
Detection and phasing of single base de novo mutations in biopsies from human in vitro fertilized embryos by advanced whole-genome sequencing
- Brock A. Peters1,2,
- Bahram G. Kermani1,
- Oleg Alferov1,
- Misha R. Agarwal1,
- Mark A. McElwain1,
- Natali Gulbahce1,
- Daniel M. Hayden1,
- Y. Tom Tang1,2,
- Rebecca Yu Zhang1,
- Rick Tearle1,
- Birgit Crain1,
- Renata Prates3,
- Alan Berkeley4,
- Santiago Munné3 and
- Radoje Drmanac1,2
- 1Complete Genomics, Inc., Mountain View, California 94043, USA;
- 2BGI-Shenzhen, Shenzhen 51803, China;
- 3Reprogenetics, Livingston, New Jersey 07039, USA;
- 4NYU Fertility Center, New York, New York 10016, USA
- Corresponding authors: bpeters{at}completegenomics.com, rdrmanac{at}completegenomics.com
Abstract
Currently, the methods available for preimplantation genetic diagnosis (PGD) of in vitro fertilized (IVF) embryos do not detect de novo single-nucleotide and short indel mutations, which have been shown to cause a large fraction of genetic diseases. Detection of all these types of mutations requires whole-genome sequencing (WGS). In this study, advanced massively parallel WGS was performed on three 5- to 10-cell biopsies from two blastocyst-stage embryos. Both parents and paternal grandparents were also analyzed to allow for accurate measurements of false-positive and false-negative error rates. Overall, >95% of each genome was called. In the embryos, experimentally derived haplotypes and barcoded read data were used to detect and phase up to 82% of de novo single base mutations with a false-positive rate of about one error per Gb, resulting in fewer than 10 such errors per embryo. This represents a ∼100-fold lower error rate than previously published from 10 cells, and it is the first demonstration that advanced WGS can be used to accurately identify these de novo mutations in spite of the thousands of false-positive errors introduced by the extensive DNA amplification required for deep sequencing. Using haplotype information, we also demonstrate how small de novo deletions could be detected. These results suggest that phased WGS using barcoded DNA could be used in the future as part of the PGD process to maximize comprehensiveness in detecting disease-causing mutations and to reduce the incidence of genetic diseases.
Worldwide, more than 5 million babies (Ferraretti et al. 2013) have been born through in vitro fertilization (IVF) since the birth of the first in 1978 (Steptoe and Edwards 1978). Exact numbers are difficult to determine, but it has been estimated that currently 350,000 babies are born yearly through IVF (de Mouzon et al. 2009, 2012; Centers for Disease Control and Prevention 2011; Ferraretti et al. 2013). That number is expected to rise, as advanced maternal age is associated with decreased fertility rates and women in developed countries continue to delay childbirth to later ages. In 95% of IVF procedures, no diagnostic testing of the embryos is performed (https://www.sartcorsonline.com/rptCSR_PublicMultYear.aspx?ClinicPKID=0). Couples with prior difficulties conceiving or those wishing to avoid the transmission of highly penetrant heritable diseases often choose to perform preimplantation genetic diagnosis (PGD). PGD involves the biopsy of one cell from a 3-d embryo or the recently more preferred method, due to improved implantation success rates (Scott et al. 2013b), of up to 10 cells from a 5- to 6-d blastocyst-stage embryo. Following biopsy, genetic analysis is performed on the isolated cell(s). Currently this is an assay for translocations and the correct chromosome copy number (Hodes-Wertz et al. 2012; Munne 2012; Yang et al. 2012; Scott et al. 2013a; Yin et al. 2013), a unique test designed and validated for each specific heritable disease (Gutierrez-Mateo et al. 2009), or a combination of both (Treff et al. 2013). Importantly, none of these approaches can detect de novo mutations.
