
Joint annotation of chromatin state and chromatin conformation reveals relationships among domain types and identifies domains of cell-type-specific expression
- Maxwell W. Libbrecht1,
- Ferhat Ay2,
- Michael M. Hoffman3,4,
- David M. Gilbert5,
- Jeffrey A. Bilmes6 and
- William Stafford Noble1,2
- 1Department of Computer Science and Engineering, University of Washington, Seattle, Washington 98195, USA
- 2Department of Genome Sciences, University of Washington, Seattle, Washington 98195, USA
- 3Princess Margaret Cancer Centre, University of Toronto, ON M5G 1L7, Canada
- 4Department of Medical Biophysics, University of Toronto, ON M5G 1L7, Canada
- 5Department of Biological Science, The Florida State University, Tallahassee, Florida 32304, USA
- 6Department of Electrical Engineering, University of Washington, Seattle, Washington 98195, USA
- Corresponding author: william-noble{at}uw.edu
Abstract
The genomic neighborhood of a gene influences its activity, a behavior that is attributable in part to domain-scale regulation. Previous genomic studies have identified many types of regulatory domains. However, due to the difficulty of integrating genomics data sets, the relationships among these domain types are poorly understood. Semi-automated genome annotation (SAGA) algorithms facilitate human interpretation of heterogeneous collections of genomics data by simultaneously partitioning the human genome and assigning labels to the resulting genomic segments. However, existing SAGA methods cannot integrate inherently pairwise chromatin conformation data. We developed a new computational method, called graph-based regularization (GBR), for expressing a pairwise prior that encourages certain pairs of genomic loci to receive the same label in a genome annotation. We used GBR to exploit chromatin conformation information during genome annotation by encouraging positions that are close in 3D to occupy the same type of domain. Using this approach, we produced a model of chromatin domains in eight human cell types, thereby revealing the relationships among known domain types. Through this model, we identified clusters of tightly regulated genes expressed in only a small number of cell types, which we term “specific expression domains.” We found that domain boundaries marked by promoters and CTCF motifs are consistent between cell types even when domain activity changes. Finally, we showed that GBR can be used to transfer information from well-studied cell types to less well-characterized cell types during genome annotation, making it possible to produce high-quality annotations of the hundreds of cell types with limited available data.
Although the mechanism of regulation of a gene by a promoter directly upstream of its transcription start site is well understood, this type of local regulation does not explain the large effect of genomic neighborhood on gene regulation. The neighborhood effect is in part the consequence of domain-scale regulation, in which regions of hundreds or thousands of kilobases known as domains are regulated as a unit (Chakalova et al. 2005; Akhtar et al. 2013; Bickmore and van Steensel 2013). Current understanding of domain-scale regulation is based on a number of domain types, each defined based on a different type of data, such as histone modification ChIP-seq, replication timing, or measures of chromatin conformation. However, as a result of the difficulty of integrating genomics data sets, the relationships among these domain types are poorly understood. Therefore, a principled method for jointly modeling all available types of data is needed to improve our understanding of domain-scale regulation.
A class of methods we term semi-automated genome annotation (SAGA) algorithms is widely used to jointly model diverse genomics data sets. These algorithms take as input a collection of genomics data sets and simultaneously partition the genome and label each segment with an integer such that positions with the same label have similar patterns of activity. These algorithms are “semi-automated” because a human performs a functional interpretation of the labels after the annotation process. Examples of SAGA algorithms include HMMSeg (Day et al. 2007), ChromHMM (Ernst and Kellis 2010), Segway (Hoffman et al. 2012), and others (Thurman et al. 2007; Lian et al. 2008; Filion et al. 2010). These genome annotation algorithms have had great success in interpreting genomics data and have been shown to recapitulate known functional elements including genes, promoters, and enhancers.
However, existing SAGA methods cannot model chromatin conformation information. The 3D arrangement of chromatin in the nucleus plays a central role in gene regulation, chromatin state and replication timing (Misteli 2007; Dekker 2008; Ryba et al. 2010; Dixon et al. 2012). Chromatin architecture can be investigated using chromatin conformation capture (3C) assays, including the genome-wide conformation capture assay, Hi-C. A Hi-C experiment outputs a matrix of contact counts, where the contact frequency of a pair of positions is inversely proportional to the positions’ 3D distance in the nucleus (Lieberman-Aiden et al. 2009; Ay et al. 2014b). Existing SAGA methods can incorporate any data set that can be represented as a vector defined linearly across the genome, but they cannot incorporate inherently pairwise Hi-C data without resorting to simplifying transformations such as principal component analysis.
We present a method for integrating chromatin architecture information into a genome annotation method. Motivated by the observation that pairs of loci close in 3D tend to occupy the same type of domain, we encourage these pairs to be assigned the same label in a genome annotation through a pairwise prior. We developed a novel computational method, called graph-based regularization (GBR), which performs inference in the presence of such a pairwise prior, and we extended the existing SAGA algorithm Segway (Hoffman et al. 2012) to implement this method.
GBR can also be used for the seemingly unrelated task of transferring information from well-studied cell types for the annotation of cell types with limited available data. Consortia such as ENCODE have characterized a small set of cell types in great detail using hundreds of genomics assays. However, due to the high cost of genomics experiments, it is feasible to perform only a few assays on any additional cell type of interest. For example, ENCODE and Roadmap Epigenomics Consortia have each performed two to 10 experiments in more than 100 cell types, and it is common for an individual laboratory to perform a small number of experiments on a particular cell type or perturbation of interest. In such settings, it is crucial to leverage information garnered from well-studied cell types to allow accurate annotation of other cell types using just a few experiments. We transfer information from well-studied cell types with GBR by using the pairwise prior that loci that were assigned the same label in many well-studied cell types should be more likely to receive the same label in a cell type of interest. Therefore, GBR makes it possible to produce high-quality annotations of the hundreds of cell types with limited available data.
Results
Chromatin domains colocalize with domains of similar activity
Previous research has shown that large chromatin domains (∼1 Mb) tend to colocalize with domains of similar activity in 3D (Lieberman-Aiden et al. 2009; Ryba et al. 2010). To further explore this trend, we compared the 1D genomics data at pairs of statistically significantly interacting loci (Supplemental Fig. 1). As expected, we found that chromatin signals were highly consistent at pairs of positions nearby in 3D. First, histone modification and replication signal values at pairs of significantly interacting loci are more highly correlated than for a rotational permutation control, which controls for the 1D pattern of the signal (Supplemental Fig. 1A). Second, Segway labels generated without using GBR from an annotation of the genome were assigned the same label much more often than a rotational permutation control (Supplemental Fig. 1B). Note that this pattern is in stark contrast to small elements (∼100 base pairs [bp]) such as promoters and enhancers, which do not, in general, cluster with elements of the same type. These observations suggest that chromatin conformation data might best be incorporated using a pairwise prior stating that a pair of positions should be more likely to receive the same label if the positions are close in 3D. Therefore, we sought to develop new methods for leveraging chromatin conformation data using this colocalization pattern.
