
Abstract
About 2% of human genetic polymorphisms have been hypothesized to arise via multinucleotide mutations (MNMs), complex events that generate SNPs at multiple sites in a single generation. MNMs have the potential to accelerate the pace at which single genes evolve and to confound studies of demography and selection that assume all SNPs arise independently. In this paper, we examine clustered mutations that are segregating in a set of 1092 human genomes, demonstrating that the signature of MNM becomes enriched as large numbers of individuals are sampled. We estimate the percentage of linked SNP pairs that were generated by simultaneous mutation as a function of the distance between affected sites and show that MNMs exhibit a high percentage of transversions relative to transitions, findings that are reproducible in data from multiple sequencing platforms and cannot be attributed to sequencing error. Among tandem mutations that occur simultaneously at adjacent sites, we find an especially skewed distribution of ancestral and derived alleles, with GC → AA, GA → TT, and their reverse complements making up 27% of the total. These mutations have been previously shown to dominate the spectrum of the error-prone polymerase Pol ζ, suggesting that low-fidelity DNA replication by Pol ζ is at least partly responsible for the MNMs that are segregating in the human population. We develop statistical estimates of MNM prevalence that can be used to correct phylogenetic and population genetic inferences for the presence of complex mutations.
One of the core challenges in evolutionary biology is to explain the distribution of mutations in time and space and harness this knowledge to make inferences about the past. When two DNA sequences have numerous differences that are spaced closely together, they are inferred to have been diverging for a relatively long time, the two lineages accumulating mutations at a steady rate since they diverged from their last common ancestor. In contrast, when two sequences have few differences that are spaced far apart, they are inferred to have diverged from a common ancestor relatively recently. This logic is the basis of a widely used class of methods that infer detailed demographic histories from the spacing between SNPs in a sample of whole-genome sequence data (Hobolth et al. 2007; Li and Durbin 2011; Harris and Nielsen 2013; Sheehan et al. 2013).
To improve the accuracy of population genetic inference from the spacing between SNPs, it will be important to assess the validity of standard assumptions about the mutational process. One such assumption is that mutations occur independently conditional on the genealogical history of the data; however, there are numerous lines of evidence that 1%–5% of SNPs in diverse eukaryotic organisms are produced by multinucleotide mutation events (MNMs) that create two or more SNPs simultaneously. If simultaneously generated mutations are regarded as independent during population genetic analysis, the ages of the clustered variants will be overestimated. This could be important not only for the inference of demographic histories but also for other endeavors such as the detection of long-term balancing selection. Closely spaced SNPs with ancient times to common ancestry can provide evidence that genetic diversity has been maintained by natural selection (Charlesworth 2006; Ségurel et al. 2012; Leffler et al. 2013), and simultaneous mutations have the potential to distort or mimic these signals.
One line of evidence for MNM comes from de novo mutations that occur in populations of laboratory organisms including Drosophila melanogaster (Keightley et al. 2009; Schrider et al. 2013), Arabidopsis thaliana (Ossowski et al. 2010), Caenorhabditis elegans (Denver et al. 2004, 2009), and Saccharomyces cerevisiae (Lynch et al. 2008), as well as de novo mutations detected by looking at human parent-child-trios (Schrider et al. 2011). The human de novo mutation rate per base per generation is somewhere between 1.0 × 10−8 and 2.5 × 10−8 (The 1000 Genomes Project Consortium 2010); assuming that mutations occur independently, it should be exceedingly rare to find two mutations within 100 kb of each other. Contrary to this expectation, trios show consistent evidence of mutations occurring in pairs ranging from 2 bp to tens of kb apart.
In yeast, there is additional evidence that MNMs are created by the activity of DNA polymerase zeta (Pol ζ), an error-prone translesion polymerase that extends DNA synthesis past mismatches and damage-induced lesions (Sakamoto et al. 2007; Stone et al. 2012). Pol ζ is also responsible for MNMs that occur during somatic hypermutation of the variable regions of mouse immunoglobulins (Daly et al. 2012; Saribasak et al. 2012). These results were established by knocking out Pol ζ in mutant yeast strains and adult mouse cells, but it has not been possible to knock out Pol ζ in live mice without destroying their embryonic viability (Bemark et al. 2000; Esposito et al. 2000; Wittschieben et al. 2000). For this reason, there is no direct experimental evidence that Pol ζ creates heritable MNMs in higher eukaryotes.
