
Enabling functional genomics with genome engineering
- 1Department of Biomedical Engineering, Duke University, Durham, North Carolina 27708, USA;
- 2Center for Genomic and Computational Biology, Duke University, Durham, North Carolina 27708, USA;
- 3Department of Orthopaedic Surgery, Duke University Medical Center, Durham, North Carolina 27710, USA
- Corresponding author: charles.gersbach{at}duke.edu
Abstract
Advances in genome engineering technologies have made the precise control over genome sequence and regulation possible across a variety of disciplines. These tools can expand our understanding of fundamental biological processes and create new opportunities for therapeutic designs. The rapid evolution of these methods has also catalyzed a new era of genomics that includes multiple approaches to functionally characterize and manipulate the regulation of genomic information. Here, we review the recent advances of the most widely adopted genome engineering platforms and their application to functional genomics. This includes engineered zinc finger proteins, TALEs/TALENs, and the CRISPR/Cas9 system as nucleases for genome editing, transcription factors for epigenome editing, and other emerging applications. We also present current and potential future applications of these tools, as well as their current limitations and areas for future advances.
Genomic research has the potential to dramatically improve medicine, agriculture, biotechnology, and our fundamental understanding of living systems. Recent advances have generated extensive annotation of genomic and epigenomic regulatory modules within chromatin (Bernstein et al. 2010; The ENCODE Project Consortium 2012; Roadmap Epigenomics Consortium et al. 2015), as well as an understanding of genomic topological architecture (Dekker et al. 2013; Pombo and Dillon 2015). However, the roles of these numerous genes, regulatory elements, epigenetic marks, and topological domains in determining overall cell function remain incompletely understood. The recent development of genome engineering technologies has enabled precise interrogation of the function of these genomic features and their causal role in gene regulation. Additionally, these tools are facilitating the translation of this genomic information into tangible benefits for biotechnology, agriculture, and human therapeutics. In this Perspective, we discuss the recent advances to the most commonly used genome engineering technologies, including synthetic zinc finger (ZF) proteins, transcription activator-like effectors (TALEs), and CRISPR/Cas9 targeting systems, and their application in a new era of functional genomics.
Genome engineering technologies
Cys2-His2 ZF domains are naturally occurring protein motifs which typically recognize three base pairs within the major groove of DNA (Pavletich and Pabo 1991; Wolfe et al. 2000). These modular ZF domains can be arrayed such that synthetic ZF DNA-binding proteins (DBPs) target a specific series of DNA triplets at unique genomic addresses (Fig. 1A; Liu et al. 1997; Gersbach et al. 2014). TALE proteins are components of plant pathogens that bind host DNA to facilitate virulence (Kay et al. 2007; Romer et al. 2007). TALEs consist of repeated DNA-binding domains containing repeat variable diresidues (RVDs), each of which recognizes a single nucleotide in target DNA (Boch et al. 2009; Moscou and Bogdanove 2009). Similar to ZFs, individual TALE RVDs can be linked in series to localize TALEs to target loci (Fig. 1B; Christian et al. 2010; Morbitzer et al. 2010; Cermak et al. 2011; Miller et al. 2011; Zhang et al. 2011). Clustered regularly interspaced short palindromic repeat (CRISPR) arrays and CRISPR-associated (Cas) proteins are components of bacterial and archaeal adaptive immune systems (Barrangou et al. 2007; Makarova et al. 2011). Unlike ZFs and TALEs, in which protein moieties dictate DNA recognition, CRISPR/Cas systems utilize RNA-mediated Watson-Crick bonding for recognition of nucleic acids.
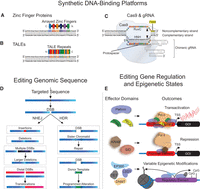
Zinc finger, TALE, and Cas9-gRNA platforms for editing genomic sequence and regulatory states. Individual zinc finger domains (A) and TALE repeats (B) that recognize unique triplets or single base pairs, respectively, can be arrayed in engineered proteins to target specific genomic sequences. (C) Cas9 in complex with a chimeric guide RNA (gRNA) can recognize a specific genomic address through complementarity between the protospacer segment of the gRNA and target DNA. The formation of this complex is dependent upon the presence of a protospacer adjacent motif (PAM). The RuvC and HNH nuclease domains of Cas9 cleave genomic DNA that matches the protospacer (i.e., the noncomplementary strand) and genomic DNA with complementarity to the protospacer (i.e., the complementary strand), respectively (indicated by black triangles). (D) Zinc fingers and TALEs fused to nuclease domains or Cas9 in complex with a gRNA can cleave targeted sequences to generate double-strand breaks (DSBs). DSB resolution through nonhomologous end joining (NHEJ) or homology-directed repair (HDR) can lead to various alterations in genomic sequence. (E) Zinc finger, TALE, or deactivated nuclease-null Cas9 (dCas9) platforms can also be fused to diverse effector domains to modify endogenous gene regulation and epigenetic states: (TSS) transcription start site; (GOI) gene of interest.