GBR expresses a pairwise prior in a SAGA method
Existing SAGA algorithms use dynamic programming algorithms to perform inference in a chain-structured Bayesian network such as a hidden Markov model. Dynamic programming algorithms such as the forward-backward algorithm can be used to perform inference efficiently in models with chain-structured dependencies; however, applying these methods in the presence of a pairwise prior that connects arbitrary pairs of positions results in inference costs that grow exponentially in the number of genomic positions. Therefore, these methods cannot be applied to genome annotation problems with millions or billions of variables. We propose a novel convex optimization framework that allows for efficient inference in this case.
The method takes as input a set of genomics data sets and weighted graphs over the genomic positions, where a large weight on a given pair of positions indicates that we have a strong prior belief that this pair should receive the same label. It outputs a probability distribution over the integer labels at each position. The method encourages pairs of positions connected by edges in the graph to be assigned the same label by minimizing a measure of dissimilarity between their output probability distributions called the Kullback-Leibler (KL) divergence.
We call this strategy of using a graph to incorporate a pairwise prior graph-based regularization (GBR) (Methods). Note that in this article we use the word “prior” in the nontechnical sense of “prior information,” not in the sense of a prior distribution for a Bayesian model. We have developed an efficient, novel alternating minimization algorithm that optimizes this objective (Supplemental Note 1). Using synthetic data, we determined that GBR outperforms alternative methods based on approximate inference, as well as existing methods for GBR (Supplemental Note 2). We then extended the SAGA method Segway to implement this algorithm (Supplemental Note 3; Hoffman et al. 2012).
Using GBR to integrate 3D structure information improves prediction of replication and topological domains
We used GBR to integrate chromatin conformation information using the pairwise prior that positions close in 3D should be more likely to be identified as the same domain type (Fig. 1A). To do this, we construct a GBR graph that connects each pair of positions with weight proportional to our statistical confidence that the positions physically interact (Methods; Ay et al. 2014a). This measure of statistical confidence controls for the bias of Hi-C for positions close in 1D, as well as biases for sequence features such as GC content and restriction site density.
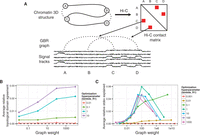
(A) Strategy for incorporating Hi-C data using GBR. (B) Effect of GBR hyperparameters on topological domain agreement. The x-axis indicates the value of the graph weight hyperparameter λG. The y-axis indicates the average over 10 annotations (one for each input histone modification data set) of the fraction improvement in topological domain label agreement by adding GBR over Segway without GBR. (C) Same as B, but y-axis indicates improvement in replication timing standard deviation explained, and average is over 29 annotations. Annotations used four labels, 10-kb resolution.
Incorporating Hi-C data using GBR has the effect of both aligning domains to regions of self-interacting chromatin and helping to determine the label of each segment. We evaluated the first effect, as a sanity check, by computing the accuracy with which our annotation predicts self-interacting regions of chromatin of size ∼1 Mb called topological domains (Dixon et al. 2012; Filippova et al. 2014). We evaluated the second effect by comparing to replication time, which is highly correlated with gene expression and chromatin state and therefore is a good proxy for domain type. In order to evaluate our performance in a variety of conditions, we ran Segway augmented with GBR separately once for each of the 29 histone modification sets available in IMR90, in each case using as input a single histone modification data set and a GBR graph based on IMR90 Hi-C data. We found that the annotation's ability to identify both topological domains (P < 10−16, t-test) and replication time (P < 10−16, t-test) was greatly improved by using GBR (Fig. 1B,C). To evaluate the degree to which an annotation matches topological domains, we computed, for each topological domain, the fraction of positions in the topological domain receiving the same label, controlling for the length and label distribution by comparing to a circularly permuted annotation (Methods). We measure the degree to which an annotation predicts replication time by the variance in replication timing explained by the annotation (Methods). The improvement from adding Hi-C with GBR was greater than the improvement achieved by instead adding another histone modification data set, for 26/30 and 27/30 histone modification data sets for topological domains and replication time, respectively. Furthermore, this improvement was consistent for a large range of hyperparameters (several orders of magnitude around optimal) (Fig. 1B,C). These results demonstrate that incorporating Hi-C data using GBR greatly improves the quality of the resulting annotation, and moreover that Hi-C is more informative for determining domain identity than most other data types.
Joint domain annotation of chromatin state and chromatin conformation captures previously described domain types
Having verified the utility of GBR for incorporating Hi-C data, we next sought to investigate domain-scale genome regulation using this method. Current understanding of domain-scale regulation is based on a number of domain types (seven, by our count), each defined based on a different type of data, such as histone modification, replication timing, or 3C-based assays (Table 1). For example, ChIP-seq on the histone modification H3K27me3 has revealed repressive domains known as facultative heterochromatin (Pauler et al. 2009; Morey and Helin 2010), and 3C-based assays have revealed regions of self-interacting chromatin known as topological domains (Dixon et al. 2012; Filippova et al. 2014). Because all of these domain types are defined using different types of data, until now it has been difficult to understand the relationships among these domain types. GBR provides a principled method for integrating all types of data into a unified annotation of domains. We therefore used Segway with GBR to create such a unified annotation in order to understand what types of domains exist and their interrelationships.
Known types of domains
We annotated the cell type IMR90 using all 30 signal data sets we had available in IMR90 and a GBR graph derived from IMR90 Hi-C data, resulting in an annotation with a median segment length of 0.4 Mb (Methods; Fig. 2A–C; Supplemental Tables 1,2). Including Hi-C into this annotation using GBR changed the label of 6% of positions relative to an annotation without GBR, meaning Hi-C has a slightly larger influence than the 1/31 = 3% difference one would expect from adding one additional data set (Supplemental Fig. 2). Because determining the optimal number of labels for an annotation remains an open problem, we specified a somewhat larger number of labels than we expected to be supported in the data (eight) and then manually merged labels that we deemed to be redundant.
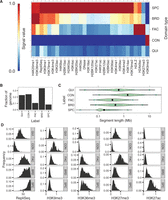
Statistics of domain annotation made with Segway augmented with GBR. (A) Heatmap of association of IMR90 domain labels to IMR90 data sets used in training. (B) Fraction of genome covered by each domain type. (C) Segment length distribution for each domain type. (D) Label-specific histograms of values for five notable data sets in IMR90.