Clusters of de novo mutations are not the only line of evidence for heritable MNMs in eukaryotes. Additional evidence can be found in patterns of linkage disequilibrium (LD) between older SNPs that segregate in natural populations. Schrider and colleagues and Terekhanova and colleagues examined pairs of nearby SNPs in phased human haplotype data and found that the two derived alleles occurred more frequently on the same haplotype than on different haplotypes (Schrider et al. 2011; Terekhanova et al. 2013). When two mutations occur independently, their derived alleles should occur on the same haplotype only 50% of the time; in contrast, MNM should always produce mutation pairs with the two derived alleles on the same haplotype. Using a different counting argument, Hodgkinson and Eyre-Walker also concluded that many SNP pairs occurring at adjacent sites were generated by a simultaneous mutational mechanism (Hodgkinson and Eyre-Walker 2010). They noted that adjacent linked SNPs outnumber SNPs 2 bp apart by a factor of two, when the two types of pairs should have equal frequency under the assumption of independent mutation.
To gather more data about the MNM process, it would be impractical to rely on de novo mutations and essential to harness LD information. Although it is easiest to classify a pair of SNPs as an MNM when the mutations are observed de novo, eukaryotes have low enough mutation rates that fewer than 1 MNM per genome is expected to occur each generation on average. Motivated by this, we use an LD-based approach to identify signatures of MNM in the 1000 Genomes Phase I data, a public repository of 1092 phased human genomes (The 1000 Genomes Project Consortium 2012). This repository is 100-fold larger than the data sets previously scrutinized for evidence of MNM, and its size confers new power to characterize the MNM spectrum.
In agreement with earlier studies of MNM, we find that patterns of LD between close-together SNPs are incompatible with mutational independence. However, the patterns are consistent with a simple mixture of independent mutations and MNMs. We leverage the size of the 1000 Genomes data set to make several novel discoveries about MNMs. First, they are enriched for transversions, with a transition:transversion ratio of 1:1 in contrast to the 2:1 genome-wide average. Second, we find that linked mutations in humans are enriched for the same allelic types recorded by Stone and colleagues in lines of yeast that have nucleotide excision repair (NER) deficiencies and thus rely heavily on Pol ζ for translesion synthesis (Stone et al. 2012). These frequent MNMs include the dinucleotide mutations GA → TT and GC → AA, as well as mutations at nonadjacent sites that produce homogeneous AA/TT derived allele pairs. Such patterns are unlikely to result from errors in the DNA sequencing process and instead suggest that normal human Pol ζ activity generates at least some of the same MNMs that are produced by Pol ζ in NER-deficient yeast (Stone et al. 2012).
Results
Simultaneous mutations can be observed directly when they occur de novo in offspring that have been sequenced along with their parents. In addition, many more MNMs can be inferred from linkage in data from unrelated individuals. Schrider and coworkers previously invoked simultaneous mutations to explain LD patterns in a phased diploid genome, observing that SNPs <10 bp apart were disproportionately likely to have their derived alleles lie on the same haplotype (Schrider et al. 2011). In the spirit of this approach, we looked at the prevalence of neighboring SNPs in the 1000 Genomes Phase I data that occur in perfect LD, meaning that the two derived alleles occur in the exact same subset of the 2184 sequenced haplotypes. We hereafter define a pair of close LD SNPs to be a pair occurring <100 bp apart in perfect LD. A few MNMs will be missed because of recombination between the mutated sites, but we estimate that fewer than 0.5% of all MNMs spanning <100 bp will be disrupted in this way (see Section S4 of the Supplemental Material).