Prokaryotes harboring type II CRISPR/Cas systems transcribe CRISPR-RNAs (crRNAs) that hybridize with trans-activating crRNAs (tracrRNAs) that complex with the Cas9 nuclease (Brouns et al. 2008; Deltcheva et al. 2011; Jinek et al. 2012; Doudna and Charpentier 2014). A single crRNA-tracrRNA chimera, known as a guide RNA (gRNA), can be designed for simplified use in engineered systems (Jinek et al. 2012). The gRNA binds to and directs the Cas9 protein to DNA through regions of crRNA complementarity (termed “protospacer” sequences). A stringent prerequisite to protospacer hybridization is the presence of a protospacer adjacent motif (PAM) in the target DNA, which flanks the region of protospacer complementarity (Fig. 1C; Mojica et al. 2009; Anders et al. 2014; Sternberg et al. 2014; Kleinstiver et al. 2015). Interactions between the PAM-proximal “seed” nucleotides in the target site and the complementary gRNA sequence are also critical drivers of Cas9 targeting. The orthogonality of various prokaryotic Cas9 proteins with differing PAM requirements can be exploited for multiplex genome engineering efforts (Esvelt et al. 2013; Ran et al. 2015).
Engineered targeting of eukaryotic genomes with ZFs, TALEs, and type II CRISPR/Cas systems has established these technologies as useful resources for present and future genome engineering endeavors. Although each of these systems has been successfully incorporated into diverse genome engineering strategies, they each have unique benefits and limitations that depend upon the particular application (Gaj et al. 2013; Carroll 2014). Other genome engineering technologies, such as meganucleases and their fusion to TALE proteins (Boissel et al. 2014; Stoddard 2014) have also been successfully applied to gene editing in eukaryotes, but have been less widely adopted in the context of functional genomics due to the complexity involved in engineering meganucleases targeted to new sequences.
Editing genome sequences with programmable nucleases
Gene targeting based on homologous recombination can introduce exogenous DNA at genomic loci (Smithies et al. 1985; Thomas et al. 1986). The efficiency of this method is dramatically enhanced in the presence of double-strand breaks (DSBs) (Rouet et al. 1994). Cells generally use two distinct pathways to resolve DSBs (Chapman et al. 2012): homology-directed repair (HDR) and nonhomologous end joining (NHEJ) (Fig. 1D). DSB resolution through NHEJ occurs by direct ligation of DSB ends or through microhomology on DSB termini (Lieber 2010). This error-prone process results in small insertions or deletions (indels) at endogenous loci. The generation of two DSBs flanking a genomic region can also lead to NHEJ-mediated chromosomal deletions (Lee et al. 2010; Carlson et al. 2012; Kim et al. 2013; Essletzbichler et al. 2014) and inversions (Carlson et al. 2012; Lee et al. 2012; Xiao et al. 2013). Programmable nucleases can also be used to create NHEJ-mediated translocations in vivo (Brunet et al. 2009; Maddalo et al. 2014). HDR uses regions of homologous DNA on sister chromatids or exogenous DNA to repair DSBs. In contrast to NHEJ, HDR is a high-fidelity process leading to largely error-free correction at DSB sites. HDR can lead to specified incorporation of sequences that are rationally designed into donor DNA templates. In many instances, this is advantageous over NHEJ, in which DSB resolution is unpredictable. However, NHEJ is active throughout the cell cycle and is the predominant DSB repair mechanism, whereas HDR occurs less frequently and is significantly down-regulated outside of S and G2 cell cycle phases. Therefore recent efforts have focused on promoting HDR by inhibition of NHEJ events to afford more precise genome editing (Chu et al. 2015; Maruyama et al. 2015).
Because NHEJ and HDR can be used to incorporate specific sequence changes into genomes, the capability to induce DSBs at target loci holds great potential for genome engineering (Carroll 2014; Kim and Kim 2014). Although ZFs and TALEs do not intrinsically cleave target DNA, they can be directly fused to the catalytic domain of the type II restriction endonuclease FokI (Li et al. 1992) to create ZF and TALE nucleases (ZFNs and TALENs). Engineering new DNA-binding specificities into the ZFs and TALEs allows programmed DSB induction (Kim et al. 1996; Christian et al. 2010). FokI domains must dimerize to cleave target DNA (Bitinaite et al. 1998; Vanamee et al. 2001), necessitating the engineering of two ZFN or TALEN monomers for each DSB. Modifications to the FokI domain have also been created to increase nuclease activity and specificity (Miller et al. 2007; Szczepek et al. 2007; Guo et al. 2010; Doyon et al. 2011b). Moreover, fusion of the FokI domain to the nuclease-inactivated Cas9 protein (dCas9) has also been used to increase the specificity of the CRISPR gene editing system (Guilinger et al. 2014b; Tsai et al. 2014). Optimizing the length of the ZF or TALE array can also increase nuclease activity and specificity (Bhakta et al. 2013; Guilinger et al. 2014a).
As an alternative to introducing DSBs and inducing DNA repair pathways, catalytic domains of site-specific recombinases can also be fused to synthetic ZFs (Akopian et al. 2003) and TALEs (Mercer et al. 2012) to excise genomic DNA segments or integrate exogenous DNA at targeted genomic sites (Gordley et al. 2009; Gersbach et al. 2011). CRISPR/Cas-based recombinase fusions have not yet been reported but may also prove useful. Additionally, transposases catalyze the rearrangement of endogenous elements and have been used for artificial genome manipulation (Ivics et al. 2009). Targeting of transposase activity with ZF, TALE, or CRISPR/Cas scaffolds is also an active area of research (Yant et al. 2007; Voigt et al. 2012; Galvan et al. 2014). Importantly, targeted recombination or transposition may reduce cellular toxicity relative to the introduction of DSBs and their subsequent resolution through NHEJ or HDR. These alternate mechanisms to genome engineering may also increase efficiencies by decoupling editing from endogenous DNA repair mechanisms.