We compared our annotation to eight types of features (Table 2). On the basis of these analyses, we merged labels that appeared redundant and assigned names to each integer label (or group of labels) that best matched our interpretation of their function. This procedure yielded five types of domains: (1) broad expression (BRD), (2) specific expression (SPC), (3) facultative heterochromatin (FAC), (4) constitutive heterochromatin (CON), and (5) quiescent (QUI). We describe the analyses that led us to these names in the following sections.
Summary of learned domain types
In order to understand how domains change state between cell types, we additionally annotated eight cell types using 12 data sets present in all eight types and a GBR graph representing common 3D contacts generated by combining the IMR90 and H1-hESC Hi-C data sets (Methods; Supplemental Table 2). This strategy of combining Hi-C data sets is motivated by the consistency of Hi-C across cell types (Supplemental Fig. 3). Again, we used eight labels and merged redundant labels, to which we assigned the same five names. We investigated the properties of these five domain types.
Repressive domains are divided into constitutive, facultative and quiescent heterochromatin
Previous studies have reported two types of repressive domains. The first type, best known as “constitutive heterochromatin” but sometimes referred to simply as “heterochromatin,” is regulated by the HP1 complex and associated with the histone modification H3K9me3 (Lachner et al. 2003). Constitutive heterochromatin is thought to repress permanently silent regions such as centromeres and telomeres. As expected, one output domain type “CON” exhibits all the known properties of constitutive heterochromatin. CON domains are associated with H3K9me3 (Fig. 2A,D), are extremely depleted for genes (Fig. 3A), are associated with low GC content and lack of evolutionary conservation (Fig. 3D,E), appear within the Hi-C eigenvector closed compartment (Fig. 3F; Methods), and cover regions that are constitutively late replicating in all cell types (Fig. 3G). CON domains are depleted both for transcription factor motifs and for transcription factor binding at motifs (Fig. 3H,I).
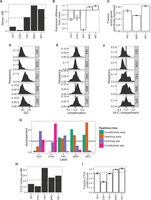
Characteristics of domain types. (A) Gene density in IMR90. (B) Gene expression relative to average over 33 cell types, averaged over eight annotations (t-test 95% confidence interval error bars). (C) Fraction of active genes also active in more than 15 other cell types, averaged over eight annotations (binomial test 95% confidence interval error bars). All gene expression data are from CAGE (Methods). (D–F) Histograms of (D) GC content, (E) conservation (PhyloP score) (Siepel et al. 2005), and (F) Hi-C compartment eigenvalues in IMR90. Positive PhyloP scores indicate evolutionary conservation; positive scores, accelerated evolution; and scores near zero, neutral evolution. Hi-C compartment values are computed according to the method of Lieberman-Aiden et al. (2009) (Methods). Positive values indicate open compartment; negative values indicate closed compartment. (G) Enrichment of each domain type with respect to IMR90 replication time (early vs. late) and replication time dynamics across cell types (constitutive vs. switching) (V Dileep, F Ay, J Sima, WS Noble, and DM Gilbert, unpubl.) (Methods). (H) CTCF motif density for each domain type. Dashed line indicates genome-wide average. (I) Fraction of CTCF motifs bound (overlapping a CTCF peak) in IMR90 (binomial test 95% confidence interval error bars). Dashed line indicates average over all motifs.
The second known type of repressive domain is best known as “facultative heterochromatin” but is also sometimes referred to as BLOCs or Polycomb-repressed chromatin (Pauler et al. 2009; Morey and Helin 2010). Facultative heterochromatin is regulated by the Polycomb complex and is associated with the histone modification H3K27me3. Facultative heterochromatin is thought to repress tissue-specific genes in cells where they are inactive. As expected, the output domain type “FAC” has all the known properties of facultative heterochromatin. FAC domains are marked by H3K27me3 (Fig. 2A,D), and they are enriched for genes (Fig. 3A), GC content (Fig. 3D), and conservation (Fig. 3E), but strongly depleted for gene expression relative to an average across cell types (Fig. 3B), indicating that FAC domains have a direct repressive effect. FAC domains are mixed between the open and closed compartments, indicating that facultative repression is independent of compartment-driven repression (Fig. 3F). However, FAC domains are almost completely absent from the annotation of the embryonic stem cell line H1-hESC, consistent with previous observations that H3K27me3 does not form domains in embryonic stem cells but rather occurs only at so-called poised or bivalent promoters (Supplemental Fig. 4; Bernstein et al. 2006).
Other semi-automated genome annotation analyses have reported a third type of repressive domain, characterized by a lack of signal from any mark, termed “quiescent domains” (Ernst and Kellis 2010; Filion et al. 2010; Hoffman et al. 2012; Julienne et al. 2013). We identified this domain type as the QUI label (Fig. 2A). Note that Segway marginalizes over missing data rather than setting the values to zero (Supplemental Note 3), so the QUI label is not simply an artifact of unmappable regions. QUI domains are highly depleted for genes (Fig. 3A) and occur in the closed compartment (Fig. 3F). QUI domains are depleted for transcription factor motifs but, unlike FAC and CON domains, are not depleted for transcription factor binding at motifs, indicating that QUI chromatin does not have a direct repressive effect (Fig. 3H,I). The mechanism behind the activity of QUI domains is unknown, but these results are consistent with a model in which QUI domains lack any activating signals but are not directly repressed.
Active domains are divided between broad and specific gene expression
Previous studies of human domains have focused on various types of repressive domains but have assigned all active chromatin to one domain category (Pauler et al. 2009; Wen et al. 2009; Julienne et al. 2013). However, studies in other organisms have reported multiple types of active domains (Filion et al. 2010; Liu et al. 2011). We therefore investigated whether our IMR90 annotation can be used to identify types of human active domains. We found that active domains in IMR90 can be split into BRD (“broad expression”) domains, characterized by transcription-associated marks such as H3K36me3, and SPC (“specific expression”) domains, characterized by regulatory marks such as H3K27ac. Both domain types are highly enriched for genes (Fig. 3A). However, while genes in BRD domains are mostly expressed across all cell types, a much larger fraction of active genes in SPC domains is expressed only in a small number of cell types (Fig. 3C). Furthermore, when a gene is in a SPC domain, that gene is expressed at a much higher level than that gene's average across cell types, suggesting that SPC domains are highly activating (Fig. 3B). In contrast, while genes in BRD domains are highly expressed, this high expression generally occurs consistently across cell types, indicating that BRD domains do not necessarily directly promote expression (Fig. 3B). Moreover, while both BRD and SPC domains are generally early replicating in IMR90, regions covered by SPC domains typically switch replication time between cell types, while regions covered by BRD domains are typically early replicating in all cell types (Fig. 3G). These results suggest a model in which genes performing housekeeping functions such as DNA repair have strong promoters but little other regulation, whereas genes specific to a given tissue are regulated by a complex web of regulatory elements, allowing the genome to specify precise conditions under which the gene is active.
