses DNase-seq data on training chromosomes and iterative machine learning methods to compute spatial profiles for each k-mer, optimizing a model in which nearby k-mer effects multiply to predict DNase-seq reads for held-out chromosomes. In this example representing a genomic region containing an NRF1 binding site, the top panel shows single base resolution predicted (black) and 5-bp smoothed observed DNase-seq data (red) across a 600-bp window. The middle panel shows the SCM-predicted spatial contribution of the top 10 k-mers in log-units and matched motifs in the legends; the teal peak corresponds to the NRF1 binding footprint. The bottom panel shows a measure of importance of each base by the k-mer starting at that position summed over the entire spatial range of k-mer influence with colored tick marks for the top 10 k-mers. Note that SCMs multiply effects of thousands of overlapping k-mers at each site, so the top k-mers do not lead to the SCM predictions in a straightforward manner. (B) Example human K562 held-out genomic region showing DNase-seq reads (red), SCM-predicted reads (black), and reads from a control model trained on IMR-90 naked DNA DNase-seq data (green) (Lazarovici et al. 2013), all smoothed at 200 bp. (C) Comparison of SCM-predicted (x-axis) and observed (y-axis) DNase-seq reads in 2-kb binned regions of K562 held-out Chromosome 14. Models were trained on K562 DNase-seq data (black) or IMR-90 naked DNA DNase-seq (red). (D) Receiver–operator curve (ROC) showing SCM predictive accuracy after binary calling of DHS in observed and predicted K562 held-out DNase-seq data. The evaluation set was balanced to 5000 positive and negative samples (uniformly taken from positive and negative sets) to avoid AUC inflation due to class imbalance.
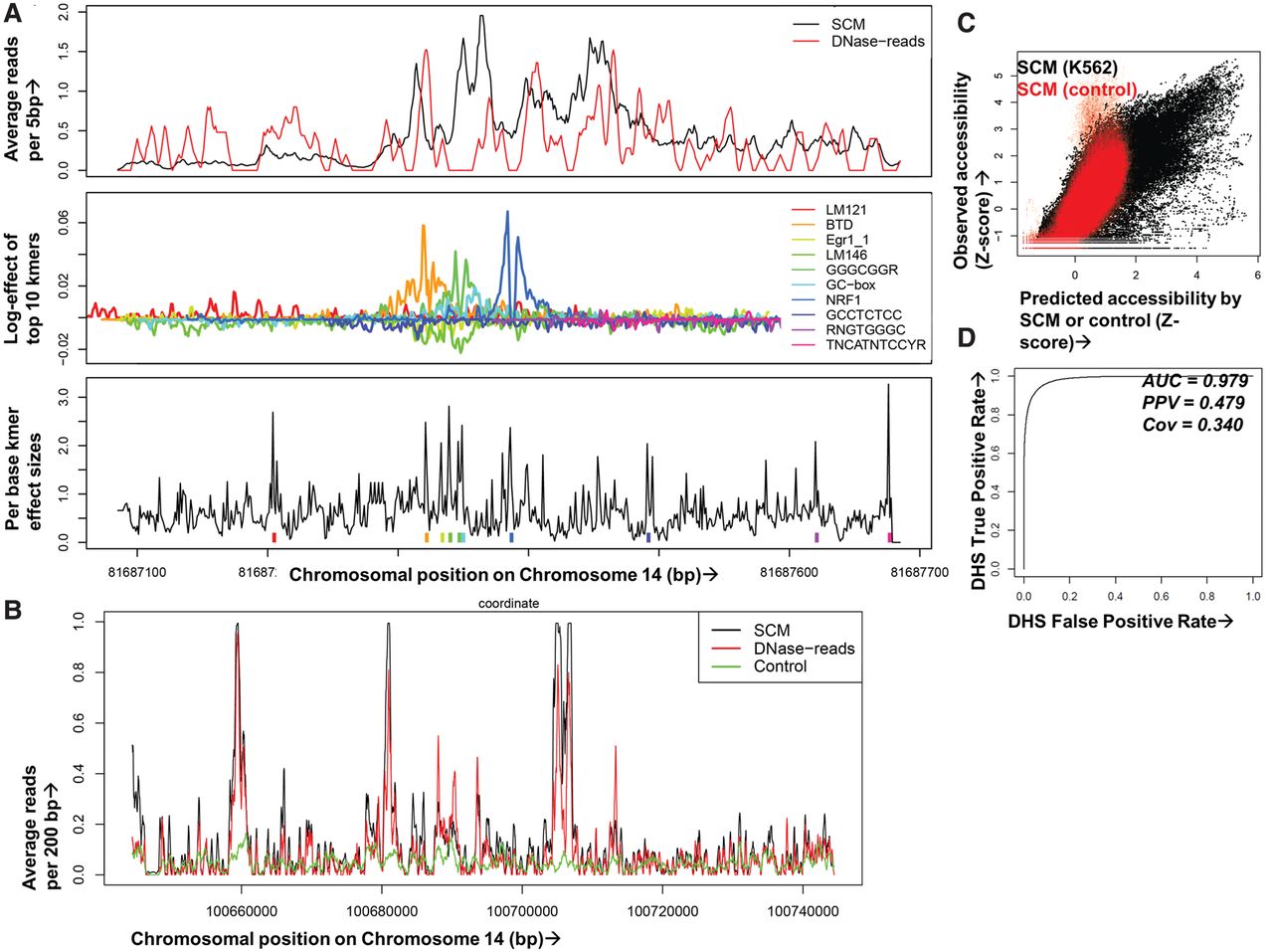
Learning the chromatin accessibility profile induced by each k-mer from hundreds of millions of examples is a challenging machine learning task. Previous approaches to learning regulatory sequences have restricted the genomic regions to a curated set (Lee et al. 2011; Ghandi et al. 2014). However, by carefully constructing our model to be tractable, we are able to avoid the use of any heuristic pruning or parameter selection and use a stochastic gradient descent algorithm (Duchi et al. 2011) to optimize the profile of every possible k-mer to predict the expected number of DNase-seq reads at every base of the genome. This optimization exercise is iterated under the influence of a penalty (L1 regularization) (Duchi et al. 2011) that acts to limit the number of k-mer profiles and the strength of each profile to avoid overfitting. SCM iteration continues under the L1 penalty until the model converges on the most accurate reproduction of the training data, and then SCM predictions of DNase-seq data are generated for held-out genomic regions to test for accuracy compared with previously unseen experimental data (Fig. 1A; see Supplemental Information for details about SCM implementation). A SCM has more than 40 million parameters, and thus several technical innovations and a parallel cloud-based implementation are required to yield practical run times (Supplemental Information). Since SCM models are convex (Supplemental Information), our gradient descent optimizer is guaranteed to find a unique solution that is insensitive to parameter initialization.