Synthetic regulation of transcription
In addition to the editing of DNA sequences, these genome engineering technologies can be used to manipulate endogenous gene expression. Early work with an engineered ZF protein directly fused to the herpes simplex viral VP16 transactivator demonstrated proof-of-principle of targeted transcriptional activation by inducing the expression of an extrachromosomal transgene in human cells (Liu et al. 1997). Subsequent studies showed that tandem repeats of VP16 were even more robust transcriptional activators than VP16 alone when linked to ZFs (Beerli et al. 1998), TALEs (Zhang et al. 2011), and nuclease-null deactivated Cas9 (dCas9) (Fig. 1E; Gilbert et al. 2013; Konermann et al. 2013; Maeder et al. 2013b; Mali et al. 2013a; Perez-Pinera et al. 2013a). Tetrameric VP16 domains (termed “VP64”) have exhibited the most widespread application as transcriptional activation domains, although larger multimers of VP16 have also been reported (Cheng et al. 2013). VP16 domains recruit cellular cofactors, such as components of the basal transcriptional machinery and chromatin remodelers (Hirai et al. 2010). Other transcriptional activation domains function similarly and have also been used in engineered transcription factors (Kim et al. 1997; Liu et al. 2001; Bikard et al. 2013; Anthony et al. 2014; Chavez et al. 2015; Konermann et al. 2015). Synergistic effects among multiple activators have been frequently observed with this class of activation domains, both when these effectors are localized in high density at adjacent sequences (Maeder et al. 2013b,c; Mali et al. 2013a; Perez-Pinera et al. 2013a,b) and when combined in cis as multimolecular complexes (Cheng et al. 2013; Chakraborty et al. 2014; Gao et al. 2014; Tanenbaum et al. 2014; Konermann et al. 2015). This synergy is likely related to enhanced subunit recruitment and/or effective decreases in cofactor dissociation rates at targeted loci.
Synthetic DBPs can also function as programmable transcriptional repressors. Localization of dCas9 near transcription start sites (TSSs) can repress active gene expression (Bikard et al. 2013; Qi et al. 2013). However, when fused to repressive domains, such as the KRAB domain (Margolin et al. 1994), the inhibitory effect of ZFs (Beerli et al. 1998), TALEs (Cong et al. 2012), or dCas9 (Gilbert et al. 2013) on transcription is markedly enhanced. Transcriptional repression by these methods is often accompanied by changes in chromatin structure (Groner et al. 2010; Kearns et al. 2015), which is likely a reflection of secondary KRAB-mediated recruitment of chromatin remodelers (Ying et al. 2015). Other domains have also been used for programmed transcriptional repression (Beerli et al. 1998; Snowden et al. 2002; Cong et al. 2012; Mahfouz et al. 2012; Gilbert et al. 2013; Konermann et al. 2013). In contrast to artificial gene activation with effectors and recruited cofactors, repression using synthetic DBPs has not been observed to function synergistically. In addition, the degree of repression by different tools varies dramatically, even within the same DNA-targeting platform and when targeting sequences in close proximity. Additional work is needed to characterize the factors that determine the potency of gene repression, which could be related to variable targeting affinities, interactions with endogenous factors, and/or local chromatin architecture.
Next generation genome engineering: epigenome editing
The ability to readily toggle epigenetic states holds tremendous value for basic research and potentially for human therapies. Efforts aimed at editing the epigenome using synthetic DBPs are rapidly evolving (Jurkowski et al. 2015). These methods can be used to provide evidence of the causality of epigenetic marks such as DNA methylation and histone subunit modifications. Furthermore chromatin-remodeling domains fused to DBPs have also expanded our ability to modulate genomic regulatory regions. For instance, transcriptional manipulation mediated by synthetic DBPs has been most well-characterized when targeted to within 300 base pairs of TSSs. However, directed modulation of distal regulatory elements, such as enhancers, has recently been shown to be possible, albeit with varying efficacy (Gao et al. 2013, 2014; Mendenhall et al. 2013; Ji et al. 2014; Frank et al. 2015; Hilton et al. 2015; Kearns et al. 2015). Enhancers represent dynamic genomic regulatory modules with differential functionality during cell lineage specification, and perturbation of enhancer function has been strongly associated with disease (Heinz et al. 2015; Roadmap Epigenomics Consortium et al. 2015). Similar to other regions of eukaryotic genomes, enhancers are controlled by dynamic epigenetic states, including methylated DNA and post-translational modification of histone subunits (Shlyueva et al. 2014; Heinz et al. 2015). Thus, epigenome editing tools for manipulating these epigenetic modifications are critical to facilitating our understanding of the links between gene regulation, development, and disease.
High levels of 5-methylcytosine (5mC) at enhancers and promoter regions are frequently correlated with transcriptional repression (Jones 2012; Schübeler 2015). Initial work with designer ZFs fused to prokaryotic DNA methyltransferases demonstrated targeted methylation of DNA in vitro (Xu and Bestor 1997; McNamara et al. 2002; Nomura and Barbas 2007; Smith and Ford 2007), on extrachromosomal (Nomura and Barbas 2007) or integrated plasmid DNA (Smith and Ford 2007), and at endogenous eukaryotic targets (Carvin et al. 2003). Furthermore, targeting of mammalian DNA methyltransferases with synthetic DBPs has established that site-specific DNA methylation at promoters can repress endogenous gene expression (Li et al. 2007; Rivenbark et al. 2012; Siddique et al. 2013). Currently, direct and targeted DNA methyltransferase activity to endogenous genes has only been applied using engineered ZF and TALE (Bernstein et al. 2015) protein scaffolds, However, it is probable that similar strategies could be adapted to CRISPR/Cas platforms.