To test this hypothesis, we computed the enrichment of Gene Ontology (GO) terms for genes in BRD and SPC domains (Gene Ontology Consortium 2000; Boyle et al. 2004). We found that genes in BRD domains were enriched for housekeeping functions such as cell cycle and DNA repair, while genes in SPC domains were enriched for IMR90-specific developmental functions such as vasculature development and stimulus response (Supplemental Tables 3, 4; Supplemental Fig. 5). In order to avoid hindsight bias, before looking at these GO term enrichments, we mixed the enriched terms with an equal number of decoy terms matched according to the number of genes associated with each term, and manually labeled which terms matched our hypothesized functions for each domain (housekeeping for BRD, IMR90-specific for SPC). We correctly identified 21/32 BRD enrichments (1/31 = 3%) and 54/64 SPC enrichments (P = 1.4 × 10−6). This demonstrates that active regions can be divided into domains of broadly expressed housekeeping genes and domains of specifically expressed developmental genes. To our knowledge, this is the first time a split between domains of BRD and SPC has been reported in human cells.
Lamina association is driven by a complex structure of domains
Previous work has shown that some repressive domains are marked with the histone modification H3K9me2, associate with the factor lamin B1, and localize to the nuclear lamina (Guelen et al. 2008; Wen et al. 2009). We found that comparing lamina association to domain annotations based on many data sets reveals a much more complex interaction than does comparison to each mark individually (Fig. 4). As expected, repressive domains (QUI and FAC) are enriched inside lamina-associating chromatin domains, while active domains are depleted. However, this analysis also reveals that CON domains are depleted immediately inside lamina-associating domain boundaries while being comparatively enriched at their centers. In contrast, FAC domains are highly enriched at lamina-associating domain boundaries while being comparatively depleted at their centers. In addition, while active domains (SPC and BRD) are depleted inside lamina-associating domains, they are highly enriched directly outside their boundaries. These observations suggest that lamina-associating domains form around a core of repressed chromatin and spread until they hit a strong active element.
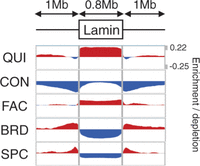
Enrichment of each domain type with respect to lamina-associating domain boundaries. The x-axis indicates position with respect to lamina-associating domains, with each domain stretched or shortened to the median length of 0.8 Mb. The y-axis indicates label enrichment or depletion [log(obs/expected)].
Developmentally consistent domain boundaries are marked by identifiable sequence elements
Previous research has shown that domain boundaries tend to be consistent between cell types even when the state of the domain changes. For example, when a region's replication time is perturbed by leukemia, the boundaries of the resulting replication domain tend to occur at the same positions as developmental replication timing domain boundaries (Ryba et al. 2012). However, the cause of these consistent domain boundaries remains unclear. We investigated the consistency of domain boundaries using our domain annotations. As expected, domain boundaries frequently occurred at consistent positions across cell types, even when the domains’ state changed (Fig. 5A). To identify these consistent domain boundaries, we combined all boundaries occurring in at least one cell type and merged boundaries within 50 kb. We defined groups of five or more boundaries as consistent (Methods; Fig. 5B). As expected, these consistent boundaries are enriched for replication domain boundaries, but many consistent domain boundaries do not overlap a replication domain boundary (Supplemental Fig. 6). We additionally found that consistent domain boundaries are highly enriched for promoters and CTCF motifs, suggesting that these elements may drive domain boundary formation (Fig. 5C,D).
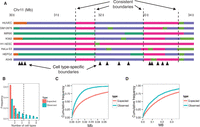
(A) Example of consistent domains. (B) Distribution of number of overlapping boundaries compared to a permutation control (binomial test 95% confidence interval error bars). Vertical dashed line denotes consistent boundary threshold. (C,D) Cumulative density of distances from consistent domain boundaries to the nearest (C) promoter and (D) distal CTCF motif. Distal CTCF motifs are defined as all CTCF motifs >5 kb from a promoter.
Using GBR to transfer information between cell types improves accuracy of predicting functional elements
GBR can also be used for the seemingly unrelated task of transferring information from well-studied cell types for the annotation of cell types with limited available data (Fig. 6A). Existing SAGA methods work well on data from a single cell type, but integrating information between cell types remains an open problem. Existing methods for using data from multiple cell types for genome annotation fail to effectively address this problem (Supplemental Note 4). We propose a novel strategy for leveraging information from well-studied cell types using the pairwise prior that if two positions received the same label in many well-studied cell types, then they should be more likely to receive the same label in the target cell type (Fig. 6A). To express this pairwise prior, we first perform a Segway annotation (without GBR) of each well-studied cell type and create a GBR graph that connects each pair of positions with weight proportional to the number of cell types in which the pair receive the same label, placing higher weight on cell types similar to the cell type of interest (Methods). We then use this graph in combination with the data sets available in the target cell type to produce an annotation of this cell type. Note that this GBR graph represents an entirely different type of information from the graphs used to represent Hi-C data in the previous sections, despite the fact that both types of data are represented as a graph.
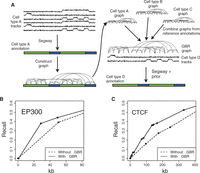
(A) Strategy for using GBR to transfer information between cell types. (B,C) Performance of predicting the locations of GM12878 (B) EP300 and (C) CTCF ChIP-seq peaks with and without using GBR to integrate information from K562. Each plot shows the fraction of elements detected as a function of the number of bases predicted (Methods). Results are shown on the test set (10 Mbp). Model training and label ordering were performed on the training set (10 Mbp). The x-axis is plotted up to 1 kb times the total number of elements. These plots can be interpreted, for example, in the context of an enhancer validation experiment, in which case it shows how many sequences would need to be tested in order to discover a certain number of enhancers.
To demonstrate the efficacy of this approach, we evaluated whether GBR improves an annotation's ability to predict enhancers and insulators. We simulated the case where the lymphoblastoid cell type GM12878 has only eight histone modifications available, a panel of data types similar to that assayed by the Roadmap Epigenomics Consortium on hundreds of human tissues (Supplemental Note 2). Because there are enough well-studied cell types to ensure that at least one reference is reasonably closely related to any cell type of interest, we used the related leukemia cell type K562 as reference. We annotated GM12878 using these eight histone modifications and a GBR graph derived from an annotation of K562 (Methods). Incorporating information from K562 this way greatly improved the accuracy with which the annotation detected enhancers and insulators (Fig. 6B,C). We evaluated the performance with which the GM12878 annotation predicts a certain type of functional element by ordering the labels by their enrichment for the element on a training set and evaluating the recall as more labels are added (Methods). The GBR annotation detects one-third of EP300 binding sites (a proxy for enhancers) (Visel et al. 2009) by predicting just 25 kb as EP300-binding, while the annotation produced without GBR predicts 43 kb before it detects this many sites (Fig. 6B). Likewise, the GBR annotation detects one-third of CTCF binding sites (a proxy for insulators) (Burgess-Beusse et al. 2002) by predicting 124 kb, compared to 241 kb without GBR (Fig. 6C). Because the algorithm was not given any knowledge of enhancers or insulators as input, it is reasonable to expect that the annotations achieve similar performance at detecting other types of functional elements, for which we do not have gold standard examples and therefore cannot evaluate against them. These results demonstrate that GBR effectively leverages information from a reference cell type and therefore provides a method for producing high-quality annotations of the hundreds of cell types with limited available data.