Advanced maternal age has long been associated with an increased risk of producing aneuploid embryos (Munne et al. 1995; Crow 2000; Hassold and Hunt 2009) and giving birth to a child afflicted with Down syndrome or other diseases resulting from chromosomal copy number alterations. Conversely, children of older fathers have been shown to have an increase in single base and short multibase insertion/deletion (indels) de novo mutations (Kong et al. 2012). Many recent large-scale sequencing studies have found that de novo variations spread across many different genes are likely to be the cause of a large fraction of autism cases (Michaelson et al. 2012; O’Roak et al. 2012; Sanders et al. 2012; De Rubeis et al. 2014; Iossifov et al. 2014), severe intellectual disability (Gilissen et al. 2014), epileptic encephalopathies (Epi4K Consortium and Epilepsy Phenome/Genome Project 2013), and many other congenital disorders (de Ligt et al. 2012; Veltman and Brunner 2012; Yang et al. 2013; Al Turki et al. 2014). Additionally rare and de novo variations have been suggested to be prevalent in patients with schizophrenia (Fromer et al. 2014; Purcell et al. 2014), and Michaelson et al. (2012) found that single base de novo mutations affect conserved regions of the genome and essential genes more often than regions of unknown function. Current targeted approaches to PGD would miss many of these important functional changes within the embryonic DNA sequence, and even a whole-genome sequencing (WGS)–based carrier screen of both parents would not enable comprehensive preimplantation or prenatal diagnoses due to de novo mutations. As more parents delay childbirth into their mid-30s and later, these studies suggest we should try to provide better diagnostic tests for improving the health of newborns. In this study, we demonstrate the use of an advanced WGS process that provides an accurate and phased genome sequence from about 10 cells, allowing highly sensitive and specific detection of single base de novo mutations from IVF blastocyst biopsies.
Results
To demonstrate the potential of WGS to analyze embryo biopsies, three sequencing libraries were made from biopsies of up to 10 cells from two individual 5-d-old blastocyst-stage embryos from the same couple. For the purpose of de novo mutation validation, two separate biopsies were removed and two separate libraries were made from a single embryo. As a control, three additional libraries were made from about 10 blood cells from unrelated anonymous donors. Libraries were made as previously described using long fragment read (LFR) technology (Peters et al. 2012), a process that only requires about 10 cells of input DNA to generate a high-quality phased genome. Blood samples from the parents and paternal grandparents were also analyzed on the Complete Genomics platform (Drmanac et al. 2010), but were made from ∼400 ng of genomic DNA and did not undergo LFR processing. Coverage was very good for all libraries with both alleles called in 88%–97% of the genome (Table 1; Supplemental Fig. 1; Supplemental Table 1). For LFR analyzed genomes, phasing rates, N50 contig lengths, and other metrics compared very favorably with previous LFR genomes (Peters et al. 2012) sequenced from ∼150 pg of high-quality isolated DNA (Table 1; Supplemental Material; Supplemental Tables 2, 3). LFR data also indicated that while we attempted to make libraries from 10 cells, only the first biopsy for embryo #1 provided that many cells; the libraries from embryo #1 biopsy 2, embryo #2, and the blood cell controls were made from three to five cells.
Comparison of sequencing performance between different genome assemblies
Assessing sensitivity and reducing false-positive variants in embryo genomes
Genomes assembled from a small number of cells and requiring highly amplified DNA, like the embryo genomes in this study, have been shown to harbor a large number of false-positive, single-nucleotide variants (SNVs) (Peters et al. 2012; Zong et al. 2012), presumably due to the error rate of polymerases. However, many of these errors can be removed using redundant haplotype information from multiple pools of DNA. The principle is simple: Errors incorporated during amplification, sequencing, and mapping in individual pools of DNA are unlikely to repeatedly occur exclusively on one parental chromosome. By linking these SNVs to the surrounding heterozygous SNPs, one can assess whether the variant is in phase with one or both haplotypes. Those SNVs that are found to be in phase with both haplotypes, an impossibility for a heterozygous variant, are likely to be sequencing or mapping errors. Conversely, those variants that are in phase with a single haplotype but are only found in a few DNA pools are likely to be polymerase errors incorporated during the early amplification steps (Peters et al. 2012). Zong et al. (2012) also used this redundancy principle by independently amplifying and deep sequencing multiple single cells and showed a reduction in false-positive errors when variants were required to be present in more than one cell.