The targeted demethylation of genomic DNA has also been used to activate gene expression using artificial DBPs. The TET family of proteins catalyzes oxidation of 5mC in eukaryotic genomes, leading to reversion to unmethylated cytosine following DNA replication (Lu et al. 2015). Direct fusion of the TET1 catalytic domain to TALEs targeting regions near endogenous human genes decreased DNA methylation, leading to increased mRNA expression (Maeder et al. 2013a). In addition, the catalytic domain of murine TET2, and to a lesser extent TET1, decreased DNA methylation when targeted to human promoter regions by engineered ZF proteins (Chen et al. 2014). Artificial localization of murine thymidine DNA glycosylase, an enzyme involved in cytosine demethylation (Cortellino et al. 2011) has also been shown to decrease DNA methylation and augment gene expression from an endogenous target promoter (Gregory et al. 2013). Collectively, these results demonstrate that the targeted manipulation of DNA methylation is possible, and cytosine methylation is functionally linked to controlling gene expression.
Certain modifications on the histone subunit tails of nucleosomes are highly correlated with genomic regulatory activity (Zhou et al. 2011; Shlyueva et al. 2014; Heinz et al. 2015). In order to take advantage of this mode of transcriptional regulation and also to develop tools to better understand its roles in gene regulation, there has been a recent emphasis on targeted perturbation of histone modifications. Acetylation at lysine residues 27 and 9 of histone subunit H3 (H3K27ac and H3K9ac, respectively) are generally enriched at loci associated with high transcriptional activity such as active promoters and enhancers. A fusion of the acetyltransferase core domain of the human EP300 protein robustly activated endogenous human genes when targeted to promoter or enhancer loci using ZFs, TALEs, and dCas9 variants (Hilton et al. 2015). This activation was accompanied by enrichment for H3K27ac at a targeted promoter and at a targeted enhancer. Notably, targeted H3K27 acetylation at the well-characterized human beta-globin HS2 enhancer using dCas9 fused to the catalytic core of EP300 also led to H3K27ac enrichment and transcriptional induction from HS2-responsive promoters. Together, these results support a model in which acetylation plays a casual role in gene activation. Furthermore, this suggests that H3K27ac enrichment at human enhancers may precede and coordinate distal H3K27ac deposition. Whether this deposition occurs through physical genomic contacts and/or other endogenous factors is the subject of ongoing study. Moreover, the direct manipulation of chromatin signatures using a chromatin acetyltransferase domain appeared to be mechanistically distinct from effectors requiring other cofactors for activity, such as VP64 (Hilton et al. 2015). Thus, improvements in these programmable epigenomic modifiers may enhance the synthetic engineering of gene activation.
The acetylation of histone subunit tails can be reversed by histone deacetylases (HDACs). Recent studies have used full proteins or truncated protein domains with HDAC activity fused to ZFs (Keung et al. 2014) or TALEs (Konermann et al. 2013) to silence gene expression. Histone subunits are also dynamically regulated through methylation and demethylation of lysine residues. The catalytic regions of histone H3K9 methyltransferases EHMT2 (also known as G9A) and SUV39H1 have been found to repress transcription and alter chromatin status at targeted promoters when fused to ZFs (Snowden et al. 2002; Falahi et al. 2013; Heller et al. 2014) and TALEs (Konermann et al. 2013). Targeted H3K4 demethylation has also been applied using TALEs or dCas9 fused to the KDM1A protein (also known as LSD1) (Mendenhall et al. 2013; Kearns et al. 2015), enabling the characterization of known and putative enhancers.
In addition to modulation of DNA methylation and histone residues, synthetic DBPs have also been used to manipulate chromosomal architecture. ZFs designed to artificially coordinate genomic looping between the HS2 enhancer of the globin locus control region and the beta-globin promoter activate gene expression in mouse cells (Deng et al. 2012) and similar designs can direct differential gene expression patterns between HS2 and globin genes in human and mouse cell lines (Deng et al. 2014). These results suggest that the physical interactions between enhancers and promoters can have a causal effect on gene expression. Although artificially generated chromosomal contacts have not been reported yet using TALEs or dCas9 platforms, similar approaches are likely feasible and would provide useful expansions to the genome engineering toolbox for rapid characterization of the role of chromatin conformation.
Specificity of ZFs, TALEs, and CRISPR/Cas9 systems
Understanding the target specificity of synthetic DBPs is central to their efficacy as biotechnological tools and therapeutics. Some studies using artificial ZFs fused to transcriptional effector domains indicate relatively high specificity for target gene modulation (Snowden et al. 2003). However, other results suggest widespread genomic interactions with these proteins that can lead to off-target transcriptional dysregulation (Falahi et al. 2013; Grimmer et al. 2014). Surveys of ZF nuclease specificities demonstrate that off-target effects can be prevalent (Cornu et al. 2008; Pattanayak et al. 2011), although this off-target activity may be mitigated by optimized design tools that minimize confounding factors such as context-dependent ZF domain effects (Isalan et al. 2001; Maeder et al. 2008; Sander et al. 2011; Gupta et al. 2012; Persikov et al. 2015).