Discussion
We introduced graph-based regularization (GBR), a method which allows probabilistic models to integrate a pairwise prior while maintaining efficient inference. We used GBR to model chromatin conformation data and thereby jointly model all available data types for the study of chromatin domains. To our knowledge, this represents the first method for integrating chromatin conformation information into SAGA methods without resorting to simplifying transformations. We showed that modeling Hi-C data with GBR improved the annotation's ability to predict replication time and topological domains. In addition, because GBR is a general method, it will likely prove useful for other applications involving dynamic Bayesian networks, such as methods for locating genes or predicting copy number.
The ability to integrate Hi-C data into an annotation allowed us to study the relationship between types of domains by integrating all available data into a single annotation (Fig. 7A). This analysis revealed a set of five domain types that encompass all previously described domain types: (1) quiescent domains, which lack any activity; (2) constitutive heterochromatin, which represses permanently silent regions and is marked with the histone modification H3K9me3; (3) facultative heterochromatin, which represses cell-type-specific regions and is marked with the histone modification H3K27me3; (4) broadly expressed domains, which cover genes that are highly expressed in all cell types; and (5) specifically expressed domains, which exhibit high regulatory activity and cover genes that are expressed in a small number of cell types.
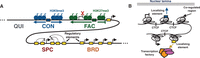
Models of (A) domain types and (B) coregulated regions.
To our knowledge, domains of SPC have not been identified previously in human cells. These domains are likely the result of complex regulatory programs designed to precisely control the condition and level of genes important for a certain cell state or function. SPC domains are similar in some ways to dense clusters of regulatory elements important for cell identity known as super-enhancers (Lovén et al. 2013; Whyte et al. 2013). However, there is only a small number of known super-enhancers (∼300), and each is much smaller than a SPC domain (∼10 kb compared to ∼200 kb). Therefore, SPC domains and super-enhancers may result from similar mechanisms, but on very different scales. However, the mechanisms underlying of both types of regions must be studied further in order to understand this relationship.
One likely mechanism of domain formation involves the spreading of heterochromatin (Weiler and Wakimoto 1995; Talbert and Henikoff 2006). Under this hypothesis, heterochromatin nucleates at silencing elements such as telomeres, repeats, or repressed promoters and sequentially assembles along chromatin. Spreading heterochromatin has been demonstrated mechanistically in Saccharomyces and Drosophila and for the SIR, HP1, and Polycomb complexes. While SIR is unique to yeast, HP1 and Polycomb have orthologs in humans that drive constitutive and facultative heterochromatin, respectively. Under the spreading hypothesis, heterochromatin can be halted by the presence of a strong active element. This halting mechanism is consistent with the observation that active domains (especially SPC) are strongly enriched directly outside of lamina-associating domains.
The consistency of domain boundaries between cell types also suggests a model in which core regions are regulated as a unit (Fig. 7B; Phillips and Corces 2009; Dixon et al. 2012). Under this hypothesis, these units self-interact as topological domains and are coregulated through availability of regulatory factors and elements such as enhancers. These coregulated units are thought to be delimited by localizing sequences, particularly CTCF sites. Under this model, each of our annotated domains is actually composed of several such neighboring coregulated regions with the same state. Therefore, while profiling a small number of cell types has allowed us to define a small number of consistent domain boundaries, profiling more cell types may lead to a complete catalog of potential boundary sites. We have described five domain types because this model allowed us to concisely summarize domain regulation, but we do not claim that this represents the “true” number of domain types. It is likely that new domain subtypes will be discovered in the future, thus increasing the number of known domain types. In addition, methods that discover the optimal number of domain types or that allow mixtures of domain types are an interesting direction for future work. To our knowledge, all existing SAGA methods require either a fixed number of labels (Day et al. 2007; Thurman et al. 2007; Lian et al. 2008; Ernst and Kellis 2010; Filion et al. 2010; Hoffman et al. 2012) or a hyperparameter that indirectly controls the number of labels (Ho et al. 2014). A method that allows for “mixed” domain labels at a given process could potentially circumvent the manual merging process that we used to reduce an eight-label model to a five-label one.
Finally, we presented a method for transferring information from well-studied cell types using GBR in order to improve the quality and interpretability of annotations of cell types with limited available data. This method enables a new strategy for understanding cell types, in which a small number of assays are performed on each cell type of interest to determine the unique characteristics of this cell type, and then Segway with GBR is used to combine these data with the large body of available information from well-studied cell types. This method has the additional benefit of matching the label semantics of the target cell types to the semantics of the reference annotations, which allows the label interpretation process to be performed automatically. Because consortia such as ENCODE and Roadmap Epigenomics are already analyzing a large number of cell types with a small number of assays each, this strategy is immediately applicable. Determining which assays are most informative as input to this strategy is an interesting question for future work.
Methods
Histone modification, open chromatin, and replication timing signal data
We acquired histone ChIP-seq, DNase-seq, and FAIRE-seq data for A549, K562, H1-hESC, GM12878, HeLa-S3, HepG2, and HUVEC from
ENCODE and for IMR90 from Roadmap Epigenomics (Bernstein et al. 2010; The ENCODE Project Consortium 2012). We used a uniform signal-processing pipeline to generate a genome-wide vector for each data set, as described by Hoffman et al. (2013). We also acquired Repli-seq data for IMR90 from ENCODE and smoothed these data using wavelet smoothing as described by Thurman et al. (2007). We applied the inverse hyperbolic sine transform 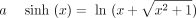 to all signal data. This transform is similar to the log transform in that it depresses the magnitude of extremely large
values, but it is defined at zero and amplifies the magnitude of small values less severely than the log transform does. This
transform has been shown to be important for reducing the effect of large values in analysis of genomics data sets (Johnson 1949; Hoffman et al. 2012).
to all signal data. This transform is similar to the log transform in that it depresses the magnitude of extremely large
values, but it is defined at zero and amplifies the magnitude of small values less severely than the log transform does. This
transform has been shown to be important for reducing the effect of large values in analysis of genomics data sets (Johnson 1949; Hoffman et al. 2012).