LFR technology allows the use of both of these strategies for removing false-positive variants as it is (1) conceptually similar to sequencing very long individual DNA molecules, making assembly of separate haplotypes possible (phasing), and (2) it uses 384 pools of DNA, ideally from 10 or more cells, allowing for many individual pools of DNA to be used in calling each variant. Using parental sequence data, we can assess the improvements in error removal through haplotype analysis. After filtering all inherited variants found in the parents and paternal grandparents, as well as common variants (likely to be inherited but false negatively called as reference in parents or grandparents), from several large databases (dbSNP, ∼400 whole genomes from the Wellderly project and a database of whole genomes from the cell lines of 54 unrelated individuals), there still remained over 100,000 SNVs called in each embryo (Supplemental Fig. 2; Supplemental Table 4). As these are not inherited, a small number (∼100) represent de novo mutations, and the remaining are most likely polymerase errors incorporated during LFR processing, sequencing errors, or mapping errors during genome assembly. Repeating the above process on only those variants found in haplotypes results in less than 2000 remaining variants (Supplemental Table 4) and, as previously shown (Peters et al. 2012), demonstrates approximately two orders of magnitude improvement in error reduction.
However, some inherited variants are unphased by LFR and would be considered errors using this strategy. To quantify this loss of sensitivity, we examined all genomic locations where one parent was called a homozygous reference and the other was called homozygous for the variant. From inheritance, each embryo must be heterozygous at these positions except in rare cases where gene conversion has taken place or where errors are made in the parental genomes. Previous studies (Drmanac et al. 2010; Roach et al. 2010, 2011) using Complete Genomics’ sequencing process with a large amount of input DNA suggest that the overall error rate for the parents should be very low and contribute little to this sensitivity calculation. Analysis of the approximately 463,000 loci that met these criteria resulted in a 5.39%–14.39% overall reduction in called heterozygous SNVs (false-negative rate) due to removal by phasing or a lack of sequence coverage in each embryo micro-biopsy (Supplemental Table 5). Not surprisingly, the genome of embryo #1 biopsy 1, generated from the most cells, had the lowest false-negative rate (5.39%).
Analysis of embryo genomes using LFR allows for de novo SNV detection and extremely low false-positive error rates
Requiring variants to be found in haplotypes removed about 100,000 false-positive SNVs; however, each embryo still had between 1000 and 2000 uninherited variants, over 10 times more than the expected number of de novo mutations (Crow 2000; Kong et al. 2012). Most of these additional variants are false-positive errors and rare family variants (not present in population databases) false negatively called reference in the parents and grandparents. A batch artifact (i.e., systematic errors incorporated during amplification and other LFR steps in embryos processed at the same time) and inheritance (i.e., real inherited variants shared between two embryos but not called in parental and grandparental genomes) removal algorithm based on comparing sequence data between individual embryos can be applied when WGS data are available for two or more embryos from a single couple. Application of this filter removed about 1000 additional variants in each of the embryo libraries in this study; however, several hundred to a thousand SNVs per embryo still remain (Table 2; Supplemental Fig. 2). In PGD, analyzing these as putative de novo variants could lead to the elimination of too many healthy embryos, and additional strategies are required to remove them.