As a first step to test the accuracy of a SCM at predicting genomic chromatin accessibility, we trained a SCM (K562) on DNase-seq data from Chromosomes 1–13 of human K562 cells. We then predicted DNase-seq data on a held-out chromosome (Chromosome 14). The SCM (K562) predictions are remarkably similar to actual DNase-seq reads (Fig. 1A,B; Supplemental Fig. 3), producing a chromosome-wide Pearson's correlation value of 0.801 between predicted and actual reads on Chromosome 14, with a range of [0.800,0.814] over Chromosomes 15–22 (Fig. 1C; Supplemental Table 1). We measured correlation after smoothing predicted and actual reads over 2000-bp windows, chosen to match the SCM window size, since actual reads are insufficiently sampled to produce accurate correlation measurements. The correlations are robust to this window choice, with Pearson's correlations of 0.738 and 0.784 for windows of 200 and 1000 bp, respectively. Despite some variation at any individual loci, the SCM model captures the overall structure of DNase I accessibility over the held-out chromosome. DNase-seq is known to have an underlying sequence preference, resulting in the possibility that a SCM model would learn the inherent sequence bias of the DNase I enzyme rather than the relationship between DNA sequence and accessibility (He et al. 2013; Lazarovici et al. 2013). In order to account for this confounder, we validate our model on DNase I hypersensitive sites (Fig. 1D, details below) as well as compare against a SCM trained on DNase-seq of purified DNA stripped of proteins (Lazarovici et al. 2013), which is far less accurate at predicting held-out chromatin accessibility with Pearson's correlation of 0.469 (Fig. 1B,C; Supplemental Table 1), showing that the SCM is not merely reading out DNase I or sequencing bias. Additionally, we tested a control model that eliminates k-mer synergism by reducing the k-mer profile size to 1 bp, resulting in each k-mer having a point effect that is then averaged over 100 bp of surrounding genomic space. This model has a Pearson's correlation of 0.409 against held-out data (Supplemental Table 1), showing the importance of the spatial profile and of k-mer synergism in predicting chromatin accessibility. The importance of spatial profile and k-mer synergism are exemplified in Figure 1A, which shows that the full SCM (top panel) predicts DNase-seq data much more accurately than the locations of k-mers with the strongest effect on chromatin accessibility (bottom panel) and the individual spatial profiles of such k-mers (middle panel). Figure 1, A and B, shows that although the SCM's accuracy decreases with smaller window sizes, it still produces quantitatively accurate predictions of DNase-seq reads (Pearsons's correlation of 0.738 at 200-bp resolution).
Although SCM differs from existing methods aimed at binary classification of hypersensitive and nonhypersensitive chromatin, we asked how SCM performance compares to four sequence-based classifiers that use either k-mer based models (gkm-svm, SeqGL) or deep learning based models (deepSEA, Basset) (Ghandi et al. 2014; Setty and Leslie 2015; Zhou and Troyanskaya 2015; Kelley et al. 2016). Although SCM is designed for quantitation and not binary prediction, SCM performs as well as the four state-of-the-art binary predictive methods on black-box binary prediction of functional genomic regions (Supplemental Fig. 2). We also find that SCM substantially outperforms these classification methods on regression tasks (Supplemental Fig. 2), which compare smoothed Pearson and Spearman correlations of predicted and observed read counts, which is expected, because these other methods are not designed for this purpose. Thus, SCM is comparable to or better than existing methods at binary classification but additionally provides a qualitatively different output of spatial read distribution prediction.
We evaluated the performance of a nonsynergistic model to test our hypothesis that sequence features operate synergistically to direct chromatin accessibility. We trained a nonsynergistic, additive model by allowing sequence effects to combine additively (implementation details are in Supplemental Material). The additive model has a chromosome-wide Pearson's correlation value of 0.74 compared to the SCM's value of 0.82, despite that both models have the same parameter size, complexity, and training procedure (Supplemental Fig. 4).
To confirm that SCM (K562) accurately predicts true chromatin accessibility, we calculated the overlap between K562 DHS (Thurman et al. 2012) and thresholded SCM-predicted peaks on Chromosome 14, finding that SCM accurately predicts 72.4% of DHS at a 1% false-discovery rate (area under ROC curve [AUC] = 0.979; PPV 0.479; TPR 0.340, under a rebalanced data set) (Fig. 1D). Among these DHS, SCM (K562) is accurate over many types of genomic regions, such as predicted enhancers, promoters, and other active chromatin types (Supplemental Fig. 5; Ernst and Kellis 2012), indicating that the SCM accurately predicts sites representing a variety of classes of predicted functional chromatin accessibility.
We next asked whether a SCM can accurately predict chromatin accessibility in additional cell types when trained on data from those cell types. We trained SCMs on DNase-seq data from 11 human data sets and three mouse data sets representing a wide range of developmental origins and including both cell lines and in vivo tissues. We found uniformly high correlation between SCM predictions and DNase-seq data across human and mouse cell types (Fig. 2A; Supplemental Table 1).