As the RVDs of each synthetic TALE repeat dictate base-pair recognition, TALEs can theoretically target any genomic sequence of interest. However, engineered TALE RVDs exhibit a quantifiable variance in nucleotide recognition as well as positional effects that can lead to localization at unintended sequences (Cermak et al. 2011; Miller et al. 2011, 2015; Mali et al. 2013a; Meckler et al. 2013; Juillerat et al. 2014). Although the frequency of generating highly active TALE nucleases is typically higher than that of ZF nucleases (Kim and Kim 2014), certain limitations exist, such as apparent difficulty targeting methylated DNA (Valton et al. 2012) and requirements for thymine bases at 5′ targeting sites (Mak et al. 2012; Lamb et al. 2013). In addition, the larger size and proclivity for recombination of repetitive sequences in TALE proteins may present difficulties in certain applications, such as viral delivery (Holkers et al. 2013), although this issue has been addressed by optimizing the codon usage of repetitive RVDs (Yang et al. 2013b). Studies of genome-wide DNA-binding, gene regulation, and chromatin remodeling suggest a high level of specificity of TALE-based transcriptional activators, although binding to off-target sites is measurable (Polstein et al. 2015).
The interaction between Cas9 and a gRNA leads to conformational changes that activate surveillance for specific target sites by Cas9 (Jinek et al. 2014; Nishimasu et al. 2014; Jiang and Doudna 2015). However, off-target interactions between dCas9 and genomic DNA have been observed in human cells, even in the absence of gRNAs (Kuscu et al. 2014; Wu et al. 2014a; O'Geen et al. 2015; Polstein et al. 2015). Although off-target binding events likely occur with dCas9-based transcriptional/epigenetic modifiers, assessments of global gene expression suggest that changes are largely restricted to the intended target sites (Gilbert et al. 2013; Perez-Pinera et al. 2013a; Hilton et al. 2015; Polstein et al. 2015). Cas9 nucleases have also been found to cause DSBs at unintended sites (Fu et al. 2013; Hsu et al. 2013; Mali et al. 2013a; Pattanayak et al. 2013), and this activity is currently thought to be related to factors including gRNA composition, chromatin accessibility, and gRNA seed/PAM sequence abundance. Several algorithms exist that allow researchers to predict potential off-target Streptococcus pyogenes gRNA binding sites and aid in optimal gRNA design (Hsu et al. 2013; Bae et al. 2014; Cradick et al. 2014; Heigwer et al. 2014; Singh et al. 2015), However, this is clearly an area where significant future research is needed.
Applications of modern genome engineering technologies
Gene knockouts
The most established application of modern genome engineering technologies is the disruption of loci through targeted nuclease activity (Fig. 2A; Urnov et al. 2010; Joung and Sander 2013; Hsu et al. 2014; Sander and Joung 2014). Genetic knockouts in eukaryotes through NHEJ-mediated disruption both in cell culture (Porteus and Baltimore 2003; Perez et al. 2008; Miller et al. 2011; Cho et al. 2013; Cong et al. 2013; Jinek et al. 2013; Mali et al. 2013b; Liao et al. 2015b) and in animals (Bibikova et al. 2002, 2003; Doyon et al. 2008; Meng et al. 2008; Cui et al. 2011; Wood et al. 2011; Hwang et al. 2013) can lead to complete loss of gene function. Moreover, genetic knockouts applied to agriculturally relevant plants and animals using genome engineering methodologies are poised to revolutionize the nutrition content and the availability of food crops and livestock (Hsu et al. 2014; Ni et al. 2014; Cyranoski 2015). In addition, these techniques to disrupt genetic information or interrogate gene function offer advantages over others, such as RNAi, which may have substantial off-target effects and incomplete abrogation of mRNA (Shalem et al. 2015). Deletions of genomic regions are also useful for removing entire genes or portions of the genome using the concurrent action of two targeted nucleases flanking the region to be excised (Lee et al. 2010; Kim et al. 2013; Essletzbichler et al. 2014; Ousterout et al. 2015b).
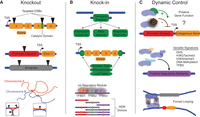
Manipulation of endogenous loci using genome engineering tools. (A) Genetic knockout of coding regions, protein catalytic domains, promoters, enhancers, or genomic contact points is possible using targeted nuclease platforms. (B) Genetic knock-in using appropriate donor repair templates can be applied to deliver various transgenic payloads, including epitope tags, regulatory components, or disrupted motifs such as mutant transcription factor binding sites (TFBSs) within cis regulatory modules, to decipher endogenous regulatory element activity. (C) Dynamic regulation of endogenous loci using nuclease-null genome engineering tools fused to effector domains can be used to interrogate gene function or the activity of putative regulatory elements enriched with varying epigenomic signatures. Additionally, these tools can be used to artificially direct physical interactions between distal endogenous loci. (DSB) double-strand break; (TSS) transcription start site; (HDR) homology-directed repair; (DHS) DNase hypersensitivity site.
The complete and precise deletion of a gene or genetic segment may obviate potential confounding factors of gene knockout with a single nuclease that might still lead to functionally active truncated or frame-shifted proteins (Shi et al. 2015). Disruption or deletion of promoter or enhancer regions could also be used to knockout or diminish gene function, and enhancer elements have been characterized and validated through such methods (Bauer et al. 2013; Li et al. 2014; Mansour et al. 2014; Zhou et al. 2014a). Deletion of boundaries between topologically associating domains can also reveal important mechanistic properties of genome structure (Nora et al. 2012; Dowen et al. 2014; Crane et al. 2015b; Lupiáñez et al. 2015). However, deletion of genomic sequences may have disadvantageous pleiotropic effects, such as unintended alterations in the native architecture of bystander regulatory elements.