Embryo de novo SNV detection and false-positive error removal
The current LFR phasing process was designed to obtain longer haplotype contigs, resulting in the incorporation of some false-positive errors. Further, using phased variants also does not allow for detection of de novo variants in the 20% of the genome that cannot be haplotyped due to regions of low heterozygosity (RLHs) (Peters et al. 2012). Fortunately, in both cases, the number of wells exclusively carrying sequence for each allele of a heterozygous variant can be used as criteria for determining the accuracy of a call. Sequence reads defining a false variant caused by amplification, sequencing, or mapping errors are unlikely to be exclusively (not co-occurring with the reference allele) found in multiple wells. Counting exclusive wells is much more informative than read counts due to the amplification bias and mapping errors that can generate many reads for the false allele. These reads would likely be located in a large number of nonexclusive wells or just a few exclusive wells with overamplification. By analyzing 10 cells aliquoted across 384 wells after DNA denaturing, we expect true variants to be found in 15 to 20 wells in regions with good coverage and in approximately five wells in the majority of low-coverage regions.
By comparing the well counts of de novo–like and random inherited variants (Supplemental Fig. 3), a well threshold of six was determined to be indistinguishable between the two variant categories. Applying this threshold reduced the detection rate of inherited and de novo SNVs to 82% in embryo #1 biopsy 1 and resulted in 94 possible de novo SNVs (82 within LFR contigs and 12 outside of contigs) (Table 2; Supplemental Table 6). Importantly, while many of these SNVs were not called in the second biopsy library, 87 (∼93%) were found to have reads supporting the variant call in at least one well. This suggests that many of these variants are real but lack sufficient read coverage to be called in biopsy 2. It should be noted that some small portion of these 87 SNVs could be inherited but undetected in the genomes of the parents, paternal grandparents, and embryo #2. The seven putative de novo SNVs not detected in the second biopsy represent some combination of false-positive errors in the LFR data of biopsy 1, false-negative errors in the LFR library of biopsy 2, and inherited SNVs not detected in biopsy 2 or the parents. Applying the same well threshold to biopsy 2 resulted in 58 de novo SNVs (48 within LFR contigs and 10 outside of contigs) called with an overall detection rate of 53%. Biopsy 2 is a smaller biopsy of only four cells and so the lower detection rate is not surprising. Of these 58 SNVs, 42 (72%) were also called in biopsy 1 (Supplemental Table 6), lending support to the overall detection rate of 82% for variants in biopsy 1 using a six-well threshold. Of the remaining 16 not called in the library of biopsy 1, 13 were found to have at least one well with read support. Overall, only three putative de novo SNVs were uniquely identified in biopsy 2. Repeating this process on embryo #2, a biopsy of similar size to embryo 1 biopsy 2 results in the identification of 50 de novo SNVs (41 within LFR contigs and nine outside of contigs) (Supplemental Table 7) and a reduction in the overall detection rate to ∼50.2%. The reduced sensitivity to detect de novo SNVs in both embryo #1 biopsy 2 and embryo #2 underlines the importance of starting from 10 or more cells in the process we have described here (Table 2; Supplemental Table 8).
Comparison of de novo SNV detection between embryo #1 biopsies can be used to measure an overall error rate for our process. An error rate of about 1.4 errors per Gb would result if all seven de novo SNVs found in biopsy 1, but not detected in biopsy 2, are attributed to false-positive errors (seven errors in 4.9 Gb of analyzable diploid genome based on 82% sensitivity). Likewise, repeating the process with the three de novo SNVs called in biopsy 2 but not found in biopsy 1 results in an error rate of ∼0.9 errors per Gb (three errors in 3.2 Gb of analyzable diploid genome based on 53% sensitivity). This range of about 0.9–1.4 errors per Gb is ∼100-fold lower than our previous sequencing study with 10 cells (Peters et al. 2012).
