
The nonessentiality of essential genes in yeast provides therapeutic insights into a human disease
- 1Key Laboratory of Gene Engineering of Ministry of Education, Collaborative Innovation Center for Cancer Medicine, School of Life Sciences, Sun Yat-sen University, Guangzhou 510275, China;
- 2State Key Laboratory of Biocontrol, School of Life Sciences, Sun Yat-sen University, Guangzhou 510275, China
- Corresponding author: hexiongl{at}mail.sysu.edu.cn
Abstract
Essential genes refer to those whose null mutation leads to lethality or sterility. Theoretical reasoning and empirical data both suggest that the fatal effect of inactivating an essential gene can be attributed to either the loss of indispensable core cellular function (Type I), or the gain of fatal side effects after losing dispensable periphery function (Type II). In principle, inactivation of Type I essential genes can be rescued only by re-gain of the core functions, whereas inactivation of Type II essential genes could be rescued by a further loss of function of another gene to eliminate the otherwise fatal side effects. Because such loss-of-function rescuing mutations may occur spontaneously, Type II essential genes may become nonessential in a few individuals of a large population. Motivated by this reasoning, we here carried out a systematic screening for Type II essentiality in the yeast Saccharomyces cerevisiae. Large-scale whole-genome sequencing of essentiality-reversing mutants reveals 14 cases whereby the inactivation of an essential gene is rescued by loss-of-function mutations on another gene. In particular, the essential gene encoding the enzyme adenylosuccinate lyase (ADSL) is shown to be Type II, suggesting a loss-of-function therapeutic strategy for the human disorder ADSL deficiency. A proof-of-principle test of this strategy in the nematode Caenorhabditis elegans shows promising results.
There are usually hundreds to thousands of essential genes in an organism (Giaever et al. 2002; Kobayashi et al. 2003; Baba et al. 2006; Harris et al. 2010). It is often assumed that essential genes execute the functions that are indispensable to cellular life (Mushegian and Koonin 1996; Koonin 2000). For example, in the bacterium Escherichia coli, the essential gene dnaA is responsible for DNA replication initiation, tufA is responsible for RNA elongation in transcription, and infA is responsible for translation initiation (Gerdes et al. 2003). The counterexamples, however, can be easily conceived. Suppose there are 10 genes encoding a stable protein complex that carries out a dispensable function in a cell. Inactivation of one of the 10 genes may cause dosage imbalance, generating a toxic intermediate that is lethal to the cell. Under this circumstance, each of the 10 genes may appear essential, while they together execute a dispensable function. Sporadic reports of such genetically conditional essentiality can be found in the literature (Zekhnov et al. 1995; Zhao et al. 1998; Delia et al. 2009; Bergmiller et al. 2012). For example, in the toxin-antitoxin system of bacteria, deletion of the antitoxin gene is often fatal because of the cellular toxicity conferred by the remaining toxin gene. The antitoxin gene would become nonessential if the corresponding toxin gene does not exist. An in silico analysis of metabolic network models tested systematically how genetic background could affect gene essentiality in E. coli and Saccharomyces cerevisiae and predicted frequent conditional essentiality (Motter et al. 2008). These data collectively suggest two types of essential genes: Type I essential genes execute “core” functions that are indispensable to the organism; Type II essential genes execute “periphery” functions that are dispensable to the organism, the lack of which, however, results in fatal side effects. Such a conceptual separation is meaningful, because inactivation of a Type I essential gene can be rescued only by restoration of the core function, while inactivation of a Type II essential gene could be rescued by further loss-of-function mutations on another gene to suppress the otherwise lethal side effects (Fig. 1). This reasoning has important implications to medical science because partial loss-of-function mutations on essential genes are involved in at least several hundred human inheritance disorders (Hamosh et al. 2005; Goh et al. 2007; Park et al. 2008). The prevailing therapeutic strategies for these diseases are to restore the lost functions that are believed to be indispensable (Garcia-Blanco et al. 2004; Maguire et al. 2008; Bidou et al. 2012). Availability of Type II essentiality suggests a possibility of using loss-of-function therapy to eliminate the disease-causing side effects in some disorders, which is conceptually much easier than the conventional gain-of-function therapy.
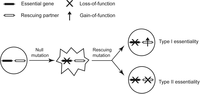
Two types of essential genes whose null phenotypes are masked by either gain of the original function (Type I) or loss of another function (Type II).