SCMs predict chromatin accessibility at base pair resolution across cell types and data types. (A) Pearson correlation coefficients on held-out Chromosome 14 DNase-seq data for SCMs trained on DNase-seq from 10 cell types. (B) Receiver–operator curve (ROC) showing predictive accuracy of a SCM trained on GM12878 DNase-seq data at predicting held-out GM12878 ATAC-seq peaks. (C) Example human GM12878 held-out genomic region showing ATAC-seq reads (black) and reads predicted from a DNase-seq trained SCM (red), both smoothed at 200 bp. (D) Example human GM12878 held-out genomic region showing 10-bp smoothed DNase-seq reads (black), SCM-predicted reads (red), and reads from a control model trained on IMR-90 naked DNA DNase-seq data (blue) surrounding two NRF1 binding sites (vertical black lines denote binding call). (E) Heatmap showing clear footprints for both DNase-seq and SCM at NRF1 binding sites on Chromosome 14.
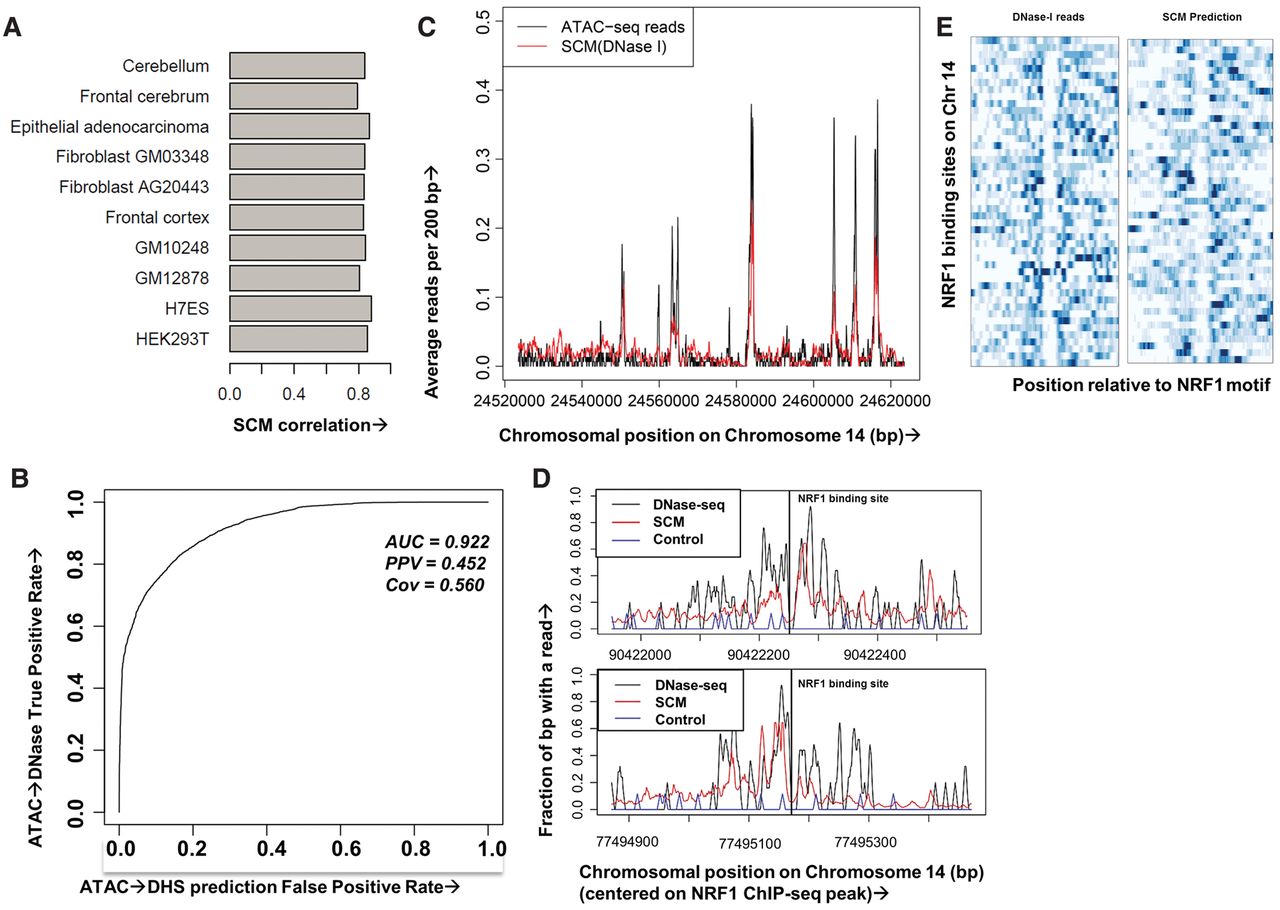
As an additional test that SCMs predict true chromatin accessibility and not DNase I bias, we analyzed data from ATAC-seq, a technique that uses transposition to map sites of chromatin accessibility (Buenrostro et al. 2013). We find that the Pearson's correlation between the raw reads derived from the two methods is 0.584 (Supplemental Fig. 6), indicating only a partial overlap in the chromatin accessibility signal calculated by these methods. A SCM trained on ATAC-seq data and tested on held-out ATAC-seq data achieves a genome-wide Pearson's correlation of 0.610 (Supplemental Fig. 7) and achieves decent predictive accuracy of thresholded peaks (AUC 0.953; PPV 0.384; TPR 0.385), revealing that a SCM is able to predict ATAC-seq data, although less accurately than it predicts DNase-seq data. We speculate that this decreased accuracy could be the result of lower ATAC-seq read counts, which could negatively impact SCM performance. We then asked whether a SCM trained on DNase-seq data could predict held-out ATAC-seq data. Because of the substantial differences in the raw signal, we focused on comparing the accuracy of a DNase I–trained SCM (K562) at predicting the locations of thresholded ATAC-seq peaks. The DNase I–trained SCM (K562) achieves an AUC of 0.922 in predicting thresholded ATAC-seq peaks (PPV 0.351; TPR 0.215) (Fig. 2B,C), indicating that the SCM is able to predict sites of accessibility identified by distinct techniques.
One feature that distinguishes SCMs from discriminative motif-finding algorithms is that SCMs generate predictions of DNase-seq data at base pair resolution. Since bound TFs are known to leave DNase I footprints when bound (Wu 1980; Hesselberth et al. 2009), we asked whether the SCM recapitulates DNase I footprints at known locations. We compared the SCM (mESC) DNase-seq predictions and actual DNase-seq data surrounding NRF1 binding sites in human GM12878 cells as determined by NRF1 ChIP-seq (The ENCODE Project Consortium 2012), finding evidence of footprints in both the predicted and actual DNase-seq data (Fig. 2D,E). Thus, SCMs are capable of generating high spatial resolution predictions of DNase-seq data, including TF footprints.
We then compared the k-mers with the strongest effects on chromatin accessibility across distinct cell types. By gauging k-mer effect size, the total SCM-predicted effect of each k-mer on surrounding chromatin accessibility, we find that the k-mers exerting the strongest effect on chromatin accessibility are highly conserved across human cell types such as between K562 and frontal cortex cells (Fig. 3A). The Pearson's correlation of all k-mer effect sizes between K562 and other cell types is typically above 0.7 (Fig. 3A; Supplemental Fig. 7). Despite the conservation of a large number of k-mers, we found that predicted read rates across cell types recapitulate similarities in DNase-seq reads (Supplemental Fig. 8), and we found a small number of cell-type–specific k-mers corresponding to the binding sequence of actively expressed proteins in a cell type (Supplemental Fig. 9).
SCMs are more accurate at predicting DNase I hypersensitive regions that are active in multiple cell types. (A) Summed k-mer effect sizes for each k-mer in SCMs trained on K562 (x-axis) versus human frontal cortex (y-axis). (B) Histogram showing the fraction of the genomic space covered by DNase I hypersensitive sites (DHS) in a single cell type that are hypersensitive in 10 other human cell types. (C) Histogram showing the average balanced accuracy of SCM DHS predictions binned according to the cell-type specificity of DHS activity.
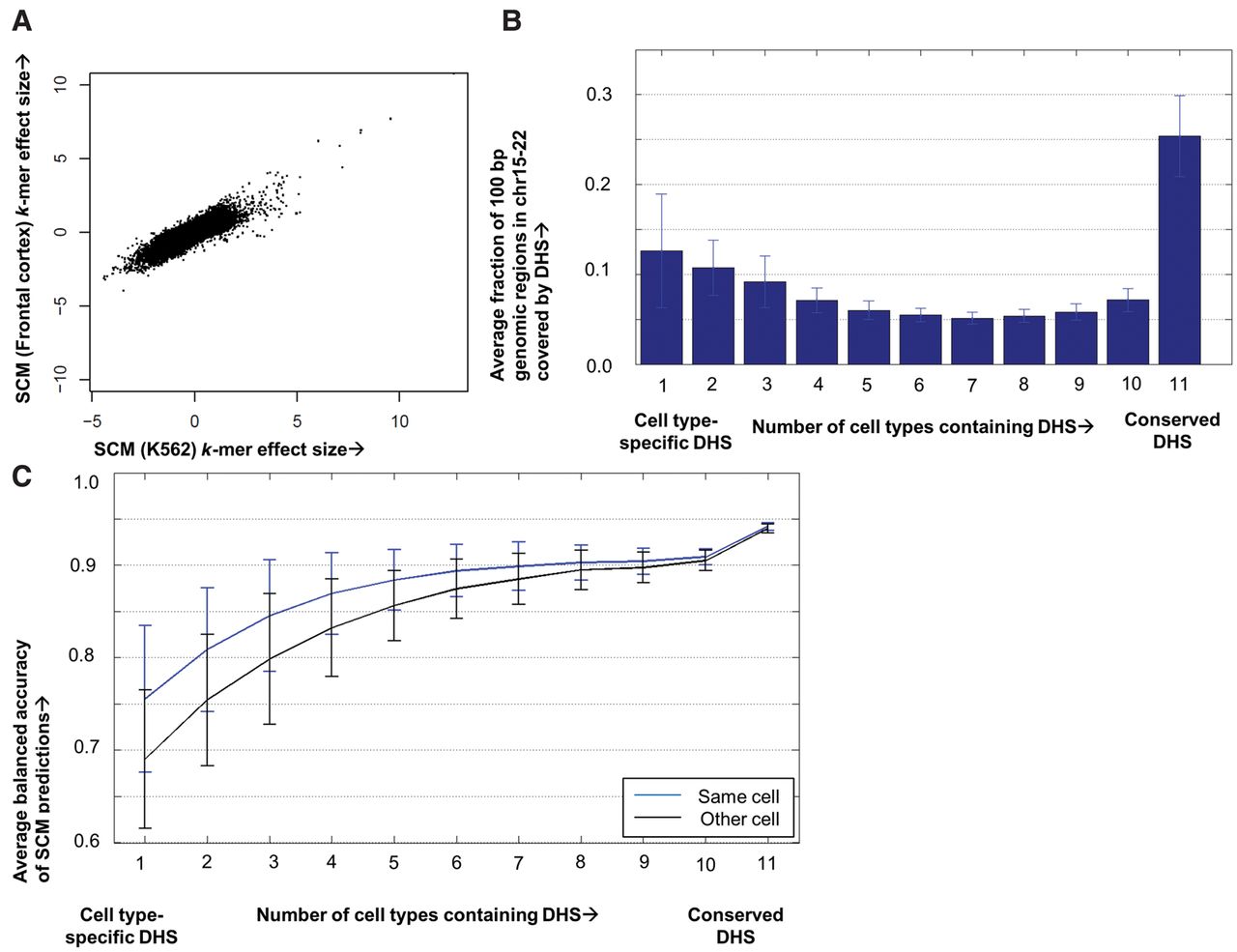
The similarity in features among SCMs trained on distinct cell types surprised us, because it is well-documented that cell-type–specific chromatin accessibility (e.g., tissue-specific enhancer activity) plays an important role in establishing cellular identity (Thurman et al. 2012; Stergachis et al. 2013; Andersson et al. 2014). In fact, it has been reported that <1% of DHS are conserved across all cell types (Thurman et al. 2012). Thus, we analyzed the cell-type specificity of DHS in our data set of 11 human cell types. To do so, we binned the raw DNase-seq data using a 100-bp smoothing window and then called DHS above a threshold of statistical significance (0.05 FDR). We then asked what percentage of the genomic space covered by DHS in a given cell type is also covered by DHS in the other 10 human cell types used in this study. We find that 25% of the genomic space (bases) covered by DHS is conserved across the 11 human data sets used in our study, and 12% is specific to that data set (Fig. 3B). These percentages are similar when Hotspot-called DHS (John et al. 2011) are used for these data sets (Supplemental Fig. 10). The fraction of hypersensitive genomic space that is cell-type specific differs from previously published numbers (Thurman et al. 2012) because of our differing definition of cell-type specificity. In our definition, we consider the fraction of a cell type's hypersensitive genomic space that is cell-type specific with respect to the other 10 data sets in our study. Prior analysis (Thurman et al. 2012) tallied DHS from all ENCODE DNase-seq data sets, calculating the cell-type specificity of regions using this larger denominator and thus leading to a lower apparent fraction of shared DHS.