Excess nearby SNPs in LD
We counted 35,620 pairs of close LD SNPs in the 1000 Genomes Phase I data with both sites passing genotype quality control and with a consistent ancestral state identifiable from a human/chimp/orang/macaque reference alignment (see Methods). Simultaneous mutations should always create SNPs in perfect LD, but we also expect some independent mutations to create SNPs in perfect LD, and we quantified this expectation by simulating data under a Poisson process model of independent mutation and recombination implemented in Hudson’s coalescent simulator ms (Hudson 2002). We simulated a total of 4.8 × 108 bp from an alignment of 2184 haplotypes under a realistic human demographic model (Harris and Nielsen 2013) and recovered 36,991 close LD SNP pairs. For comparison, we also simulated 1.8 × 108 bp of data under the standard neutral coalescent with constant effective population size N = 10,000, recovering 36,202 close LD SNP pairs.
As shown in Figure 1, the distribution of distances between close LD SNPs was quite different in the simulated versus real data, with the real data containing about fivefold more adjacent SNPs in LD and a decaying excess of SNPs separated by up to 20 bp in LD. In contrast, the simulations under different demographic models produced similar distributions of close LD SNPs.
Nearby SNPs in LD: 1000 Genomes Phase I data vs. simulation under mutational independence. When we simulated 2184 haplotypes under a realistic demographic model, we observed ∼37,000 SNP pairs in LD separated by <100 bp in a sample of total length 4.8 × 108 bp. Their spacing was distributed almost uniformly between 1 and 100 bp. We observed much less uniformity in the distribution of distances between SNP pairs in LD in the 1000 Genomes data, with an extreme excess of SNPs in LD at 1–2 bp and a less extreme excess of SNPs at distances up to 20 bp apart. (Note that the axes are logarithmically scaled, making exponential curves appear concave downward.)