To assess the generality of Type II essentiality, we here examined the model organism yeast S. cerevisiae. We reasoned that Type II essential genes may become nonessential in a few individuals of a large population of yeast cells since the potential rescuing mutations may occur spontaneously. By sequencing genomes of the few individuals with such rescuing mutations, we could identify the mutations and consequently know the genes that interact with the focal essential genes. Indeed, we obtained 17 such gene pairs where inactivation of an essential gene is rescued by loss-of-function mutations on another gene, revealing a total of five Type II essential yeast genes. Of particular interest was the gene encoding adenylosuccinate lyase, an enzyme in the purine de novo synthesis pathway. Partial loss-of-function mutations on this gene in humans cause adenylosuccinate lyase deficiency (ADSL; OMIM 103050), a rare Mendelian disorder with mental retardation and seizures as typical symptoms (Georges and Van den Berghe 1984; Jaeken et al. 1988; Van den Berghe et al. 1997). Accordingly, we suggested a loss-of-function therapeutic strategy to suppress the phenotypes of ADSL deficiency, and a proof-of-principle test of the strategy in the nematode C. elegans showed promising results.
Results
The rationale of using spontaneous mutations to reveal Type II essentiality
We started with the Yeast Tet-promoters Hughes Collection (yTHC) that is composed of 800 strains; in each strain the endogenous promoter of an essential gene is replaced by a tetracycline promoter (Mnaimneh et al. 2004). In principle, for a given strain the expression of the focal essential gene will be shut off upon the addition of doxycycline to the growth medium (Dox+ medium), resulting in no viable cells. However, if the focal essential gene is Type II whose expression shut-off can be rescued by inactivation of other genes, there might be some viable individuals among a large number of cells because such rescuing mutations can occur spontaneously despite at a low rate. In other words, we may simply plate a certain number of cells of a yTHC strain onto an agar plate supplied with doxycycline. Some viable clones would appear if the focal essential gene is Type II.
Using simulation, we estimated the number of cells required for capturing the spontaneous rescuing mutations that can mask Type II essentiality. We considered only critical loss-of-function mutations that include severe missense mutations, nonsense mutations, and frame-shift indels. We started from a single cell and ended with a population of a given size N. The probability that at least one critical loss-of-function mutation occurs during the population expansion was estimated for each of the ∼6000 yeast genes (Methods). Up to ∼86% of the yeast genes could reach a probability of >80% when N = 6 × 107, and no apparent increase was observed with larger Ns (Fig. 2A). The remaining genes with a lower probability of acquiring a critical loss-of-function mutation in the population of N = 6 × 107 were mostly of short length (Fig. 2B), and it seemed difficult to achieve a satisfactory detection probability for the ∼15% yeast genes with a length of <800 base pairs. At any rate, the simulation results suggested a reasonably high probability of capturing the potential rescuing mutations if N = 6 × 107 yeast cells are tested.
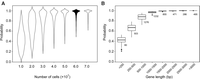
The probability of individual genes with at least one critical loss-of-function mutation during the population expansion from a single cell. (A) The probability distribution of the ∼6000 yeast genes (y-axis) as a function of the ending population size N (x-axis). The probabilities have no apparent increase after the N reaches 6 × 107. (B) The probability distribution (y-axis) as a function of the gene length (x-axis) when N = 6 × 107. The number of genes in each length category is shown next to the box.
Characterization of Type II essentiality
Among the ∼800 yTHC yeast strains, some can grow well on the Dox+ medium. This is possibly because the Tet promoter is not 100% shut off, confounding the above strategy of characterizing Type II essential genes. To circumvent this problem, for each yTHC stain we first tested 104–105 cells and obtained 280 strains, each showing no single colony on Dox+ agar (Supplemental Table S1). Only these strains are further examined. For each of the 280 strains, we tested ∼6 × 107 cells on Dox+ agar plates and observed a few colonies on some plates (Fig. 3A; Methods). There are two possibilities underlying this observation: Mutations that invalidate the Tet-off system occurred such that the focal essential gene expresses normally on the Dox+ agar; alternatively, mutations that mask the focal gene essentiality occurred. In cases of the second possibility, we should be able to remove physically the focal essential genes from the genomes of the clones observed on the Dox+ plates. We carried out the corresponding gene deletions using homologous recombination and confirmed that, for five essential genes ADE13, ERD2, MIM1, SEC14, and RER2, there are viable clones on the Dox+ plates explained by the second possibility (Methods).