Of the 110 putative de novo mutations detected in both biopsies from embryo #1, of which 100 are expected to be real de novo assuming about 10 are errors (Supplemental Table 6), 58 were found on paternal chromosomes and 35 were located on maternal chromosomes (Fig. 1A). An additional 17 could not be phased because either they fell outside of LFR contigs (16) or parental phasing data was ambiguous along the LFR contigs (one). Interestingly, on Chromosome X there are three de novo mutations within 7 bp of each other that all come from the maternal chromosome. These could represent a potential short-read mapping error, but manual inspection of the reads showed clear support for the de novo events. Further, they are supported by at least two wells in both biopsies and not by any reads in embryo #2, despite good read coverage. The most likely explanation is that these mutations represent a single de novo event. Multibase de novo events, similar to this, have previously been described (Schrider et al. 2011; Campbell et al. 2012; Michaelson et al. 2012; Iossifov et al. 2014). The total number of ∼100 de novo mutations and the observation that more are paternally inherited are in agreement with prior analyses (Crow 2000; Conrad et al. 2011; Kong et al. 2012) and lend further support to the assertion that most of these are real de novo mutations. The process of creating de novo mutations is responsible for all of the inherited variation we see in human genomes; therefore, true de novo mutations should have a nucleotide change profile similar to that of all inherited SNVs. Examination of all de novo and inherited variants in embryo #1 confirmed this to be the case (Fig. 1B). No de novo mutations were found to be coding in either biopsy from embryo #1 (Supplemental Table 6), but two coding changes in the genes ZNF266 and SLC26A10, both potentially damaging, were found in embryo #2 (Supplemental Table 7). However, it is unclear if there would be any detrimental effect to the health of a child born with these variants.
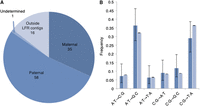
Characteristics of de novo SNVs in embryo #1. After filtering, 110 putative de novo SNVs (including about 10 errors) were identified in embryo #1. (A) Phasing enabled the parent of origin to be determined for 93 of the de novo SNVs. Similar to previous studies, almost twice as many de novo SNVs came from the father as compared to the mother. (B) Specific nucleotide changes for de novo (dark blue) and inherited (light blue) were plotted by frequency. Frequencies of nucleotide changes were similar between de novo and inherited, as would be expected for true de novo SNVs. 95% confidence interval error bars were computed using a one sample proportions test, allowing for Yates’ continuity correction using R software (R Core Team 2014). The error bars suggest that the small differences observed between de novo and inherited are insignificant.
Exon deletions can be detected using haplotype information
Detecting variations at single base resolution is critical for a truly comprehensive PGD test, but just as important is proper quantification of gains and losses of multibase regions of the genome. Currently this is performed as a PGD procedure using array comparative genomic hybridization (aCGH) technologies or, less frequently, low-coverage, next-generation sequencing data (Hou et al. 2013; Wells et al. 2014). These technologies are useful for detecting large copy number changes (>6 Mb) in an economical fashion and could be combined with WGS to first remove embryos with obvious large structural variations from further analysis. Further, whole-genome sequence carrier screening of parents could be used to discover many smaller copy number variations that, combined with sequence data from the embryo, could be used as additional screening criteria. However, small de novo copy number variations in the embryo would still be missed, and there are currently no technologies available for detecting these variants. We hypothesized that LFR haplotype information could be used to improve the detection of small heterozygous deletions in spite of the bias from DNA amplification from 10 cells. As a demonstration, we attempted the detection of exon deletions between 20 and 2000 bp in length.
To achieve this, long DNA fragments from each well were assembled in silico, the parental origin was determined, and coverage was measured at intervals of 20 base pairs (bp) across fragments mapping to gene coding regions. Exons were considered heterozygously deleted in regions with sufficient coverage overall but with zero coverage from one parent (Supplemental Methods). This method of analysis detected six deletions of small exons in embryo #1 biopsy 1 (Fig. 2; Supplemental Table 9), but in embryo #1 biopsy 2 and embryo #2, too many potential exon deletions were detected. Unfortunately, this makes it impossible to measure the true sensitivity of this method. However, five of the six deleted exons were confirmed by analyzing fragment coverage in the parental genomes. As we have mentioned previously, libraries made from five or fewer cells have less redundant long DNA fragment coverage, resulting in too many regions with a stochastic loss of coverage from one parental chromosome. Until improvements in LFR library processing are made, this method of detecting exon deletions can only be reliably used on biopsies of 10 or more cells. Regardless, this demonstrates the potential of using haplotype information to detect challenging types of genomic variation, and with many more libraries made from 10 cells or more, it will be possible to characterize the performance of this process.