We acquired transcription factor ChIP-seq data from ENCODE. Peaks were called for each factor using MACS using an IDR threshold of 0.05 (Zhang et al. 2008; Landt et al. 2012).
We acquired CAGE expression data for 33 cell types from GENCODE (Harrow et al. 2012).
The full list of data sets used is available in Supplemental Table 1.
Hi-C data
We used publicly available Hi-C data sets for two human cell lines (IMR90 and H1-hESC) (Dixon et al. 2012). We processed raw paired-end libraries with a pipeline that combines reads from two replicates per cell line, maps these reads, extracts the read pairs for which each end maps uniquely, and removes potential PCR duplicates. We then partitioned the human genome into a collection of nonoverlapping 10-kb windows and assigned each end of a read pair to the nearest 10-kb window midpoint. This process yielded a 303,641 × 303,641 whole-genome contact map. These contact maps consisted of both intra- and interchromosomal contacts and contained only ∼0.3% nonzero entries. We assigned statistical confidence estimates to these contact counts using the method Fit-Hi-C, which jointly models the random polymer looping effect and technical biases (Ay et al. 2014a). First, we applied the bias correction method ICE to the contact map to estimate a bias associated with each 10-kb locus, after eliminating all loci that have <50% uniquely mappable bases (Imakaev et al. 2012). Second, using these computed biases and raw contact maps as input, we estimated a P-value of interaction for each pair of 10-kb loci with nonzero contact counts (P-value was set to one for pairs with zero contacts). We used a slightly modified version of the original Fit-Hi-C algorithm, which handles inter- as well as intrachromosomal contacts and omits the refinement step for fast computation. Because Fit-Hi-C normalizes for 1D genomic distance, the majority of significant contacts were at long distances (Supplemental Fig. 7). Note that while the data sets we used have insufficient coverage to identify many high-confidence contacts at 10-kb resolution, Segway with GBR aggregates information over ∼400 kb in order to make each domain call, so individual high-confidence interactions are not necessary.
We computed the genome chromatin compartment using eigenvalue decomposition on the normalized contact maps of IMR90 and H1-ESC cell lines at 1-Mb resolution as described by Lieberman-Aiden et al. (2009). For each chromosome, we calculated the Pearson correlation between each pair of rows of the intrachromosomal contact matrix and applied eigenvalue decomposition to the correlation matrix. Similar to Lieberman-Aiden et al. (2009), we used the second eigenvector in cases where the first eigenvector values were either all positive or all negative to define the compartments. We used average GC content to map signs of eigenvectors to either open (higher GC content) or closed chromatin compartments.
Graph-based regularization
In a SAGA method, we are given a set of vertices V that index a set of n = |V| random variables XV = {X1,…,Xn} and a probability distribution parameterized by θ, pθ(XH,XO). Different SAGA methods employ different distributions pθ. GBR could be applied to any probabilistic model, but in this work we use the Segway model (Supplemental Methods) because it can handle real-valued and missing data, and it can use nongeometric segment length distributions. We denote random variables with capital letters (e.g., XH) and instantiations of variables with lower-case letters [e.g., xH ∈ domain(XH)]. We use capitals to denote sets and lowercase letters to denote values (e.g., Xh for h ∈ H).
Training the model involves a set of observed data 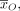 where a subset of variables O ⊆ V is observed and the remainder H = V/O are hidden. The maximum likelihood training procedure optimizes the objective
where a subset of variables O ⊆ V is observed and the remainder H = V/O are hidden. The maximum likelihood training procedure optimizes the objective
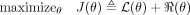 (1)
where
(1)
where
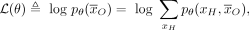 (2)
where ℛ(θ) is a regularizer that expresses prior knowledge about the parameters. Many regularizers are used in practice, such
as the ℓ2 or ℓ1 norms, which encourage parameters to be small or sparse, respectively.
(2)
where ℛ(θ) is a regularizer that expresses prior knowledge about the parameters. Many regularizers are used in practice, such
as the ℓ2 or ℓ1 norms, which encourage parameters to be small or sparse, respectively.
Dynamic programming algorithms such as the forward-backward algorithm can be used to perform inference in SAGA models, because all such existing models have dependencies in the form of a chain. That is, the variables associated with position i depend only on the variables associated with positions i − 1 and i + 1. Examples of such chain-structured models include hidden Markov models and dynamic Bayesian networks. However, these dynamic programming algorithms do not apply if a pairwise prior is added to the model, since the prior may have an arbitrary structure. Several techniques have been proposed to handle models with arbitrary structure (Supplemental Note 5). However, none of these techniques are optimal for expressing a pairwise prior.
Therefore, we instead employ a novel strategy based on posterior regularization (Ganchev et al. 2010) to integrate this prior. This is done by introducing an auxiliary joint distribution q(XH), placing a regularizer on q(XH), and encouraging q to be similar to pθ through a KL divergence penalty. The regularizer is
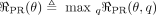 (3)
(3)
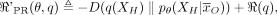 (4)
where
(4)
where  is the KL divergence
is the KL divergence
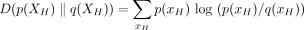 and
and 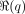 is a posterior regularizer that expresses prior knowledge about the posterior distribution. KL divergence measures the dissimilarity
of probability distributions, such that
is a posterior regularizer that expresses prior knowledge about the posterior distribution. KL divergence measures the dissimilarity
of probability distributions, such that 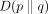 is zero if the distributions are identical and can be arbitrarily large if they are not. Several posterior regularizers have
been proposed in the past, such as those that require posteriors to satisfy constraints in expectation (Ganchev et al. 2010).
is zero if the distributions are identical and can be arbitrarily large if they are not. Several posterior regularizers have
been proposed in the past, such as those that require posteriors to satisfy constraints in expectation (Ganchev et al. 2010).
We propose a new type of posterior regularizer that expresses a pairwise prior (Fig. 8). We are given a weighted, undirected regularization graph over the hidden variables GR = (H, ER), where ER ⊆ H × H is a set of edges with non-negative similarity weights 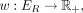 such that a large w(u, v) indicates that we have a strong belief that Xu and Xv should be similar. (We describe how we generate this graph in the next two sections.) For a distribution p(XH), let
such that a large w(u, v) indicates that we have a strong belief that Xu and Xv should be similar. (We describe how we generate this graph in the next two sections.) For a distribution p(XH), let 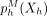 indicate the marginal distribution over Xh,
indicate the marginal distribution over Xh, 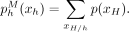 Let λG be a hyperparameter controlling the strength of regularization. The posterior regularizer is
Let λG be a hyperparameter controlling the strength of regularization. The posterior regularizer is
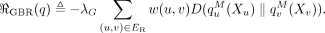 (5)
Thus the full objective is
(5)
Thus the full objective is
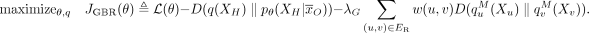 (6)
We term this strategy of adding graph-based penalties graph-based regularization (GBR).