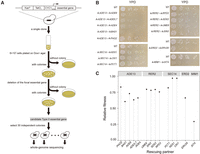
Identification of Type II essential genes and their rescuing partners. (A) A flowchart of the experimental procedure. (B) The colony size of each of the double gene deletion segregants. Cells diluted in gradient were spread on a YPD plate and incubated for 48 h, except for the ΔMIM1 + ΔSIT4, which was incubated for 72 h. Each panel shows a Type II essential gene and its rescuing partners. (C) The fitness of each double gene deletion segregant relative to the wild type in YPD.
We focused on the five essential genes and expanded the plating experiment to obtain for each strain 30 independent clones wherein the focal essential genes can be deleted. To map the potential rescuing mutations, we sequenced the genomes of the 30 × 5 = 150 clones and compared them with that of their parental clone (Methods). There were often quite a few de novo mutations found in a clone. For the 30 clones with the same focal essential gene, independent mutations found in the same gene(s) are likely to be the ones that mask the focal gene essentiality. Indeed, we found a dozen genes each enriched with mutations (Table 1). Interestingly, these mutations appear to be mostly loss-of-function, suggesting that inactivation of these genes may mask the otherwise lethal effect resulting from inactivation of the focal essential gene, a phenomenon characteristic of the Type II gene essentiality (Supplemental Table S2).
Identification of putative Type II essential genes and their rescuing partners
To test this we carried out the corresponding gene deletions. We started with BY4743, a diploid yeast strain with the wild-type genotype unless otherwise stated, and deleted one copy of the focal essential gene and one copy of the putative rescuing partner gene (Methods). Analysis of the haploid gametes of the diploid double heterozygous deletion mutants help confirm whether the focal essential genes are indeed Type II. For example, ∼80 gametes of the double heterozygous deletion mutant ADE13/Δade13+PHO2/Δpho2 were genotyped and the numbers of gametes with genotypes of ADE13+PHO2, ADE13+Δpho2, Δade13+PHO2, and Δade13+Δpho2 are 33, 23, 0, and 30, respectively. The largely normal growth of the mutant Δade13+Δpho2 indicates that deletion of PHO2 masks well the lethal effect of deleting ADE13. Overall, we obtained 14 cases in which inactivating one of the five essential genes is rescued by inactivation of another gene (Table 2). Comparison to the genetic interaction data curated by BioGRID (Stark et al. 2006) showed that 11 out of the14 cases are novel, and three interactions, including SEC14-KES1, SEC14-CKI1, and SEC14-PCT1, have been reported before (Li et al. 2002). The rescuing effects are generally strong since the growth rates of the double deletion mutants in the rich medium YPD are often over 70% of the wild-type rate (Fig. 3B,C).
The number of haploid segregants of each type produced by the corresponding diploid double heterozygous deletion mutants
Mechanisms underlying the Type II essentiality
Gene Ontology (GO) analysis showed that the five Type II essential genes and their rescuing partners often have the same GO terms (Table 1). For example, the essential gene ERD2 and its nonessential partner ERV29 are both involved in “regulation of endoplasmic reticulum (ER) to Golgi vesicle-mediated transport.” ERD2 is responsible for the recycling of vesicle-associated proteins between the ER and Golgi, and suppression of ERD2 leads to abnormal Golgi structure due to the accumulation of too many proteins in the Golgi (Hardwick et al. 1992; Townsley et al. 1994). Interestingly, ERV29 is responsible for transporting proteins from the ER into the Golgi. This suggests a likely mechanism of the ERD2-ERV29 interaction: Suppression of ERD2 causes detrimental protein accumulation in the Golgi, which is alleviated by suppressing ERV29 to slow down the transport of proteins into the Golgi. In line with this, it has been shown that overexpression of SED1, SED4, SED5, and SEC12, four genes responsible for transporting proteins out of the Golgi, can rescue the otherwise lethal effect of deleting ERD2 (Hardwick et al. 1992).