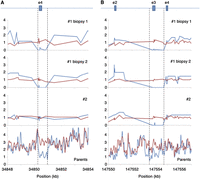
Detection of heterozygous deletions of small exons. LFR haplotype information can be used to separate coverage for each allele. Normalized coverage from each LFR haplotype for embryo #1 biopsies 1 and 2 and embryo #2, as well as 50-bp read coverage windows for both parents, were plotted (blue indicates father; red, mother). (A) A heterozygous deletion of ∼500 bp in the gene TTC23L removing all of exon 4 and part of the intron on either side in both biopsies of embryo #1 and the father was detected. (B) A heterozygous deletion of ∼1000 bp in the gene SPINK14 removing all of exon 3 and parts of the intron on either side was identified in all three biopsies. Coverage for the parents is more difficult to interpret in this region, but it appears that again the father has less coverage.
Discussion
In this paper we demonstrate that advanced WGS, using LFR for haplotype data and enhanced accuracy, can confidently call ∼95% of the embryonic genome, starting with about 10 cells (∼66 pg of DNA). Without using parental WGS to impute missing variants or remove errors, which would also remove true de novo mutations, we demonstrate very high specificity with only a few called errors per genome and an overall 15% loss of sensitivity in the high-confidence SNV detection versus standard WGS from nanogram amounts of genomic DNA. This enables accurate calling of ∼82% of de novo SNVs, the majority of which are also placed into haplotypes, allowing compound heterozygosity analysis with inherited variants or assignment of imprinted status as in Prader-Willi syndrome (Schaaf et al. 2013). Further, we show that the number of false-positive SNVs accumulated as a result of amplifying DNA from a small number of cells, without some form of error reduction such as LFR or limiting WGS to calling only parental variants, dramatically reduces the accuracy of WGS approaches. LFR allows these low error rates (fewer than 10 false SNVs per genome) and detection of most de novo point mutations despite starting with only five to 10 cells and performing 20,000-fold MDA amplification, which introduces about 100,000 DNA mutations per sample. Starting with fewer cells results in lower sensitivity in order to achieve the required specificity for PGD. Thus, we strongly recommend preparation of 10 cell biopsies to maximize sensitivity and specificity of detection of all genetic defects, including de novo mutations.
This is the first demonstration that a large majority of single base, de novo mutations, which cause a disproportionally high percentage of genetic defects (Michaelson et al. 2012), can be detected in PGD. We expect that short de novo indels would be efficiently detected with this barcoding process by using sequencing data and software that allows mapping and well-counting of reads for indel alleles. Barcoding reads that belong to longer genomic fragments, potentially with thousands of distinct barcodes, and the error reduction process as described in this study provide a fundamental solution for accurate and phased WGS from IVF biopsies (and other scarce samples), applicable for both current massively parallel short-read technologies and future longer read, single-molecule sequencing technologies. That said, additional clinical studies with many more samples are required to further demonstrate the promises of this type of analysis for PGD.