Gene knock-ins
When appropriate donor DNA repair templates are provided, HDR can lead to defined integration events such as the inclusion of epitope tags, reporter genes, and regulatory units, such as LoxP sites, at endogenous loci (Fig. 2B; Hockemeyer et al. 2009, 2011; Doyon et al. 2011a; Yang et al. 2013a). These methodologies allow for epitope-based detection of proteins and interacting partners for which high quality or specific antibodies are not available, as well as tracking of cellular proteins in real time on a single-cell basis. In addition, conditional alleles are useful in instances in which genetic knockout results in embryonic lethality or when assessments of gene function at different developmental stages and lineages are desired. HDR-mediated introduction of natural genetic variation into an isogenic background can also be used to elucidate the in vivo contributions of specific regulatory elements and DNA-interacting proteins. For instance, the targeted alteration of specific transcription factor binding site motifs in otherwise intact loci could reveal the functional contribution of transcription factor binding to regulatory element activity. This approach could also be extended to dissect cis regulatory modules in which several transcription factor binding sites are putatively involved in regulatory specificity (Hardison and Taylor 2012; Hnisz et al. 2015).
Dynamic regulation of genomic activity and conformation
In addition to nuclease-mediated disruption or deletion, genes and associated regulatory regions can also be dynamically manipulated using targeted ZF, TALE, or dCas9-based transcription factors or epigenome editing tools (Fig. 2C). This approach is particularly useful for avoiding the stochastic and cell-type– and cell-cycle–dependent DNA repair pathways involved in nuclease-mediated genome editing. Such methods can elucidate potential endogenous gene function without exogenous overexpression or permanent sequence disruption. These tools are also especially applicable in targeting the multitude of putative regulatory regions that contain epigenetic hallmarks correlated with activity in certain settings, such as differentially active enhancers (Shlyueva et al. 2014; Farh et al. 2015; Heinz et al. 2015; Roadmap Epigenomics Consortium et al. 2015; Leung et al. 2015). For example, recent efforts have identified signatures, such as DNase hypersensitivity, H3K27ac, and H3K4 methylation that are associated with active enhancers and promoters. However, validation of the potential functionality of these elements is a major scientific bottleneck to our understanding of the epigenetics of gene regulation. Selective and targeted writing or erasure of appropriate epigenetic modifications can establish the causality of respective marks in determining gene expression and may also define their relevance in organismal development and in cellular responses to distinct stimuli. Furthermore, targeted activation or suppression of regulatory loci involved in lineage specification or reprogramming could have enormous biotechnological utility (Gao et al. 2013; Chakraborty et al. 2014; Ji et al. 2014; Chavez et al. 2015). Additionally, programmed looping to connect distal genomic regions can serve to define the function of specific genomic contacts (Deng et al. 2014). Ultimately, the most comprehensive and valuable definitions of regulatory element functionality will use several independent approaches, including sequence disruption and manipulation of activity across varied biological contexts.
Genome engineering to model disease and develop therapeutics
ZFs, TALEs, and the CRISPR/Cas9 system are also important tools for understanding and modeling disease. In Mendelian disorders, in which single gene products are implicated in disease or development, nuclease-aided disruption or deletion can be used to determine causal relationships between genes and phenotypes (Soldner et al. 2011; Toscano et al. 2013). In addition, specific SNPs or corrections can be introduced into coding regions to identify and validate variants associated with disease (Kiskinis et al. 2014; Wienert et al. 2015). SNPs in noncoding elements, such as enhancers, are also associated with many diseases; hence, similar modeling and validation methods outside of coding regions will also be extremely useful. Notably, genome engineering is unique in its ability to perturb regulatory elements in their endogenous genomic context in contrast to other tools, such as RNAi and small molecules that modulate mRNA and protein activity. Large-scale genomic models of complex diseases, based on genome-wide associations, could also be created with these strategies in order to discriminate germane versus immaterial genetic variance (Fig. 3A; Bauer et al. 2013). Some disease phenotypes are also the result of fusion proteins generated during aberrant genomic translocations (Bunting and Nussenzweig 2013). Programmable nucleases could also be used to insert deleterious fusion proteins at endogenous loci to quantify causal effects and develop drug targets or to recapitulate chromosomal translocations to mimic disease (Maddalo et al. 2014).
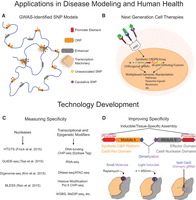
Outlook for genome engineering technologies. (A) Genome editing technologies can be used to incorporate genome-wide association study (GWAS)-identified single nucleotide polymorphisms (SNPs) into models of complex diseases and to ascertain casual disease variants. (B) Human cells can be programmed using genome engineering to produce the next generation of advanced cell therapies. (C) Techniques designed to assess potential off-target activity of programmable nucleases and transcriptional/epigenetic modifiers will facilitate technology improvements. (D) Increased control of genome engineering tools can be achieved with chemical and optogenetic regulation to develop highly articulated systems with increased specificity and dynamic properties.