Reproducing this latter analysis from all 11 data sets, we find that 7% of total DHS space is common to all cell types, whereas 42% of total DHS space is unique to only one data set (Supplemental Fig. 10). For the purposes of our work, we believe the overlap of DHS space from the lens of one data set as compared to all others (25% conserved in all 11 data sets, 12% cell-type specific) to be the most relevant statistic, because we are interested in the accuracy of SCMs on a single target cell type.
When a SCM is trained on one cell type, it can predict chromatin accessibility in a different cell type to the extent that the accessibility or underlying logic behind the accessibility are conserved between these cell types. Given the similarity among SCMs trained on different data sets, we asked whether SCMs were better at predicting conserved DHS than cell-type–specific DHS. We plotted SCM accuracy at predicting DHS binned by their conservation across the 11 human data sets. We found that SCMs are most accurate at predicting conserved DHS (94% balanced accuracy on DHS shared among all data sets) and less accurate at predicting cell-type–specific DHS (75% balanced accuracy at cell-type–specific DHS) (Fig. 3C). We then asked whether SCMs were better at predicting their own cell-type–specific DHS than they were at predicting cell-type–specific DHS from distinct cell types. We found that SCMs did in fact predict their own cell-type–specific DHS more accurately than they predicted cell-type–specific DHS of other cell types (Fig. 3C). We propose two possible (and not mutually exclusive) rationales for the poorer performance of SCMs on cell-type–specific DHS than on conserved DHS. One possibility is that the logic governing conserved DHS is better modeled by SCMs, and highly specific DHS may utilize a more conditional logic. A second possibility is that cell-type–specific DHS are on average weaker, more sparse, and more subject to noisy data, impeding SCMs from learning their features. Nevertheless, the majority of cellular chromatin accessibility appears to be predicted by a SCM.
Thus far, we have shown that SCMs perform well at quantitative predictions of genome-wide DNase-seq reads. However, sequence duplication between training chromosomes and held-out chromosomes or redundancy in genomic DNA induced by evolutionary selection pressure could allow high predictive accuracy with an overfit model that would not generalize to novel sequence. To this end, we sought to test SCM accuracy at the prediction of the accessibility of a diverse library of novel sequences in a controlled chromatin context.
To test SCM predictive accuracy on a wider range of DNA sequences, we developed Single Locus Oligonucleotide Transfer (SLOT), a novel high-throughput platform that allows the interrogation of the chromatin accessibility of a library of synthetic sequences in a controlled chromatin context. We optimized CRISPR genome editing (Cong et al. 2013; Mali et al. 2013) to maximize homologous recombination in mESCs (Supplemental Fig. 11), achieving site-specific insertion of 175-bp sequences in 20%–50% of alleles, a substantial improvement over previously published results (Cong et al. 2013; Mali et al. 2013; Findlay et al. 2014).
We designed a library of 12,000 175-bp DNA sequences to test the SCM's ability to predict chromatin accessibility of any DNA sequence in a controlled chromatin context. We developed a de Bruijn graph technique to construct novel DNA sequences with a wide range of SCM-predicted chromatin accessibility levels (Supplemental Fig. 11). Most library sequences are highly divergent from any genomic DNA sequence (Supplemental Fig. 11). Each sequence in the library is flanked by PCR primers allowing PCR amplification with tailed PCR primers for site-specific genome insertion and contains a unique barcode allowing unambiguous identification by short-read next-generation sequencing (Supplemental Fig. 11). The 100 bp in the middle of this primer-flanked template varies in each of the 12,000 sequences, and we designate this 100-bp region a DNA “phrase” because it contains a small set of sequence elements that alter the chromatin accessibility of the otherwise identical locus of integration.
We performed SLOT to integrate our library of DNA phrases with diverse predicted chromatin opening properties into a genomic locus that resides in natively inaccessible chromatin. By performing DNase hypersensitivity analysis on a pool of phrase-integrated mESCs followed by deep sequencing of phrase barcodes, we obtain quantitative information on the relative accessibility of each of the phrases in this defined chromatin environment (Fig. 4A). The phrases identified in the genomic DNA of technical replicates are highly concordant, indicating our ability to accurately quantify phrase abundance using SLOT (Supplemental Fig. 11), and we confirmed that barcodes were matched to full phrase sequences through full phrase sequencing of genomically integrated phrases (Supplemental Fig. 11). Off-target integration is rarely detected and eliminated from downstream analysis by our library preparation pipeline that includes locus-specific PCR amplification (Supplemental Information). We use genomic positive and negative control primers to ensure enrichment of DNase hypersensitive DNA before sequencing the pool of phrases.
SCMs predict sequence-dependent chromatin accessibility in a high-throughput SLOT screen. (A) In SLOT, a library of 12,000 175-bp DNA sequences containing 100-bp variable phrases is PCR amplified to add a 67-bp homology arm to each end. mESCs stably expressing a guide RNA targeting a natively heterochromatin region are co-electroporated with the DNA library, Cas9, and additional guide RNA, resulting in a population of cells in which 20%–50% of alleles at a single genomic locus have phrase incorporation. Comparison of phrase representation between genomic DNA and DNase I hypersensitive DNA reveals the subset of phrases encoding chromatin accessibility in this controlled genomic context. (B) Fraction of phrases, grouped by their overall SCM-predicted chromatin accessibility (y-axis), that are DNase I hypersensitive in a SLOT assay (x-axis). The units shown are only appropriate for trend comparison.