In addition to separating inherited from de novo mutations, there are many other benefits of having parental WGS in addition to phased embryonic genomes, such as the ability to detect uniparental heterodisomy (Handyside et al. 2010), which is impossible to do without knowing parental haplotypes. Additionally, parental sequence data can help impute the ∼15% of inherited variants that are detected with lower confidence by LFR and improve phasing in RLHs that in turn helps in phasing and verifying more de novo mutations. Because the cost of WGS is expected to further decrease with technology improvements and broader use, and using parental WGS as the ultimate genetic test (Drmanac 2012) also allows implementation of genomic medicine for parents, we believe that future reproductive medicine should include advanced phased WGS of couples (or parents-to-be, serving as a carrier screen) and of IVF or prenatal embryos.
Use of information gleaned from accurate and complete WGS of IVF embryos as PGD must be limited to known, or novel but obviously disruptive, mutations and variants. As is the case currently for whole-genome analysis of adults and children, some variants with unwanted detrimental effects cannot be reported until there is more precise knowledge of which disrupted genes can be tolerated and which nonsynonymous variants are not disruptive to protein function. Otherwise, all embryos could appear affected and rendered unusable. Thus, with the WGS accuracy demonstrated here and future improvements, the factors limiting completeness and sensitivity of PGD are shifted from genome reading to genome interpreting, including interpretation of combinations of causative and protective genetic variants in the context of signaling and regulatory pathways. This adds additional pressure to improve our genomic knowledge by analyzing millions of human genomes, epigenomes, and transcriptomes with detailed phenotypic information through even more efficient massively parallel nucleic acid analysis systems that are currently under development.
One important detail to consider in the potential use of WGS as a PGD test is that these experiments and analyses currently take months to perform (but eventually we expect this to be reduced to less than a week); therefore, it is necessary to freeze the embryos during analysis. This is perfectly acceptable, and indeed preferable, as embryos frozen using current vitrification techniques show similar or better pregnancy rates when compared to fresh embryos, since frozen embryos are transferred to a more receptive uterus unaffected by the hormones needed to produce multiple eggs for IVF (Zhu et al. 2011; Shapiro et al. 2013). Further, freezing embryos after biopsy is commonly performed in other current PGD techniques (Schoolcraft et al. 2010; Colls et al. 2012). Additionally, the analyses performed in this paper are much improved when carried out on about 10 cells biopsied from a day 5-6 blastocyst. Detection of de novo variants would be extremely difficult from only one cell of a day 3 embryo due to excessively high error rates and the inability to remove those errors using redundant haplotyping. Importantly, recent studies have demonstrated that culturing embryos to day 5 selects against some chromosome abnormalities that block development (Ata et al. 2012), thus decreasing the total number of embryos that would need to undergo testing and reducing the cost of this procedure. Finally, an efficient simple screen for aneuploidy and detectable CNVs using cost-effective conventional PGD techniques would likely be employed prior to WGS to remove ∼50% of embryos analyzed, further decreasing the cost of this type of analysis by only focusing on two to three remaining embryos without gross genetic defects. This type of two-step PGD test could potentially prevent most of the severe genetic diseases in IVF newborns.
We have previously demonstrated the importance of haplotype information in identifying inactivated genes and removing false-positive SNVs (Peters et al. 2012). In this study, we have expanded our use of LFR data to detect hemizygous short exon deletions and analyze the mitochrondrial genome (Supplemental Materials; Supplemental Tables 10–12). Moreover, we demonstrate how LFR well data can be used to dramatically reduce false-positive errors and allow for the detection of single base de novo mutations from a small number of cells. These analyses, combined with recent ENCODE annotations of regulatory sequences and a rich list of population variants obtained by high-quality WGS on a large number of unrelated individuals, create a very powerful genome-wide prediction tool. These types of analyses can be expanded from comprehensive IVF embryo testing to any tissue in which about 10 cells are available, opening the door to noninvasive prenatal genetic testing using circulating fetal cells and cancer screening from circulating tumor cells or microbiopsies. Furthermore, our results indicate that practically error-free comprehensive WGS of individual genomes can be obtained without expensive variant validation by applying LFR on 10 or more easy-to-obtain blood cells, resulting in the ultimate genetic test (Drmanac 2012) that can be stored and used during a person’s entire life.