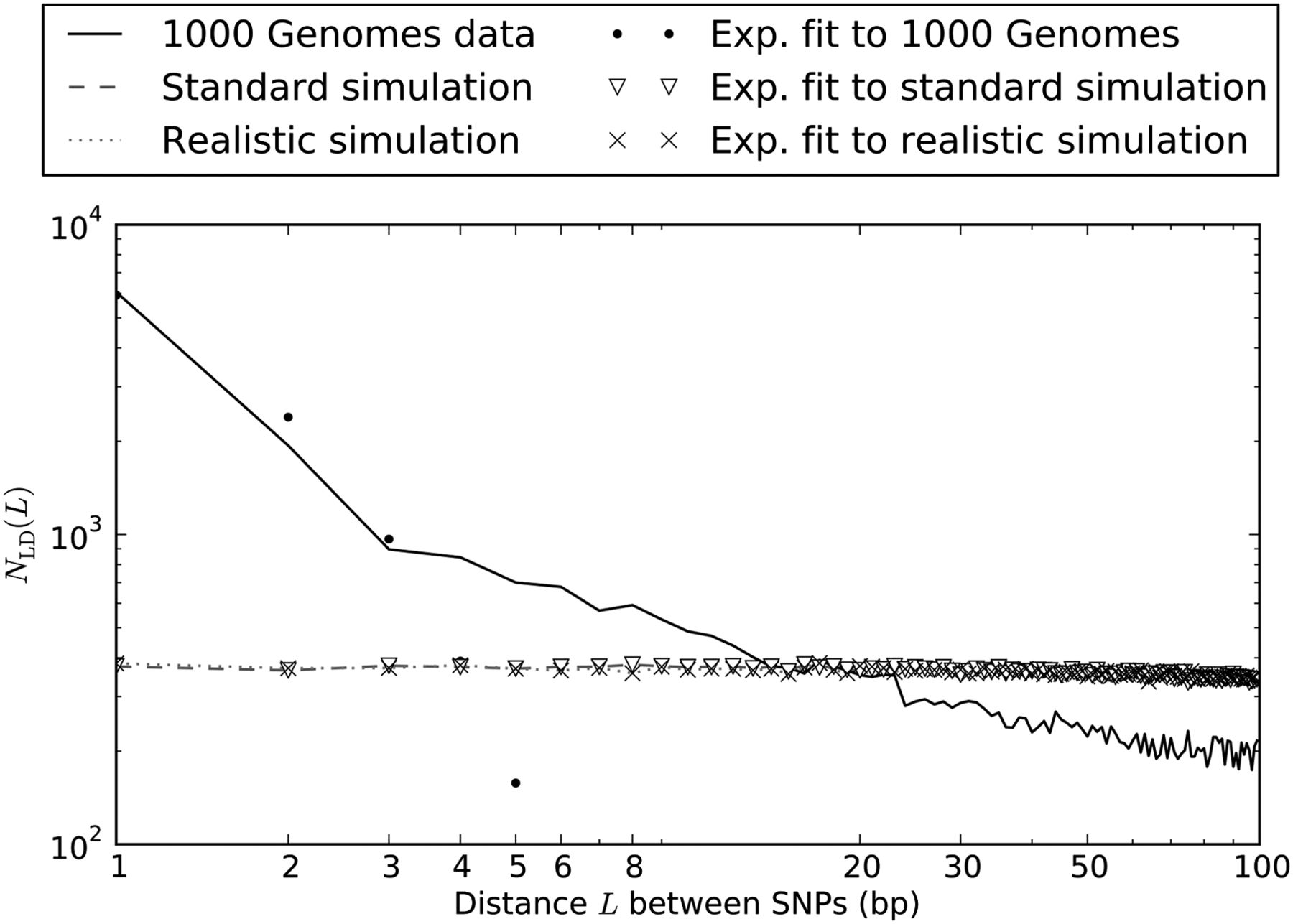
Under the coalescent with independent mutation, the abundance of SNP pairs L bp apart in LD should decline approximately exponentially with L for small values of L (see Supplemental Material Section S1), and we find this to hold for the simulated data in Figure 1. In contrast, the optimal least-squares exponential fit is a poor approximation to the abundance distribution of close LD SNP pairs in the 1000 Genomes data, which we denote by NLD(L). A possible explanation is that close LD SNPs are produced by a mixture of two processes, a point-mutation process that is accurately modeled by the coalescent and an MNM process that is not.
Close LD SNPs have unusual transition/transversion frequencies
To our knowledge, no previous work has addressed whether MNMs have the same transition:transversion ratio as ordinary mutations. However, there is abundant evidence that different DNA polymerases produce mutations with different frequencies of ancestral and derived alleles. To investigate this question, we measured the fractions of linked SNP pairs at distance L that are composed of transitions, transversions, and mixed pairs (one transition plus one transversion). We denote these fractions 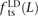 ,
, 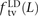 , and
, and 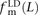 . We also measured the analogous fractions
. We also measured the analogous fractions 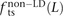 ,
, 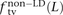 , and
, and 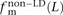 of transitions, transversions, and mixed pairs among SNPs not found in perfect LD.
of transitions, transversions, and mixed pairs among SNPs not found in perfect LD.