A more intriguing case is the essential gene ADE13, which encodes the enzyme adenylosuccinate lyase (ADSL) and has five nonessential rescuing partners revealed by the above screening. These genes are all related to de novo purine biosynthesis. A close examination of the de novo purine biosynthesis pathway showed that, with the exception of ADE13, all genes in the pathway are nonessential with negligible fitness reduction upon deletion (Giaever et al. 2002; Qian et al. 2012). This suggested that the process of de novo purine biosynthesis per se is dispensable to the yeast cell in the rich medium YPD, so the essentiality of ADE13 must be due to factors unrelated to purine production. We hypothesized that deletion of the enzyme gene results in accumulation of its substrates, which might be toxic to the cell and thus fatal to the yeast. If this hypothesis is true, deletion of the upstream genes to block the production of S-AMP and SAICAR, the two substrates of ADE13, should be able to mask the effect of ADE13 deletion (Fig. 4A).
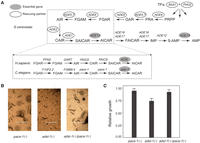
A loss-of-function therapy for the ADSL deficiency. (A) A schematic presentation of the purine de novo synthesis pathway. The pathway is nearly identical among yeast, nematodes, and humans. The filled ovals mark the essential gene and the unfilled ovals show its Type II rescuing partners. (B) The worms were fed with vectors producing the double-stranded RNAs each silencing adsl-1, pacs-1, and adsl-1/pacs-1, respectively. Knockdown of pacs-1 masks well the phenotypic defects of adsl-1 knockdown. Scale bars, 1000 μm. (C) The relative growth of the F1 worms from Day 3 to Day 6 compared to a negative control wherein the worms were fed with an empty vector. The worms of adsl-1(−)/pacs-1(−) grow significantly better than those of adsl-1(−) (P < 1.4 × 10−3; Welch's t-test). Error bar, 1 SEM.
Consistent with this hypothesis, ADE4, ADE5,7, ADE6, BAS1, and PHO1, the five rescuing partners, are all upstream of ADE13 in the pathway. Specifically, ADE4, ADE5,7, and ADE6 encode enzymes directly involved in the de novo purine biosynthesis, and BAS1 and PHO1 are two transcription factors regulating the pathway (Denis et al. 1998). However, our screening provides no evidence that the other three upstream genes in the pathway, namely ADE8, ADE2, and ADE1, can mask the effect of ADE13 deletion. Notably, a previous study showed that inactivation of ADE2 can mask the lethal effect of ADE13 deletion in the medium YPD (Zekhnov and Domkin 2000). We thus used target gene deletions and confirmed that deleting each of the three genes is able to mask the otherwise lethal effect of ADE13 deletion. This result well supported the above substrate accumulation hypothesis for explaining the Type II essentiality of ADE13. ADE13 catalyzes two reactions with S-AMP and SAICAR as substrates, respectively (Van den Bergh et al. 1993). It is unclear which one is responsible for the lethality of ADE13 deletion. If S-AMP is the toxin, deletion of ADE12, the gene encoding the enzyme directly responsible for its production, should mask the effect of ADE13 deletion. Alternatively, deletion of ADE1, the gene encoding the enzyme phosphoribosylaminoimidazole carboxylase (PAICS), should mask the effect of ADE13 deletion if SAICAR is the toxin. We successfully obtained mutants of the genotype Δade13+Δade1 but failed to obtain the genotype Δade13+Δade12 after analyzing 17 gametes of the diploid double heterozygous deletion mutant ADE13/Δade13+ADE12/Δade12 (P < 0.003; χ2 test), suggesting that accumulation of SAICAR, rather than S-AMP, explains the lethal effect of ADE13 deletion.
Loss-of-function therapy for the loss-of-function ADSL disease
The purine de novo biosynthesis pathway is evolutionarily conserved among various lineages including yeast, nematodes, and mammals (Fig. 4A; Kanehisa and Goto 2000). Importantly, partial loss-of-function mutations on the human gene encoding the enzyme ADSL cause ADSL deficiency, a rare Mendelian disease with mental retardation and seizures as typical symptoms (Georges and Van den Berghe 1984; Jaeken et al. 1988; Van den Berghe et al. 1997). To date, there is no treatment with proven clinical efficacy for this disease despite a variety of therapeutic attempts (Jaeken et al. 1988; Salerno et al. 1998, 2002; Ciardo et al. 2001; Jurecka et al. 2008). For such essential gene-related loss-of-function diseases, a gain-of-function therapeutic strategy is predominant. However, recognition of the Type II essentiality of ADSL suggests a loss-of-function therapeutic strategy to suppress the phenotypes of ADSL deficiency, which is conceptually much easier than the conventional gain-of-function strategy to restore the lost function. We decided to test this idea in the nematode C. elegans. The nematode genes encoding the two enzymes ADSL and PAICS are adsl-1 and pacs-1, respectively. RNA interference (RNAi) was used to mimic the loss of function of the two genes in C. elegans (Methods).