(6)
We term this strategy of adding graph-based penalties graph-based regularization (GBR).
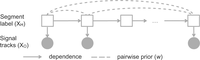
GBR model. Squares and circles denote discrete and continuous random variables, respectively. Filled-in and unfilled shapes denote observed and unobserved variables, respectively.
GBR optimization
We have developed a novel algorithm for efficiently optimizing JGBR in q. This algorithm alternates between using a method for probabilistic inference such as the forward-backward algorithm and
applying a message passing algorithm over the regularization graph GR. In the inference step, the model receives evidence from the message passing step in the form of a “virtual evidence” distribution,
 , over each variable h. These virtual evidence distributions are used in conjunction with the original SAGA model to compute a posterior distribution
over the labels using any algorithm for probabilistic inference on dynamic Bayesian networks, such as belief propagation or
the forward-backward algorithm.
, over each variable h. These virtual evidence distributions are used in conjunction with the original SAGA model to compute a posterior distribution
over the labels using any algorithm for probabilistic inference on dynamic Bayesian networks, such as belief propagation or
the forward-backward algorithm.
In the message passing step, the algorithm updates rM to minimize the KL penalties in the objective function JGBR. This message passing step is itself performed using an alternating optimization algorithm, which passes messages over the regularization graph GR. This algorithm is similar to one originally developed for the field of semi-supervised learning (Subramanya and Bilmes 2011).
The inference and message passing steps are iterated until convergence. These two updates are linear in the number of variables (for chain-structured models, which include all existing SAGA methods) and linear in the degree of the regularization graph, respectively. The algorithm exhibits monotonic convergence, similar to the EM algorithm. We derive the algorithm for optimizing JGBR and prove its convergence in Supplemental Note 1.
GBR graph for incorporating Hi-C data
When we are using GBR to incorporate Hi-C data, we are given a matrix of contact P-values 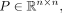 generated from a matrix of contact counts as described above. To remove noise and decrease the degree of the graph, we removed
all contacts with uncorrected P-value P > 10−6 and multiplied the remaining P-values by 106, similar to a Bonferroni correction. Note that due to the large number of hypotheses, performing a full Bonferroni correction
would result in very few contacts. Moreover, the graph weights allow the algorithm to take into account the strength of each
connection, so the choice of 106 was made for computational, not statistical, reasons. We computed the weights as
generated from a matrix of contact counts as described above. To remove noise and decrease the degree of the graph, we removed
all contacts with uncorrected P-value P > 10−6 and multiplied the remaining P-values by 106, similar to a Bonferroni correction. Note that due to the large number of hypotheses, performing a full Bonferroni correction
would result in very few contacts. Moreover, the graph weights allow the algorithm to take into account the strength of each
connection, so the choice of 106 was made for computational, not statistical, reasons. We computed the weights as
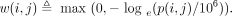 (7)
As with the graph for transferring information between cell types, the multiplicative scale of the weights is arbitrary, since
it is controlled by the graph weight hyperparameter λG. We used only intrachromosomal contacts for forming the GBR graph. To produce a GBR graph representing cell type–consistent
chromatin conformation used in the domain annotation of eight cell types, we added the edge weights from the IMR90 and H1-hESC
Hi-C GBR graphs.
(7)
As with the graph for transferring information between cell types, the multiplicative scale of the weights is arbitrary, since
it is controlled by the graph weight hyperparameter λG. We used only intrachromosomal contacts for forming the GBR graph. To produce a GBR graph representing cell type–consistent
chromatin conformation used in the domain annotation of eight cell types, we added the edge weights from the IMR90 and H1-hESC
Hi-C GBR graphs.
GBR graph for annotation of multiple cell types
When we are using GBR to transfer information about cell type A to improve annotation of cell type B, we are given an annotation 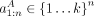 of cell type A, produced without GBR. We construct a GBR graph from this annotation by connecting each pair of positions that received the
same label in aA with an undirected edge of weight 1. Note that the weight is arbitrary, since it is scaled by the regularization parameter
λG. To mitigate the problem of quadratic growth in the degree of this graph, we randomly subsampled this graph such that each
node had an outgoing degree 17 ≈ loge(n). We chose this graph degree because a randomly subsampled graph with n logen edges has the same connected components as the full graph with high likelihood (Erdös and Rényi 1960), and our experiments on synthetic data (not shown) showed that the sparse graph performed similarly to a complete graph.
of cell type A, produced without GBR. We construct a GBR graph from this annotation by connecting each pair of positions that received the
same label in aA with an undirected edge of weight 1. Note that the weight is arbitrary, since it is scaled by the regularization parameter
λG. To mitigate the problem of quadratic growth in the degree of this graph, we randomly subsampled this graph such that each
node had an outgoing degree 17 ≈ loge(n). We chose this graph degree because a randomly subsampled graph with n logen edges has the same connected components as the full graph with high likelihood (Erdös and Rényi 1960), and our experiments on synthetic data (not shown) showed that the sparse graph performed similarly to a complete graph.
Circular permutation
As a null model for several experiments, we performed a circular permutation of the genome along each chromosome arm as follows.
We randomly choose a translation fraction θ ∈ [0,1]. For each coordinate i ∈ {1…n} within a chromosome arm that spans the range [a,b), we translate i to t(i), where
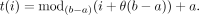 (8)
To circularly permute a genome feature, such as an annotation or a Hi-C contact map, we translate each element from position
i to t(i). Thus, when a circularly permuted feature is compared to an unpermuted feature, all positional correspondence between permuted
and unpermuted features are removed, but each feature's spatial patterns are preserved. In each case, we performed this permutation
200 times and report the average over all permutations. If the feature includes any centromere- or telomere-defined elements,
we remove these as a preprocessing step.
(8)
To circularly permute a genome feature, such as an annotation or a Hi-C contact map, we translate each element from position
i to t(i). Thus, when a circularly permuted feature is compared to an unpermuted feature, all positional correspondence between permuted
and unpermuted features are removed, but each feature's spatial patterns are preserved. In each case, we performed this permutation
200 times and report the average over all permutations. If the feature includes any centromere- or telomere-defined elements,
we remove these as a preprocessing step.