Programmable DBPs can also be applied for genome-wide phenotypic screening. This is especially relevant for CRISPR/Cas9-based screens, owing to the relative ease of multiplexing by simply using Cas9 in tandem with libraries of gRNAs that can be synthesized at high throughput (Shalem et al. 2015). Cas9 nuclease-based knockout screens have been recently used in combination with both positive and negative selection strategies in mammalian cell lines. These techniques have revealed genes that are essential for certain cell states and sensitivity to toxins or drugs (Koike-Yusa et al. 2014; Shalem et al. 2014; Wang et al. 2014; Zhou et al. 2014b; Chen et al. 2015). In addition, nuclease-inactivated dCas9-based repressors and activators can be used in loss-of-function and gain-of-function screens, respectively (Gilbert et al. 2014; Konermann et al. 2015). Importantly, these screening strategies have not only validated previously implicated genes, but have also identified novel drivers of selected cellular phenotypes. The relative ease of construction of these screening platforms should make these technologies broadly useful to numerous research laboratories investigating multiple different pathologies and phenotypes.
The advent of modern genome engineering tools has also stimulated persistent and warranted optimism in the field of gene therapy. Knockout of pathological genes with Mendelian phenotypes is possible using ZFs, TALEs, and Cas9 nucleases. Similar approaches could also be used to correct deleterious copy number variation. The knockout of portions of genes or reading frame correction is also a useful strategy for pathologies in which truncated genetic variants provide amelioration, such as Duchenne muscular dystrophy (Li et al. 2015a; Ousterout et al. 2015a). Furthermore, as methods for HDR-mediated gene correction and integration continue to improve, the replacement of causative SNPs at coding regions and regulatory elements may become routine (Genovese et al. 2014; Crane et al. 2015a; Hoban et al. 2015). Similarly, targeted addition of exogenous genetic payloads may also become a valuable tool to correct loss-of-function, provide dosage compensation, or create novel cellular phenotypes (Li et al. 2011; Genovese et al. 2014). Engineered DBPs may also have utility as vaccination and immunotherapeutic agents. For instance, targeted disruption of viral entry molecules, such as CCR5 for HIV, have been shown to be efficacious as a prophylactic measure (Holt et al. 2010; Tebas et al. 2014). Viral loads have also been decreased using nucleases targeted to other viral genomes (Kennedy et al. 2014, 2015; Wang and Quake 2014; Liao et al. 2015a). In addition, genome engineering efforts to enhance human T cell immunotherapy have demonstrated substantial promise (Provasi et al. 2012; Torikai et al. 2013; Beane et al. 2015) and may pave the way toward customized patient-specific prophylactics and therapies.
The therapeutic use of ZFs, TALEs, and Cas9 as artificial transcriptional/epigenetic modifiers also has tremendous potential. The precise genetic repression of detrimental genes may be a useful strategy to mitigate pathology, such as repressing the huntingtin gene for Huntington's disease (Garriga-Canut et al. 2012). Similarly, targeted activation of aberrantly silenced endogenous loci may provide therapeutic benefits (Lara et al. 2012). Additionally, the manipulation of irregular epigenetic modifications or genomic contacts could be useful to prevent or correct disease states. Notably, in contrast to permanent nuclease-based sequence modification, synthetic transcriptional or epigenetic modifications are dynamic, tunable, and reversible. In certain circumstances, limited durations or transmission of artificial manipulation may be preferable. Furthermore, the versatility and multiplex capacity of these genome engineering tools combined with the recent advances in synthetic eukaryotic genetic circuit designs (Khalil et al. 2012; Slusarczyk et al. 2012; Esvelt et al. 2013; Farzadfard et al. 2013; Daringer et al. 2014; Kabadi et al. 2014; Kiani et al. 2014; Nielsen and Voigt 2014; Nissim et al. 2014; O'Connell et al. 2014; Li et al. 2015b; Zalatan et al. 2015) may lead to the next generation of cell therapies (Fig. 3B).
Future outlook
Genome engineering tools have been widely implemented for editing and understanding eukaryotic genomes. However, technological improvements are still needed to fulfill the potential of these technologies. Ideal genome engineering tools would have completely predictable effects, lack toxicity, be easily designed and constructed, and be easily deliverable with high efficiency in vitro and in vivo. Although ZFs, TALEs, and CRISPR/Cas9 platforms have propelled the field of genome engineering, they still suffer potential limitations in all of these areas. The comprehensive characterization and optimization of targeting specificities and in vivo delivery parameters of modern genome engineering tools are arguably the most pressing concerns (Kim and Kim 2014; Cox et al. 2015).
The successful application of genome engineering in diverse organisms demonstrates that the technologies are effective with limited negative side effects in vivo, However, the translation of these tools to clinical settings requires a greater resolution of any off-target effects than has previously been possible. Some examples of the possible off-target activity of the ZF, TALE, and Cas9-based platforms have been published (Perez et al. 2008; Fu et al. 2013; Hsu et al. 2013; Mali et al. 2013a; Pattanayak et al. 2013; Guilinger et al. 2014a; Kim and Kim 2014). An important recent advance has been the development of unbiased approaches for detecting off-target effects with a much greater level of sensitivity than previous methods (Fig. 3C; Gabriel et al. 2011; Frock et al. 2015; Kim et al. 2015; Ran et al. 2015; Tsai et al. 2015). The use of large sets of negative control gRNAs in high-throughput CRISPR/Cas9-based screening applications is also enabling reproducible and highly quantitative analyses of off-target gRNA activity (Gilbert et al. 2014; Sanjana et al. 2014). However, despite the importance of these advances, their detection limits are still restrained by the accuracy of current DNA sequencing technologies, and this will be a critical area for improvement moving forward.