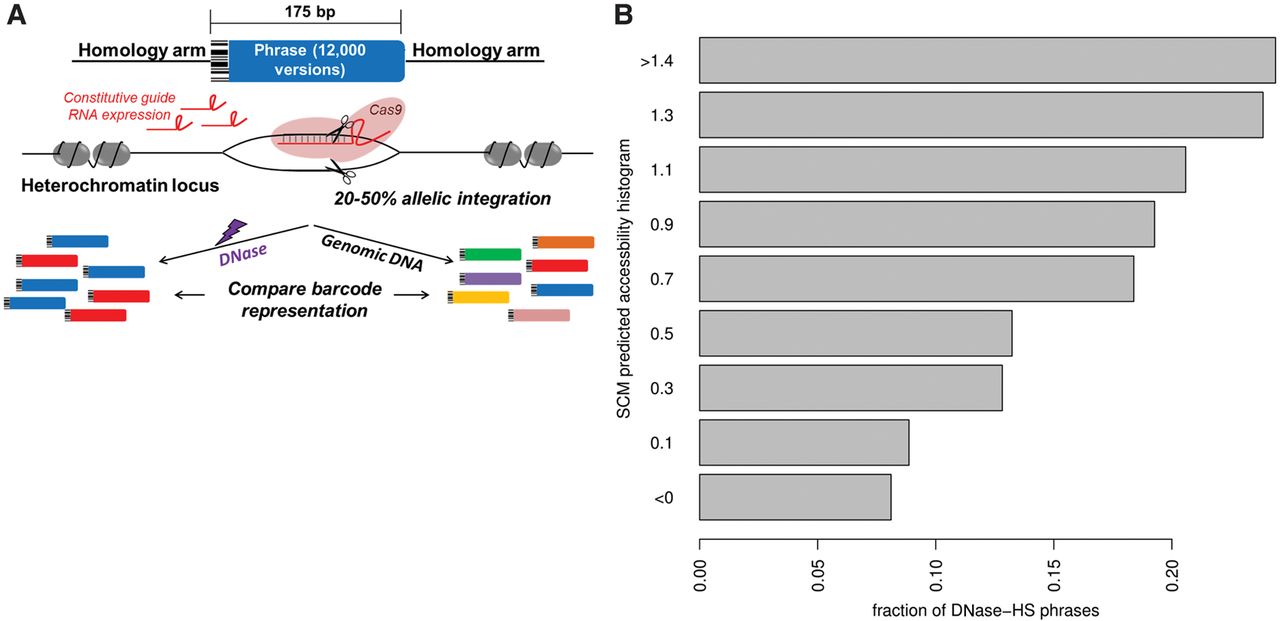
Barcode sequencing of DNase I hypersensitive phrases reveals an association between groups of phrases predicted by the SCM to promote open chromatin and those which are overrepresented in our assay (Fig. 4B; Supplemental Fig. 11). SCM is also weakly predictive when predicting individual phrases as a binary classification task without grouping of similar phrases; the degraded performance arises from noise in the individual phrase measurements (AUC = 0.60) (Supplemental Fig. 11). SLOT allows targeting the same DNA library to any genomic locus, and we have obtained similar relationships between library sequence DNase I hypersensitivity and SCM predictions in a second locus (Supplemental Fig. 11). Thus, the SCM predicts the chromatin accessibility in a uniform chromatin context of a set of sequences that often bear no resemblance to genomic DNA sequences, demonstrating that the SCM does not simply memorize DNase I hypersensitive sequences.
The ability of SCMs to accurately predict chromatin accessibility in both native and high-throughput test environments suggests that a SCM could describe a DNA-embedded logic for accessibility. Thus, we asked whether we could interpret a SCM to reveal the underlying biological paradigms driving chromatin accessibility. The fully trained mESC SCM (mESC) uses around 20,000 of the 87,380 initial k-mers to model chromatin accessibility, and models with fewer k-mers have decreased correlation with held-out data (Supplemental Fig. 12). Our recent work implicated a class of pioneer TFs in opening chromatin and two other TF classes, settler TFs and migrant TFs, in responding to preexisting chromatin accessibility (Sherwood et al. 2014). To determine whether pioneer TFs also play a role in SCM chromatin accessibility prediction, we compared a set of the 200 k-mers with the strongest SCM-predicted chromatin opening across both mESC and hESC to randomly selected k-mers with no SCM-predicted chromatin opening function, finding that the strongest SCM k-mers are highly enriched in similarity to known pioneer TF motifs (Fig. 5A). Thus, SCMs are interpretable and consistent with previous research into chromatin accessibility.
Chromatin accessibility arises from synergistic interactions, largely among pioneer TFs. (A) Enrichment of the 200 k-mers with strongest mESC SCM effect sizes in similarity to pioneer, settler, and migrant TF motifs. (B) Example position weight matrix (PWM) motifs derived from clustering the 500 k-mers with strongest mESC SCM effect size. Below the PWM are merged spatial k-mer effect sizes for all k-mers contributing to the motif within ±1000 bp of the k-mer in hESC (red), mESC (blue), and K562 (green), showing the common effects of k-mers in these cell types. Names above correspond to high-confidence database matches with TF motifs when known, and known pioneer TFs are denoted. (C,D) NRF1 ChIP-seq reads from 1-kb regions surrounding above-threshold NRF1 motif matches on held-out Chromosome 18 (y-axis) plotted versus NRF1 PWM strength (C, x-axis) or SCM (NRF1 ChIP)-predicted ChIP reads in the region (D, x-axis). Pearson's correlation coefficients are shown above each plot. (E) Receiver–operator curve (ROC) showing predictive accuracy of a SCM trained on NRF1 ChIP-seq data at predicting held-out NRF1 ChIP-seq peaks.