Topological domain agreement
To evaluate the degree to which an annotation A matches a set of topological domains, we computed the number cd,ℓ of bases by which domain d is covered by label ℓ. We then computed 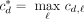 to be the number of bases covered by the highest-coverage label for domain d, and divided
to be the number of bases covered by the highest-coverage label for domain d, and divided 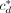 by the length of d to produce
by the length of d to produce 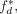 the fraction of d covered by its plurality label. The agreement
the fraction of d covered by its plurality label. The agreement 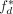 takes its maximum value of one if the domain d is covered by exactly one annotation label. We computed the raw genome-wide agreement
takes its maximum value of one if the domain d is covered by exactly one annotation label. We computed the raw genome-wide agreement
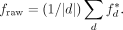 This raw genome-wide agreement fraw can be improved simply by increasing the length of segments and decreasing the number of labels. Therefore, we circularly
permuted A to form Ap and used this permuted annotation to compute
This raw genome-wide agreement fraw can be improved simply by increasing the length of segments and decreasing the number of labels. Therefore, we circularly
permuted A to form Ap and used this permuted annotation to compute 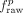 . Finally, we computed the topological domain agreement
. Finally, we computed the topological domain agreement 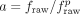 as the ratio of unpermuted and permuted raw agreements. This normalized agreement is large when the annotation has small
segments that exactly match the topological domains and is small when the annotation's segments are not correlated with topological
domains.
as the ratio of unpermuted and permuted raw agreements. This normalized agreement is large when the annotation has small
segments that exactly match the topological domains and is small when the annotation's segments are not correlated with topological
domains.
Signal variance explained
To evaluate the similarity between a genome-wide signal vector and a genome annotation, we use the following measure, which
we term the variance explained (VE). We are given a genome annotation with k labels 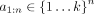 and a vector
and a vector 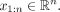 We compute the signal mean over the positions assigned a given label ℓ as
We compute the signal mean over the positions assigned a given label ℓ as
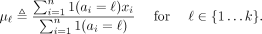 (9)
We define a predicted signal vector
(9)
We define a predicted signal vector 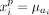 and compute the prediction error as
and compute the prediction error as 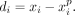 We compute the residual standard deviation of the signal vector as
We compute the residual standard deviation of the signal vector as
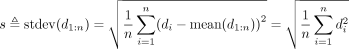 (10)
The last equality holds because
(10)
The last equality holds because 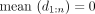 by construction. We define the VE for annotation a and signal vector x as
by construction. We define the VE for annotation a and signal vector x as 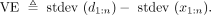 VE is bounded by the range
VE is bounded by the range 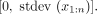 VE is a measure of the extent to which a genome-wide signal data set and annotation are similar, where higher values indicate
better agreement.
VE is a measure of the extent to which a genome-wide signal data set and annotation are similar, where higher values indicate
better agreement.
Genomic element prediction
We form a classifier for a set of genomic elements based on an annotation using the following strategy. We are given a genome
annotation with k labels 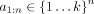 and a set of positions
and a set of positions 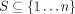 , which represent some set of elements of interest such as enhancers or CTCF binding sites. Define
, which represent some set of elements of interest such as enhancers or CTCF binding sites. Define 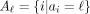 to be the positions annotated by label ℓ. To avoid biases caused by differing-size elements, we assume that each element
occupies just 1 bp. In the case of larger elements (such as MACS-called TF binding sites, which are ∼200 bp), we define each
element as the middle base pair of the range.
to be the positions annotated by label ℓ. To avoid biases caused by differing-size elements, we assume that each element
occupies just 1 bp. In the case of larger elements (such as MACS-called TF binding sites, which are ∼200 bp), we define each
element as the middle base pair of the range.
For each label, we compute the predictive precision of label ℓ as
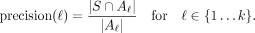 (11)
We rank the labels in decreasing order of their precision on a training set to get an order
(11)
We rank the labels in decreasing order of their precision on a training set to get an order 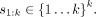 Using this ordering, we form k predictors,
Using this ordering, we form k predictors,
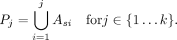 The true positives and false positives of a predictor P are
The true positives and false positives of a predictor P are 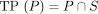 and
and 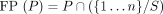 , respectively. The predictors are in order of decreasing stringency; that is,
, respectively. The predictors are in order of decreasing stringency; that is, 
We can trace out the full sensitivity–specificity tradeoff (such as for an ROC or PR curve) by interpolating between each
successive pair of predictors. To interpolate between a pair of predictors Pj and Pj+1, we form an interpolated predictor Pj,j+1,θ by sampling each position 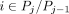 with probability θ ∈ [0,1]. The expected number of true positives and false positives of an interpolated predictor
with probability θ ∈ [0,1]. The expected number of true positives and false positives of an interpolated predictor 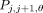 can be shown to be
can be shown to be
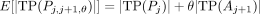 (12)
and
(12)
and
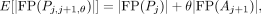 (13)
respectively. We report our performance using a test set disjoint from the training set used to order the labels.
(13)
respectively. We report our performance using a test set disjoint from the training set used to order the labels.
Developmental replication domains
In order to evaluate the replication timing dynamics of different types of domains, we used a four-label (constitutive early/late, switching early/late) annotation of the human genome using published replication timing data for 16 different human cell types (gathered by V Dileep, F Ay, J Sima, WS Noble, and DM Gilbert, unpubl.). This annotation first windowed replication timing data into 40-kb bins and then determined for each window whether it replicates early (RT value > 0.5) or late (RT value < −0.2) in all cell types. Such windows with consistent timing profiles across all cell types were labeled as “constitutively early” and “constitutively late,” respectively. The remaining windows either were labeled as switching or were left unlabeled. Switching windows are determined as those with an absolute value of replication timing larger than 0.5 in all cell types but with an opposite sign than others in at least one cell type. Switching windows that are early and late replicating in IMR90 were labeled as switching early and switching late, respectively.
Consistent domain boundaries
When we annotated domains in eight cell types, we found that domain boundaries were shared between annotations much more often than would be expected by chance. To identify developmentally consistent domain boundaries, we first formed a list of all segment boundaries that occurred in at least one cell type. For each boundary, we computed the number of cell types with boundaries within 50 kb. We formed a set of representative by greedily selecting the boundary with the most nearby boundaries as a representative, removing all boundaries near the representative from the list, and repeating the process until no two boundaries in the list were within 50 kb of one another. While this problem is an instance of the NP-hard set cover problem, the greedy approach is guaranteed to result in a constant-factor approximation of optimal (Nemhauser et al. 1978). This yielded a set of 13,906 boundary groups, each >50 kb from all other groups. We defined the 2967 boundary groups composed of at least five boundaries as consistent boundaries.
Data access
Domain annotations and code for Segway with GBR are available as Supplemental Material and online at http://noble.gs.washington.edu/proj/gbr.
Acknowledgments
This work was supported by NIH awards U41HG007000 and R01ES024917 and by the Princess Margaret Cancer Foundation.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.184341.114.
- Received September 13, 2014.
- Accepted February 6, 2015.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