The protein engineering of nuclease domains to improve activity and specificity will also continue to be a focus area, particularly as the methods for computational design and experimental selection of proteins with novel properties improve. For example, engineered “nickases” have been developed to stimulate HDR but minimize off-target indels generated by the creation of double-strand breaks by nucleases, but the nickase-mediated HDR frequencies are generally lower (Doyon et al. 2011b; Miller et al. 2011; Kim et al. 2012; Wang et al. 2012; Mali et al. 2013a; Ran et al. 2013; Guilinger et al. 2014b; Tsai et al. 2014; Wu et al. 2014b). Protein engineering may also be used to alter the specificity of sequence recognition requirements of DNA-binding proteins, such as the directed evolution of Cas9 variants to recognize alternate PAMs (Kleinstiver et al. 2015) or of TALEs to change the requirement of a 5′ thymine (Lamb et al. 2013). Continued efforts to generate effective guidelines for optimal gRNA design will also be extremely important for future Cas9-based technologies (Briner et al. 2014; Cho et al. 2014; Doench et al. 2014; Fu et al. 2014; Chari et al. 2015; Farboud and Meyer 2015; Singh et al. 2015; Xu et al. 2015). For example, the development of algorithms that incorporate biological phenomena into optimal gRNA design are an important area for future work (Singh et al. 2015). Finally, the continued development of targeted recombinases and transposases could eventually supplant nuclease-based genome editing if the challenges of efficiently and specifically targeting highly active versions of these proteins to new sites can be overcome.
The specificity of the genome engineering technologies is also pertinent for transcriptional and epigenetic modification using these tools. Although genome-wide measurements of gene expression and chromatin structure suggest low frequencies of off-target effects (Hilton et al. 2015; Polstein et al. 2015), a thorough and controlled assessment of unintended epigenetic modifications is still necessary for many of the newly described effector domains. The duration and heritability of these synthetically deposited modifications should also be the subject of future investigation (Kungulovski et al. 2015). Optimal concentrations and spatiotemporal control by chemical or optogenetic regulation will also aid in mitigating any inadvertent effects (Fig. 3D; Polstein and Gersbach 2012, 2015; Konermann et al. 2013; Davis et al. 2015; Nihongaki et al. 2015; Wright et al. 2015; Zetsche et al. 2015). In addition, the direct delivery of purified ZFs, TALEs, or preassembled Cas9/gRNA complexes can minimize off-target activity (Gaj et al. 2012; Chen et al. 2013b; Ru et al. 2013; Kim et al. 2014; Ramakrishna et al. 2014; Liu et al. 2015; Zuris et al. 2015), which may further safeguard against aberrant effects.
Despite these areas of potential optimization and improvement, genome engineering technologies are clearly ready to have a significant impact on genomics and medicine. Functional characterization of the epigenetic modifications associated with gene regulation is now possible using these tools. This will allow high resolution functional annotation and indexed categorization of genomic regulatory elements. The assignment of functional data to these regulatory regions will also benefit from analyses across lineages and cell states, thereby providing regulatory atlases across genomic space and organismal development. Ongoing improvements in delivery systems for primary cells and tissues will continue to facilitate this work (Kabadi et al. 2014; Ran et al. 2015; Zuris et al. 2015). Similarly, the manipulation of epigenetic states at specific loci will also allow determinations of causal effects of these marks and the role of epigenomic signatures at specific regulatory elements in disease and lineage specification.
The precise perturbation of genomic contact points may also be useful to determine the functionality of physical connections and to understand the reported stochasticity associated with these interactions (Kind et al. 2013; Nagano et al. 2013). Imaging of genomic loci using engineered DBPs is also possible (Chen et al. 2013a; Miyanari et al. 2013), and this could provide an independent approach to demonstrate colocalization of genomic regions in real time. In addition, directed localization of specified genomic regulatory elements to nuclear regions associated with repression, such as the nuclear lamina (Amendola and van Steensel 2014; Pombo and Dillon 2015), could aid in validating the functional relevance of nuclear subcompartmentalization. Therefore the challenge of mapping the complex and dynamic four-dimensional genome is positioned to be a major future research area enabled by these new genome engineering technologies.
The functional characterization of the expanding sets of putative regulatory regions is nontrivial but will be facilitated by modern genome engineering. Likewise, determination of the causal variants in complex non-Mendelian diseases could be accomplished using precise recapitulation of the variants in otherwise isogenic cell cultures and animal models. Developing custom genomic models will continue to enable drug development and models of potential drug resistance (Kasap et al. 2014; Smurnyy et al. 2014). This will facilitate rapid identification of drugs with therapeutic efficacy and realize the potential of personalized and precision medicine by connecting genomics, therapeutic targets, and disease phenotypes. Modern genome engineering platforms are now established as indispensable research tools for diverse areas of biotechnology, and promising areas for their direct and indirect application to improving human health are rapidly expanding. This new era of genomic understanding is likely to continue to create new possibilities for genome research for the foreseeable future.
Competing interest statement
I.B.H. and C.A.G. are inventors on patent applications related to genome engineering. C.A.G. is a scientific advisor to Editas Medicine, a company engaged in therapeutic development of genome engineering technologies.
Acknowledgments
We thank Gregory Crawford and Timothy Reddy for helpful discussions on the topics of this Perspective and Matthew Gemberling and Sandi Wong for critical reading of the manuscript and helpful comments. This work was supported by US National Institutes of Health (NIH) grants R01DA036865, U01HG007900, R21AR065956, P30AR066527, UH3TR000505, and an NIH Director's New Innovator Award (DP2OD008586), and a National Science Foundation (NSF) Faculty Early Career Development (CAREER) Award (CBET-1151035).
Footnotes
-
Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.190124.115.
-
Freely available online through the Genome Research Open Access option.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵