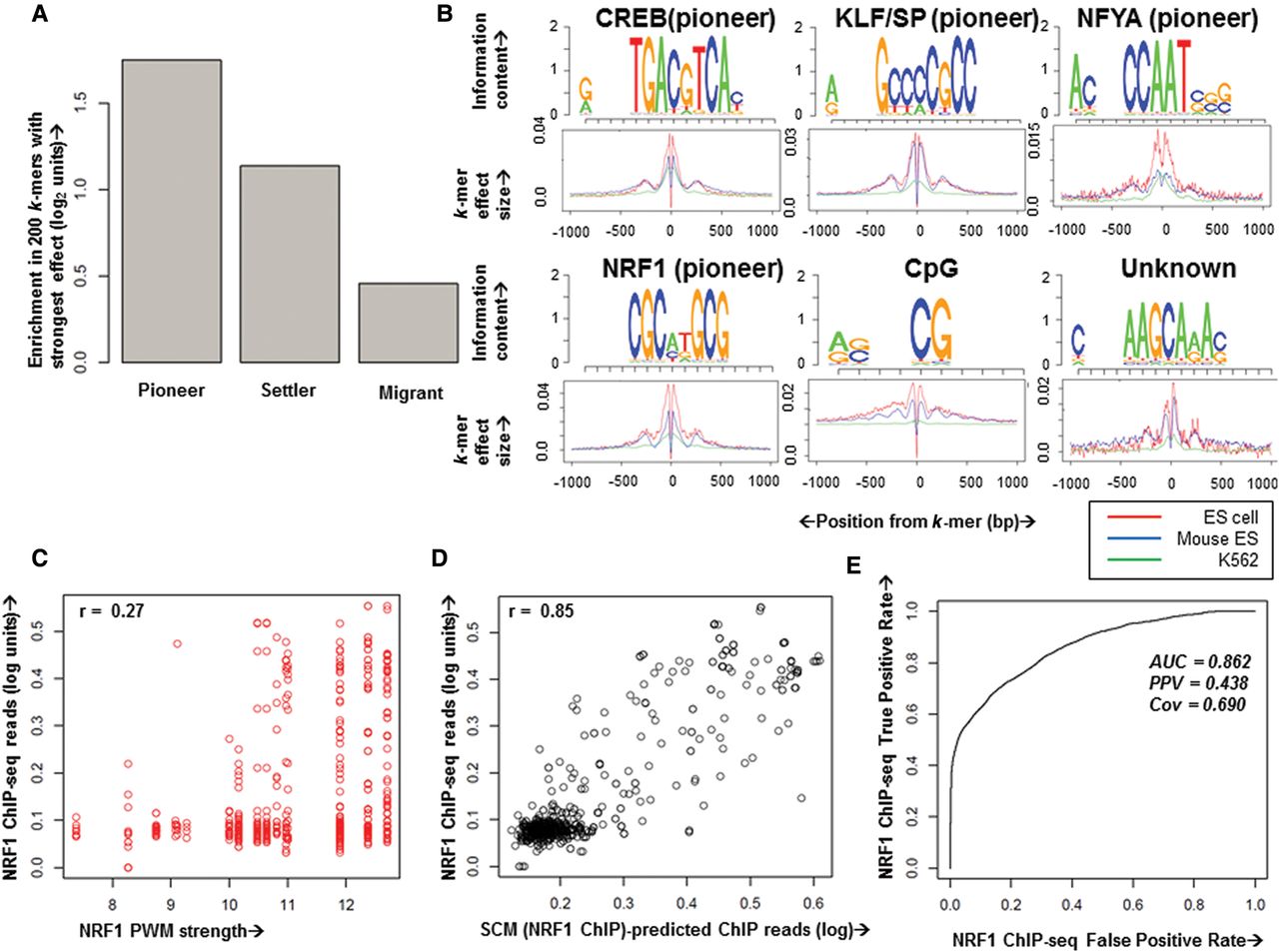
To explore the DNA sequence determinants of chromatin accessibility in more depth, we performed clustering of the top 200 k-mers in the SCM (hESC and mESC) and found that many of the strongest SCM k-mers can be clustered into position weight matrix (PWM) motifs (Fig. 5B; Supplemental Fig. 12). SCM-predicted spatial DNase-seq read patterns surrounding these motifs reveal a profile of increased surrounding hypersensitivity with a central footprint (Fig. 5B; Supplemental Fig. 12), recapitulating the stereotypical behavior of TF motifs (Sherwood et al. 2014). The majority of these PWMs show activity in SCMs trained on DNase-seq data from two human and one mouse cell types (K562, hESC, and mESC) (Fig. 5B; Supplemental Fig. 12), indicating the robustness of the PWMs across data sets and species. Some motifs extend past 8 bp, suggesting that chromatin accessibility-determining elements can be longer than the SCM's maximal k-mer length and are modeled by the SCM as collections of truncated versions (Fig. 5B; Supplemental Fig. 12). Thus, without any curation of the task, SCMs are capable of recapitulating TF motifs with spatial profiles, which is not possible with discriminative motif-finding approaches.
In addition to motifs that are highly similar to known pioneer TF motifs (Fig. 5B), SCM-identified motifs suggest a role for CpG in affecting chromatin accessibility (Fig. 5B), and still other motifs do not match known TF motifs and may represent novel TF motifs or sequences with as yet unknown roles in coordinating chromatin accessibility (Fig. 5B; Supplemental Fig. 12). Notably, canonical promoter motifs like the TATA box (Lenhard et al. 2012) are not found, which suggests that chromatin accessibility may be uncoupled from RNA polymerase recruitment. The lack of contribution of such promoter motifs to chromatin accessibility is consistent with recent computational analysis showing that canonical promoter motifs are degenerate and not statistically enriched at promoter sites (Siebert and Söding 2014). Thus, the DNA determinants underlying chromatin accessibility comprise only a subset of all possible k-mers, many of which are pioneer TF binding motifs.
Our finding that SCMs predict chromatin accessibility through modeling synergistic interactions led us to ask whether SCMs could accurately model pioneer TF binding decisions. To evaluate pioneer TF binding logic, we collected ChIP-seq data for the strong pioneer TF NRF1 (Sherwood et al. 2014) in mESCs. As expected, NRF1 binding is enriched at sites containing strong NRF1 motifs (Fig. 5C); however, even the strongest NRF1 motifs are only bound a small fraction of the time (Sherwood et al. 2014); thus, there is only weak correlation between NRF1 PWM strength and NRF1 binding (Fig. 5C). We then trained a SCM to predict NRF1 ChIP-seq reads using the same approach as for DNase-seq SCMs. Predicted reads from this SCM (NRF1 ChIP) in the 1-kb regions surrounding held-out NRF1 motifs are highly concordant with actual NRF1 ChIP-seq reads (Fig. 5D; Supplemental Fig. 13), suggesting that a SCM can accurately quantitate NRF1 binding (correlation over motif matches: 0.85; whole genome: 0.638). The SCM (NRF1 ChIP) accurately predicts the majority of held-out NRF1 ChIP locations after binary peak calling (AUC = 0.862; PPV = 0.438; TPR = 0.690) (Fig. 5E). As further validation that the SCM (NRF1 ChIP) accurately predicts NRF1 binding logic, we performed a SLOT screen integrating a library containing a wide variety of native and synthetic phrases containing NRF1 motifs into a single genomic locus. The SCM (NRF1 ChIP) accurately predicts which of the 12,000 phrases bind NRF1 in this controlled genomic context, whereas NRF1 motif strength does not (Supplemental Fig. 13). We posit that pioneer TF binding logic is in fact predictable: Undetectably weak interactions between pioneer TFs and individual binding motifs can be reinforced through a synergistic logic, resulting in an apparently binary set of bound and unbound loci genome-wide, thus contributing predictably to genomic chromatin accessibility.
Discussion
In sum, we have shown a method for predicting chromatin accessibility from DNA sequence. We have developed a computational algorithm, SCM, capable of learning a synergistic set of rules governing genome-wide chromatin accessibility. A SCM provides a range of insights and capabilities. It provides a base pair resolution estimate of how specific genomic sequences direct chromatin accessibility, and the effect profiles of these sequences are interpretable (Fig. 5B). The local, nonspecific, synergistic logic among short DNA sequences modeled by SCMs is capable of consistently achieving over 0.8 Pearson's correlation with DNase-seq data across a diverse class of human and mouse cell types. This logic predicts not only native accessibility but also accessibility of non-native sequences in a controlled chromatin context (Fig. 4B). Examination of key k-mers showed a correspondence to known pioneer motifs, and we demonstrated that the same synergistic logic was able to predict the binding of the pioneer TF NRF1. SCM code and output data are available for use with any DNase-seq data set.
The SLOT method we developed is independently valuable, because it enables the integration of a defined library of DNA sequences into any target genomic locus, such that the effect of the integrated sequence can be measured in a common genomic sequence context. In this work, we showed that the synergistic DNA logic uncovered by SCMs predicts which sequences are sufficient to open chromatin and enable NRF1 binding when inserted into two genomic sites with natively inaccessible chromatin. To do so, we paired SLOT with DNase I hypersensitivity and ChIP assays. The SLOT assay could additionally be paired with assays for DNA methylation, histone modification, or gene expression to gain insight into how DNA sequence encodes these functions in controlled genomic contexts as well as elucidating cell-state–specific chromatin accessibility and gene regulation when applying the same DNA library across different cell states. In addition, SLOT could be used to engineer specific genomic sites to have a desired level of chromatin accessibility.