Body length was used to assess the effects of knocking down the C. elegans genes. We considered the average increase of body length of the worms from Day 3 to Day 6 after their birth. As expected, knocking down the essential gene adsl-1 alone resulted in a substantial reduction of body growth (P < 10−11; Welch's t-test), while knocking down the nonessential gene pacs-1 showed no significant difference (P > 0.05; Welch's t-test) from the negative control, wherein no genes are intended to be knocked down (Fig. 4B,C; Methods). Interestingly, the effect of knocking down simultaneously the two genes is largely the same as that of knocking down pacs-1 alone. This result indicated that the phenotype of ADSL deficiency in the nematode C. elegans is nearly fully masked by a further loss-of-function perturbation on the gene pacs-1. Given the identical purine de novo biosynthesis pathway in C. elegans and humans, the therapeutic strategy for the ADSL deficiency demonstrated in C. elegans has the potential to be effective in humans.
Discussion
In this study, we suggested two types of gene essentiality: The Type I can be rescued/masked only by gain-of-function perturbations, while the Type II can be rescued/masked by loss-of-function perturbations. This proposition is different from the previously proposed concept of genetically conditional essentiality that states nothing about the way of rescuing (Zekhnov et al. 1995; Zhao et al. 1998; Gerdes et al. 2006; Rancati et al. 2008; Vernon et al. 2008; Delia et al. 2009; Bergmiller et al. 2012). Taking advantages of spontaneous mutations, a useful tool for probing the gene network (Szamecz et al. 2014; Liu et al. 2015), as well as the affordable whole-genome sequencing, we carried out a systematic screening in the yeast S. cerevisiae and identified five Type II essential genes each with 1–8 rescuing partners. This is a preliminary screening, and there are good reasons to expect many more Type II essential genes. First, only one fourth of the yeast essential genes were examined because of the leakiness of the Tet promoter used in the yTHC stock, and for each gene, only ∼6 × 107 cells were tested. We found that plating more cells produced viable colonies that are primarily due to mutations knocking down the Tet system. Second, the mutation rate varies among genes by orders of magnitude, so our strategy that was designed based on the average mutation rate became invalid for detecting the rescuing mutations on the genes with an intrinsically low mutation rate. This point was well demonstrated in the case of ADE13 that has eight rescuing partners; our first-round screening identified only five of the eight, and the remaining three were suggested by reasoning and validated by target gene deletions. Third, a recent study reported that up to ∼10% of the yeast essential genes are dispensable in a newly evolved genetic status (Liu et al. 2015). Although the resulting aneuploidy precludes the authors from identifying the specific genes responsible for the essentiality turnover, it won't be surprising if a further study shows a large proportion of them as Type II essential. Interestingly, only one of the five Type II essential genes identified in this study was found conditionally dispensable in that study, suggesting that both screenings are far from saturation.
It is worth noting again that the Type II essentiality of the yeast gene ADE13 has been reported by a previous study (Zekhnov and Domkin 2000), although, to the best of our knowledge, the understanding has not been applied to thinking of therapy for human disease. The clinical difficulty for Mendelian diseases is, in great part, due to the fact that many of the diseases are due to loss-of-function mutations on essential genes. The basic therapeutic principle for such essential gene-related diseases has been to restore the lost functions (Garcia-Blanco et al. 2004; Maguire et al. 2008; Bidou et al. 2012), which is extremely difficult to achieve. Because inventing a compound to inactivate a function is much simpler than inventing a compound to restore a function, using loss-of-function perturbations to mask the loss-of-function effects of Type II essential genes suggests a conceptually much easier therapeutic strategy for such human diseases. Therefore, studies of the nature of essential genes not only deepen our understandings of the gene network but could also help in medical practice.
Methods
Yeast strains
The Yeast Tet-promoters Hughes Collection (yTHC) was purchased from GE Healthcare Dharmacon. A total of 800 essential genes are included in the collection. For each essential gene, a kanR-tetO7-TATA cassette was used to replace the endogenous promoter of the gene in the haploid strain R1158 (URA3::CMV-tTA MATa his3-1 leu2-0 met15-0). The diploid yeast strain used in this study is BY4743 (MATa/α his3Δ1/ his3Δ1 leu2Δ0/ leu2Δ0 lys2Δ0/ lys2Δ0 metΔ15/met15 ura3Δ0 /ura3Δ0).