Methods
Blastocyst biopsy
Following conventional ovarian stimulation and egg retrieval, eggs were fertilized by intracytoplasmic sperm injection (ICSI) to avoid sperm contamination in the PGD test. Following growth to day 3, embryos were biopsied using fine glass needles and one cell was removed from each embryo. Each blastomere was added individually to a clean tube, covered with molecular-grade oil, and shipped on ice to Reprogenetics for PGD. Following the clinical PGD testing and embryo transfer, unused blastocyst-stage embryos were donated to the NYU Fertility Center and shared with Reprogenetics for use in developing new PGD testing modalities. Patients were informed of the research and all work was undertaken with full approval by an IRB from the NYU Fertility Center. Up to 10 cells were biopsied from each embryo, frozen, and shipped to Complete Genomics for advanced WGS analysis.
LFR libraries
Briefly, isolated cells from each blastocyst were lysed, and DNA was alkaline denatured with the addition of 1 μL of 400 mM KOH/10 mM EDTA. After 1 min, thio-protected random 8-mers were added to denatured DNA. The volume was brought to 400 μL by addition of dH2O, and 1 μL was aliquoted into each well of a 384-well plate. Long genomic fragments in each well were amplified ∼20,000-fold using a modified multiple displacement amplification (Dean et al. 2002; Peters et al. 2012) and fragmented to ∼500 bp. A unique 10-base barcode was ligated to all fragments in each well, and all barcoded fragments were pooled and analyzed on Complete Genomics DNA nanoarray sequencing platform (Supplemental Methods; Supplemental Fig. 1; Drmanac et al. 2010) and phased using a method designed for analyzing low-read coverage from each initial long DNA fragment (0.5×) (Peters et al. 2012). Genomic data were mapped and phased as previously described (Drmanac et al. 2010; Carnevali et al. 2012; Peters et al. 2012).
Single-pixel imaging
The current Complete Genomics platform uses patterned arrays of DNA nano-balls (Drmanac et al. 2010) with a spacing of 600 nm center to center. A single 1″ × 3″ microscope slide has ∼4 billion DNA spots. To take advantage of the patterned DNA grid for fast imaging, a CCD camera is aligned with the DNA arrays so that each spot is read with one CCD pixel for each of four colors. This yields the theoretical maximum imaging efficiency for massively parallel genomic sequencing. At ∼70 bases per spot with a 60% total yield, one array generates >50× coverage of a human genome per slide (4B spots × 0.6 yield × 70 bases/spot/3 Gb genome).
Data access
Read and mapping data have been submitted to the database of Genotypes and Phenotypes (dbGaP; http://www.ncbi.nlm.nih.gov/gap/) under study ID phs000858.v1.p1.
Competing interest statement
Employees of Complete Genomics have stock options in the company. Complete Genomics has filed several patents on this work.
Acknowledgments
We acknowledge the ongoing contributions and support of all Complete Genomics employees, in particular the many highly skilled individuals that work in the libraries, reagents, and sequencing groups that make it possible to generate high-quality, whole-genome data.
Author contributions: B.A.P., R.D., and S.M. conceived the study. S.M., A.B., and R.P. collected, biopsied, and performed standard PGD analysis on the embryos. B.A.P., D.M.H., R.Y.Z., and M.A.M. developed the laboratory processes and made the libraries for sequence analysis. B.G.K., B.A.P., N.G., M.A., R.T., R.D., O.A., and Y.T.T. performed analyses. B.C. curated all of the data. B.A.P., B.G.K., S.M., and R.D. coordinated the study. B.A.P., B.G.K., M.A.M., S.M., and R.D. wrote the paper. All authors contributed to revision and review of the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.181255.114.
Freely available online through the Genome Research Open Access option.
- Received July 10, 2014.
- Accepted January 5, 2015.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