Although highly accurate, our model is unable to predict every accessible site in the genome. These overlooked sites may not occur frequently enough in our training data to impact learning of model likelihood, may use a more combinatorial logic of specific cofactor interactions, or may involve nonlocal chromatin state spreading. Specifically, differences in chromatin accessibility among cell types play a large role in determining cellular identity (Thurman et al. 2012; Stergachis et al. 2013), and some of the coding mechanisms regulating cell-type–specific chromatin accessibility remain to be determined. Because chromatin accessibility is a common feature of promoters, enhancers, and other gene regulatory elements, it remains to be seen whether the functional differences among these subclasses of elements are encoded using the logic we identified or whether the DNA encodes a distinct set of sequences layered on top of the accessibility logic to distinguish among regulatory element subclasses (McVicker et al. 2013). Additionally, emerging techniques that permit live tracking of gene regulation (Stasevich et al. 2014) will be important to shed new light on the dynamic process by which protein–DNA interactions govern chromatin accessibility. Unraveling the codes underlying gene regulation will aid efforts to guide stem cell differentiation and reprogramming and to explain disease-associated noncoding sequence variation.
Methods
The SCM model is a type of regularized generalized linear model (GLM). We introduce the motivation and inference procedure for SCM briefly. The Supplemental Material contains a more detailed exposition of our framework. Our goal is to produce a predictive model of sequence to a quantitative, integer-valued trait measured per base on the genome.
The design of our algorithm is guided by the following goals:
Predictive model: Our model should predict traits that can be held out and evaluated for goodness of fit. This makes the overall problem well-defined and easy to evaluate.
Parameter independence: The model should not have any performance-influencing parameters. All parameters that can be set should be set as large as memory and computation time allows.
Tractable runtime: The model should run in less than several days for any number of experiments on the human genome.
Interpretable parameters: The output parameters should be interpretable as the local effects of a k-mer.
Theoretical grounding: The model should provide reasonable theoretical guarantees on model recovery and prediction capacity.
In the paper, we used a maximum k-mer length of 8, which was the maximum that would fit in memory in an Amazon EC2 c3.8xlarge instance. Larger k-mers tested on a larger memory machine did not perform substantially better than 8-mers.
Notation and genome representation
Throughout, we assume that the genome consists of one large chromosome with coordinate 0 to N. In practice, we will construct this by concatenating chromosomes with the telomeres acting as a spacer. The variable K represents the maximum k-mer length considered; the model fits all k-mers from 1, …, K. The variable M represents the influence of each k-mer.
The regularization parameter η is a scalar representing our belief about the sparsity of the problem.
Whenever possible, we will use i for genomic coordinate, k for k-mer length, and j for coordinate offset from the start of a k-mer.
The input variable c is a vector of length N representing counts and ci represents the read-count observed at base i.
The latent variable λ is a vector of length N representing the current estimate for c using θ.
θk is the parameter matrix of size 4k × 2M associated with the set of all k-mers.
The variable gk is a mapping from genomic coordinate i to the k-mer starting at i. The k-mer for gk is represented as an integer that maps to rows of θ such that the gkth row of θk is the effect of a k-mer starting at coordinate i.
For instance, is the 4-mer starting at coordinate i. If this is ATCG, then the row must be the effect that ATCG exerts on its neighbors.
The special parameter θ0 is used to set the average read rate of the genome globally.
Model setup
The problem we solve is a regularized Poisson regression. We would like to maximize the following:
The intermediate variables λ are defined by:
Naive inference algorithm
We describe a simple method for fitting this model for expository purposes. The actual method uses several acceleration techniques described in the Supplemental Material. Due to the convexity of regularized Poisson regression, these additional tricks do not change the global optimum of the model.
1. Given current iterate θ, calculate current λ for all bases by
2. Given current λ, calculate the per base gradient vector
3. Propagate the errors back to the parameter θ. Let s be the integer index corresponding to a k-mer. Then the gradient of this k-mer s with offset j is
and
4. Update the current parameter with stepsize alpha:
5. Update the constant offset
6. Apply the proximal operator for L1 regularization
This algorithm is prohibitively slow, with an iteration runtime of O(NMK + 4KM). In practice, contribution from NMK dwarfs that of 4KM since the gradient computation is cache incoherent and N ≈ 3 × 109, which is much greater than 4KM ≈ 6 × 104. Accelerated methods for inference using this model are described in Supplemental Material.
There are two free parameters (α and η). The value for η is set via grid-search over values of η using held-out sets starting with the maximal feasible η. This maximum is calculated analytically as the maximal η for which all k-mers are nonzero. We discuss setting α in Supplemental Material.
Cell culture
Mouse embryonic stem cell culture was performed according to previously published protocols (Sherwood et al. 2014). Undifferentiated 129P2/OlaHsd mouse ES cells were maintained on gelatin-coated plates feeder-free in mES media composed of Knockout DMEM (Life Technologies) supplemented with 15% defined fetal bovine serum (FBS) (HyClone), 0.1 mM nonessential amino acids (Life Technologies), Glutamax (Life Technologies), 0.55 mM 2-mercaptoethanol (Sigma), 1X ESGRO LIF (Millipore), 5 nM GSK-3 inhibitor XV, and 500 nM UO126. Cells were regularly tested for mycoplasma. Genetic manipulations to stem cell lines are described below.
DNase-seq
DNase-seq was performed as described previously (Sherwood et al. 2014). Between 10 million and 100 million cells were digested with 60–100 units of DNase I (Promega) per 107 nuclei. Using E-Gel SizeSelect Agarose 2% gels (Life Technologies), 50–125 bp hypersensitive DNA was collected. Library preparation and Illumina HiSeq were performed by the MIT BioMicroCenter.
ChIP-seq
ChIP was performed according to the “Mammalian ChIP-on-chip” protocol (Agilent) using a polyclonal antibody against NRF1 antibody (ab34682, Abcam) and Protein G Dynabeads (Life Technologies). Between 10 million and 100 million cells were used for each experiment. qPCR using positive and negative control primers was performed to ensure ChIP enrichment. Library preparation and Illumina HiSeq were performed by the MIT BioMicroCenter.
Single Locus Oligonucleotide Transfer (SLOT)
A library of 175-bp oligonucleotide sequences containing 100-bp variable phrases was designed with the following common features: flanking primer sequences distinct from any genomic DNA sequence, a unique DNA barcode distinct from all other barcodes at Levenshtein distance = 2, and a common internal primer past the barcode (see Supplemental Fig. 8) from Broad Technology Services. This library was amplified using primers that add 67-bp homology arms to each end using NEBNext High-Fidelity 2× PCR Master mix (New England Biolabs), because we found that this polymerase minimized library amplification bias. Homology arms were designed to flank two genomic CRISPR guide RNA sequences in genomic regions with no surrounding DNase-seq activity in mESC.
PCR-amplified libraries were electroporated along with Cas9 expression plasmid and sgRNA expression plasmid into mESCs constitutively expressing a locus-specific sgRNA. For the experiments described in this work, we electroporated 107 mESC with 20 µg of each component DNA, achieving 20%–50% allele frequency in all three loci. Library-integrated mESCs were grown for 7–21 d after electroporation before DNase I hypersensitivity analysis, and care was taken to maintain high pool complexity by splitting at high density.