Estimating the number of cells required for capturing rescuing mutations
We ran simulations to estimate the probability of observing critical loss-of-function mutations on a gene. Here, critical
loss-of-function mutations refers to severe missense mutations, nonsense mutations, and frame-shift indels. The probability
(P′) of having at least one critical loss-of-function mutation on a gene was estimated using the formula:
 where µi = 0.02 × 10−9 is the small (1, 2, or 3 bp) indel mutation rate per site per generation (Lynch et al. 2008), q = 0.83 is the fraction of small indels that are not multiples of 3 bp (Zhang and Webb 2003), L is the length of the focal gene, µs = 0.33 × 10−9 is the substitution mutation rate per site per generation (Lynch et al. 2008), n is the assigned number (here, n = 0) of mutation events on the gene, Nu and Ns are the number of nonsense and missense mutation sites of the gene, respectively,
and r = 0.05 is the fraction of missense mutations that are severe (Lang and Murray 2008). In this formula,
where µi = 0.02 × 10−9 is the small (1, 2, or 3 bp) indel mutation rate per site per generation (Lynch et al. 2008), q = 0.83 is the fraction of small indels that are not multiples of 3 bp (Zhang and Webb 2003), L is the length of the focal gene, µs = 0.33 × 10−9 is the substitution mutation rate per site per generation (Lynch et al. 2008), n is the assigned number (here, n = 0) of mutation events on the gene, Nu and Ns are the number of nonsense and missense mutation sites of the gene, respectively,
and r = 0.05 is the fraction of missense mutations that are severe (Lang and Murray 2008). In this formula,
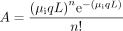 represents the Poisson probability of having n critical indel mutations, and
represents the Poisson probability of having n critical indel mutations, and
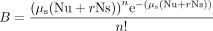 represents the Poisson probability of having n critical substitution mutations. When n = 0, P′ = 1−A*B is the probability of having at least one critical loss-of-function mutation. The Poisson probability (P) of having at least one individual with critical loss-of-function mutations on a given gene in a population of N individuals is
represents the Poisson probability of having n critical substitution mutations. When n = 0, P′ = 1−A*B is the probability of having at least one critical loss-of-function mutation. The Poisson probability (P) of having at least one individual with critical loss-of-function mutations on a given gene in a population of N individuals is
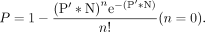
Screening for rescuing mutations
The yTHC strains were streaked onto YPD agar plates (1% yeast extract, 2% peptone, 2% dextrose, 2% agar), and for each strain, a single colony (parental clone) was picked and inoculated in YPD liquid overnight. Approximately 6 × 107 cells were collected and spread onto YPD agar plates supplied with 10 µg/mL doxycycline (Dox+ plate) and incubated at 30°C for 3–4 d. For some strains, a few colonies appeared on the Dox+ plate. To check if those clones grew because of the turnover of essentiality of the focal essential genes in a mutated genetic background, we deleted the focal essential genes in the clones using homologous recombination. Five essential genes can be deleted. For each of the five cases, we carried out 30 screening experiments and obtained 30 independent mutant clones that grew on Dox+ plates. For each mutant clone, the focal essential gene was deleted to confirm the turnover of gene essentiality.
Genome sequencing and mutation calling
A total of 150 mutant clones (30 × 5) and five parental clones were subject to whole-genome sequencing to identify mutations that are potentially able to mask the lethal effects of deleting the focal essential genes. For each clone, genomic DNA was extracted from ∼108 yeast cells using the Dr. GenTLE (from Yeast) High Recovery kit (Clontech, #9082). DNA fragments of ∼400–600 bp were collected for library construction, and samples were sequenced using an Illumina HiSeq 2000 with a paired-end (PE) 125 strategy. Approximately 8 million reads were generated for each library, corresponding to an average sequencing depth of ∼80×. Sequence reads were aligned to the yeast genome by Bowtie 2 (Langmead and Salzberg 2012) with default parameter settings, and duplicated reads were removed by Picard tools (http://picard.sourceforge.net). Single-nucleotide mutations and indels were called on the Genome Analysis Toolkit (GATK) platform (Margulies et al. 2005) with default settings.