DNase I hypersensitivity analysis was performed mostly according to our previously published protocol (Sherwood et al. 2014) with several differences. Immediately after nuclear extraction, 5%–10% of nuclei were reserved for genomic DNA isolation to serve as a control. The remaining nuclei were treated with 70–90 units of DNase I per 107 cells. After DNA purification, E-gel size-selection was performed to isolate 125–275 bp DNA, a size range that accommodates the minimal size required to amplify with locus-specific and internal primers (see Supplemental Fig. 8). qPCR using positive and negative control primers was performed to ensure enrichment of DNase-hypersensitive DNA. Then, we performed a three-step library preparation to allow Illumina deep-sequencing analysis of barcode representation (see Supplemental Fig. 8). For the experiments reported in this work, we used 70-bp single-end Illumina MiSeq, performed by the MIT BioMicroCenter. Full phrases were also sequenced from genomic DNA using a similar library preparation strategy as above but using the flanking primer instead of the internal primer to amplify locus-integrated phrases. These samples were sequenced using 150 bp paired-end MiSeq.
Data access
Sequencing data and associated fitted models from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE80105. SCM source code and data are available in the Supplemental Material and at http://scm.csail.mit.edu.
Acknowledgments
We thank the MIT BioMicro Center, Tarjei Mikkelsen, Broad Technology Laboratories, Koichi Kawakami, and Feng Zhang for reagents and technical assistance, and Richard Maas for help with the manuscript. We acknowledge funding from The National Institutes of Health 5UL1DE019581, RL1DE019021, 1K01DK101684-01, 1U01HG007037, and 5P01NS055923 to D.K.G.; and the Harvard Stem Cell Institute's Sternlicht Director's Fund award and Human Frontier Science Program grant to R.I.S.
Author contributions: Experiments were designed by T.H., R.I.S., and D.K.G. DNase-seq and SLOT experiments were designed and conducted by R.I.S., A.A.B., B.J.M.E., and S.S. SCM was designed and implemented by T.H. and D.D.K. Computational analysis was performed by T.H., D.D.K., H.Y., N.R., T.J., and D.K.G. The manuscript was prepared by R.I.S., T.H., and D.K.G.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.199778.115.
References
- ↵Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, 2014. An atlas of active enhancers across human cell types and tissues. Nature 507: 455–461.
- ↵Bailey TL. 2011. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 27: 1653–1659.
- ↵Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. 2008. High-resolution mapping and characterization of open chromatin across the genome. Cell 132: 311–322.
- ↵Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218.
- ↵Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823.
- ↵Duchi J, Hazan E, Singer Y. 2011. Adaptive subgradient methods for online learning and stochastic optimization. J Mach Learn Res 12: 2121–2159.
- ↵The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74.
- ↵Ernst J, Kellis M. 2012. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods 9: 215–216.
- ↵Findlay GM, Boyle EA, Hause RJ, Klein JC, Shendure J. 2014. Saturation editing of genomic regions by multiplex homology-directed repair. Nature 513: 120–123.
- ↵Gaulton KJ, Nammo T, Pasquali L, Simon JM, Giresi PG, Fogarty MP, Panhuis TM, Mieczkowski P, Secchi A, Bosco D, 2010. A map of open chromatin in human pancreatic islets. Nat Genet 42: 255–259.
- ↵Ghandi M, Lee D, Mohammad-Noori M, Beer MA. 2014. Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput Biol 10: e1003711.
- ↵Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. 2007. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res 17: 877–885.
- ↵Gualdi R, Bossard P, Zheng M, Hamada Y, Coleman JR, Zaret KS. 1996. Hepatic specification of the gut endoderm in vitro: cell signaling and transcriptional control. Genes Dev 10: 1670–1682.
- ↵He X, Samee MA, Blatti C, Sinha S. 2010. Thermodynamics-based models of transcriptional regulation by enhancers: the roles of synergistic activation, cooperative binding and short-range repression. PLoS Comput Biol 6: e1000935.
- ↵He HH, Meyer CA, Hu SS, Chen MW, Zang C, Liu Y, Rao PK, Fei T, Xu H, Long H, 2013. Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat Methods 11: 73–78.
- ↵Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, 2009. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods 6: 283–289.
- ↵Huggins P, Zhong S, Shiff I, Beckerman R, Laptenko O, Prives C, Schulz MH, Simon I, Bar-Joseph Z. 2011. DECOD: fast and accurate discriminative DNA motif finding. Bioinformatics 27: 2361–2367.
- ↵John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, Hager GL, Stamatoyannopoulos JA. 2011. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet 43: 264–268.
- ↵Kelley DR, Snoek J, Rinn JL. 2016. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res 26: 990–999.
- ↵Lazarovici A, Zhou T, Shafer A, Dantas Machado AC, Riley TR, Sandstrom R, Sabo PJ, Lu Y, Rohs R, Stamatoyannopoulos JA, 2013. Probing DNA shape and methylation state on a genomic scale with DNase I. Proc Natl Acad Sci 110: 6376–6381.
- ↵Lee D, Karchin R, Beer MA. 2011. Discriminative prediction of mammalian enhancers from DNA sequence. Genome Res 21: 2167–2180.
- ↵Lenhard B, Sandelin A, Carninci P. 2012. Metazoan promoters: emerging characteristics and insights into transcriptional regulation. Nat Rev Genet 13: 233–245.
- ↵Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339: 823–826.
- ↵McVicker G, van de Geijn B, Degner JF, Cain CE, Banovich NE, Raj A, Lewellen N, Myrthil M, Gilad Y, Pritchard JK. 2013. Identification of genetic variants that affect histone modifications in human cells. Science 342: 747–749.
- ↵Setty M, Leslie CS. 2015. SeqGL identifies context-dependent binding signals in genome-wide regulatory element maps. PLoS Comput Biol 11: e1004271.
- ↵Sherwood RI, Hashimoto T, O'Donnell CW, Lewis S, Barkal AA, van Hoff JP, Karun V, Jaakkola T, Gifford DK. 2014. Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat Biotechnol 32: 171–178.
- ↵Siebert M, Söding J. 2014. Universality of core promoter elements? Nature 511: E11–E12.
- ↵Song L, Zhang Z, Grasfeder LL, Boyle AP, Giresi PG, Lee BK, Sheffield NC, Gräf S, Huss M, Keefe D, 2011. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res 21: 1757–1767.
- ↵Soufi A, Donahue G, Zaret KS. 2012. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 151: 994–1004.
- ↵Stasevich TJ, Hayashi-Takanaka Y, Sato Y, Maehara K, Ohkawa Y, Sakata-Sogawa K, Tokunaga M, Nagase T, Nozaki N, McNally JG, 2014. Regulation of RNA polymerase II activation by histone acetylation in single living cells. Nature 516: 272–275.
- ↵Stergachis AB, Neph S, Reynolds A, Humbert R, Miller B, Paige SL, Vernot B, Cheng JB, Thurman RE, Sandstrom R, 2013. Developmental fate and cellular maturity encoded in human regulatory DNA landscapes. Cell 154: 888–903.
- ↵Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, 2012. The accessible chromatin landscape of the human genome. Nature 489: 75–82.
- ↵Veitia RA. 2003. A sigmoidal transcriptional response: cooperativity, synergy and dosage effects. Biol Rev Camb Philos Soc 78: 149–170.
- ↵Weintraub H, Groudine M. 1976. Chromosomal subunits in active genes have an altered conformation. Science 193: 848–856.
- ↵Wu C. 1980. The 5′ ends of Drosophila heat shock genes in chromatin are hypersensitive to DNase I. Nature 286: 854–860.
- ↵Zaret KS, Carroll JS. 2011. Pioneer transcription factors: establishing competence for gene expression. Genes Dev 25: 2227–2241.
- ↵Zhou J, Troyanskaya OG. 2015. Predicting effects of noncoding variants with deep learning–based sequence model. Nat Methods 12: 931–934.