Identification of Type II essentiality using gene deletion
LEU2 and URA3 were used as the marker genes to delete one copy of a focal essential gene and one copy of its rescuing partner, respectively, in the diploid strain BY4743 using homologous recombination. Approximately 1 × 107 cells of the resulting diploid double heterozygous deletion mutants were cultured at 25°C in 1 mL of medium containing 10 g/L potassium acetate and 50 mg/L zinc acetate for sporulation. Five days later, ∼200 µL of cell suspension was centrifuged. The pellet was resuspended in 200 µL potassium phosphate buffer (67 mM KH2PO4; pH 7.5) supplied with 25 U Zymolyase (ZYMO Research), and incubated at 30°C for 30 min with gentle shaking to digest the cell wall of the yeast cells. After the incubation, the suspension was subject to a dry bath at 55°C for 15 min to kill diploid cells, and then spread on YPD plates to get haploid colonies. For each case, ∼100 clones were selected for PCR-based genotyping to derive the segregation pattern.
To estimate the relative fitness of double gene deletion segregants, we cultured each mutant in YPD liquid and compared their growth rates with that of the wild-type yeast BY4741. Specifically, cells were cultured in YPD liquid with an initial OD600 (optical density) of ∼0.05. The OD was measured every 1.5 h until the growth nearly ceased, and cell numbers were then derived from the OD values. We calculated the average doubling time between every two consecutive time points of the exponential growth phase. The relative fitness of a mutant is the ratio of the average doubling time of the mutant to that of the wild type.
RNAi in C. elegans
A C. elegans strain (rrf-3−/−) was maintained at 16°C on NGM plates seeded with E. coli OP50 but fed with strain HT115 containing L4440-based plasmids for RNAi (RNA interference) experiments (WormBook). We used RNAi to mimic loss-of-function mutations on genes adsl-1 and pacs-1 in C. elegans. Two single-gene-interference vectors, adsl-1/GFP and pacs-1/GFP, for knocking down adsl-1 and pacs-1, respectively, and a double-gene-interference vector, adsl-1/pacs-1, for knocking down the two genes simultaneously, were constructed. We fused the gene with the coding sequence of GFP in the single-gene-interference vectors to achieve a similar length of insert as the double-gene-interference vector, which ensures comparable RNAi efficiency for the same genes in different vectors. The L4440 empty vector was used as a negative control. The empty L4440 vector and the vectors with the above-mentioned inserts were transformed into the HT115 strain, then grown in LB medium with 100 µg/mL ampicillin at 37°C. On the next day, bacteria were 1:100-transferred into fresh LB medium at 16°C, and 1 mM IPTG was added to induce dsRNA expression. The bacteria were then harvested and transferred onto NGM plates containing IPTG. At least 10 three-day-old F0 worms were seeded onto an HT115 containing plate for each RNAi experiment. All F0 worms were killed at Day 0 when their eggs were first observed on the plate, and the body length of all F1 worms on the plate was measured on Day 3 and Day 6, respectively, using the software ImageJ (https://imagej.nih.gov/ij).
Quantitative PCR
Total RNA was extracted from the worms using TRIzol (Life Technologies, #15596-026). Two micrograms of total RNA were treated with RNase-Free DNase Set (QIAGEN, #79254) and reverse transcribed to cDNA using the PrimerScript RT reagent kit (Takara, #RR037A). The qPCR experiments were performed using SYBR Premix Ex Taq II (Takara, #RR820B) and a Roche LightCycler 480 apparatus. The act-1 gene was used as the reference gene. The expression level of the gene adsl-1 reduced by 64.4% and 61.1% in worms subject to adsl-1/GFP and adsl-1/pacs-1 treatment, respectively; the expression reduction of pacs-1 was 62.1% and 58.2% in worms subject to pacs-1/GFP and adsl-1/pacs-1 treatment, respectively.
Data access
The sequence data from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under accession number SRP075591.
Acknowledgments
The C. elegans and E. coli strains used in the RNAi experiment are gifts of Dr. X. Wang at National Institute of Biological Sciences, Beijing. This work was supported in part by Guangzhou Jun-He Biotechnology Co., Ltd. and a research grant from the Guangzhou Municipal Government (#11BppZLjj2050035 to X.H.).
Author contributions: X.H. and P.C. designed the study; P.C., D.W., and Z.Z. conducted the yeast experiments and data analysis; H.C. conducted the RNAi experiments and data analysis; P.C. and X.H. wrote the paper.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.205955.116.
- Received February 21, 2016.
- Accepted July 19, 2016.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