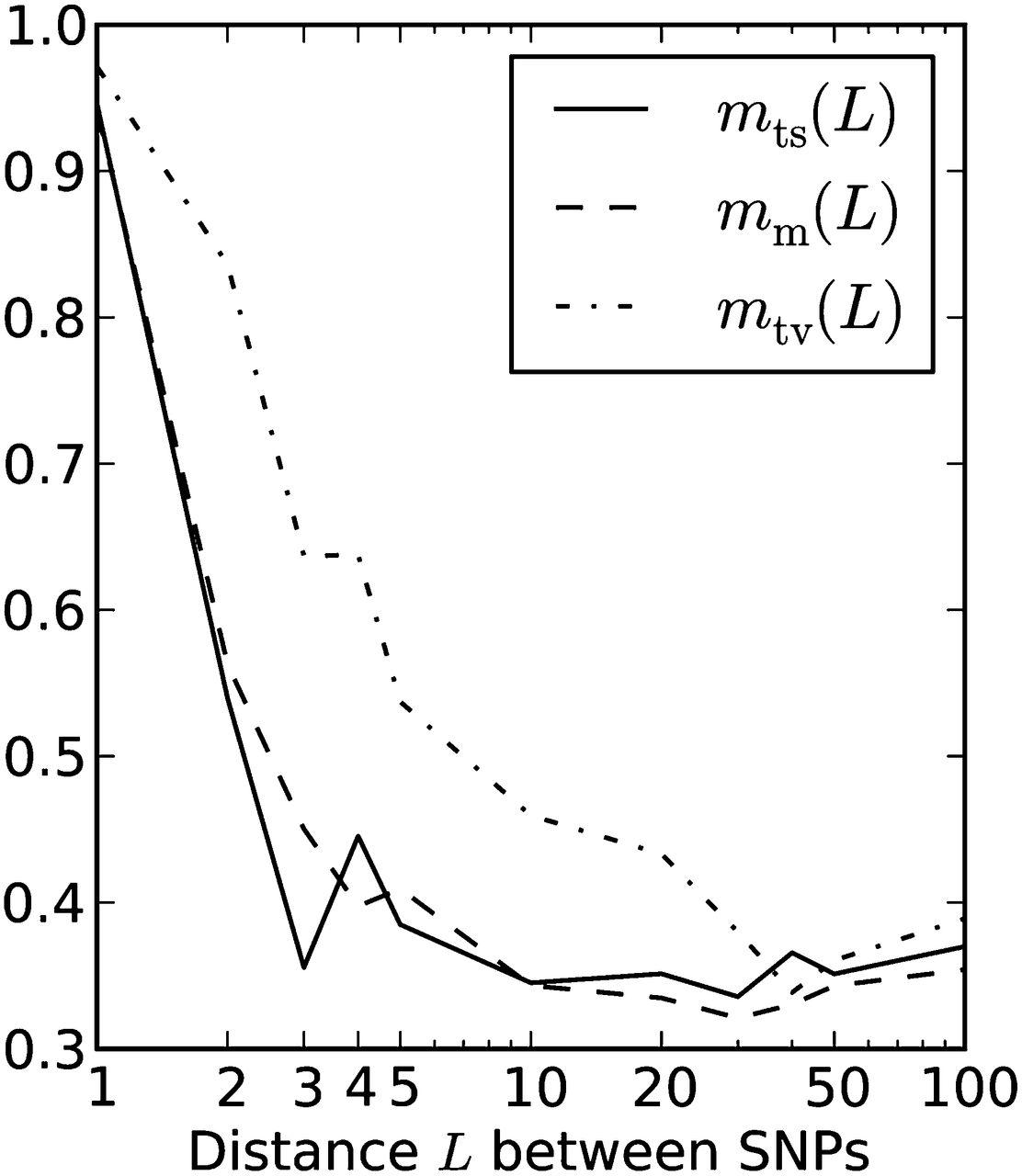
In human genetic variation data, transitions are approximately twice as common as transversions (Kimura 1980). If the two mutation types of a SNP pair were chosen independently, we would therefore expect that fts = 0.662 = 0.44, ftv = 0.332 = 0.11, and fm = 2 × 0.66 × 0.33 = 0.45. These predictions are very close to 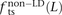 ,
, 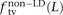 , and
, and 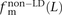 for L between 2 and 100 (Fig. 2). For L = 1,
for L between 2 and 100 (Fig. 2). For L = 1, 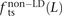 is larger than expected because of the elevated transition rate at both positions of CpG sites.
is larger than expected because of the elevated transition rate at both positions of CpG sites.
The relationship between LD and the transition:transversion ratio. In this figure, the solid black line plots the fraction of SNP pairs in LD that consist of two transitions. The fraction increases quickly as a function of the distance L between SNPs, asymptotically approaching the fraction of SNP pairs not in LD that consist of two transitions (solid gray line). The fraction of SNP pairs not in LD that consist of two transitions is nearly constant as a function of L except for an excess of adjacent transition pairs resulting from double mutation at CpG sites. Although transversion pairs make up just over 10% of unlinked SNP pairs, they account for >40% of adjacent SNPs in perfect LD and ∼20% of SNPs in LD at a distance of 10 bp apart.
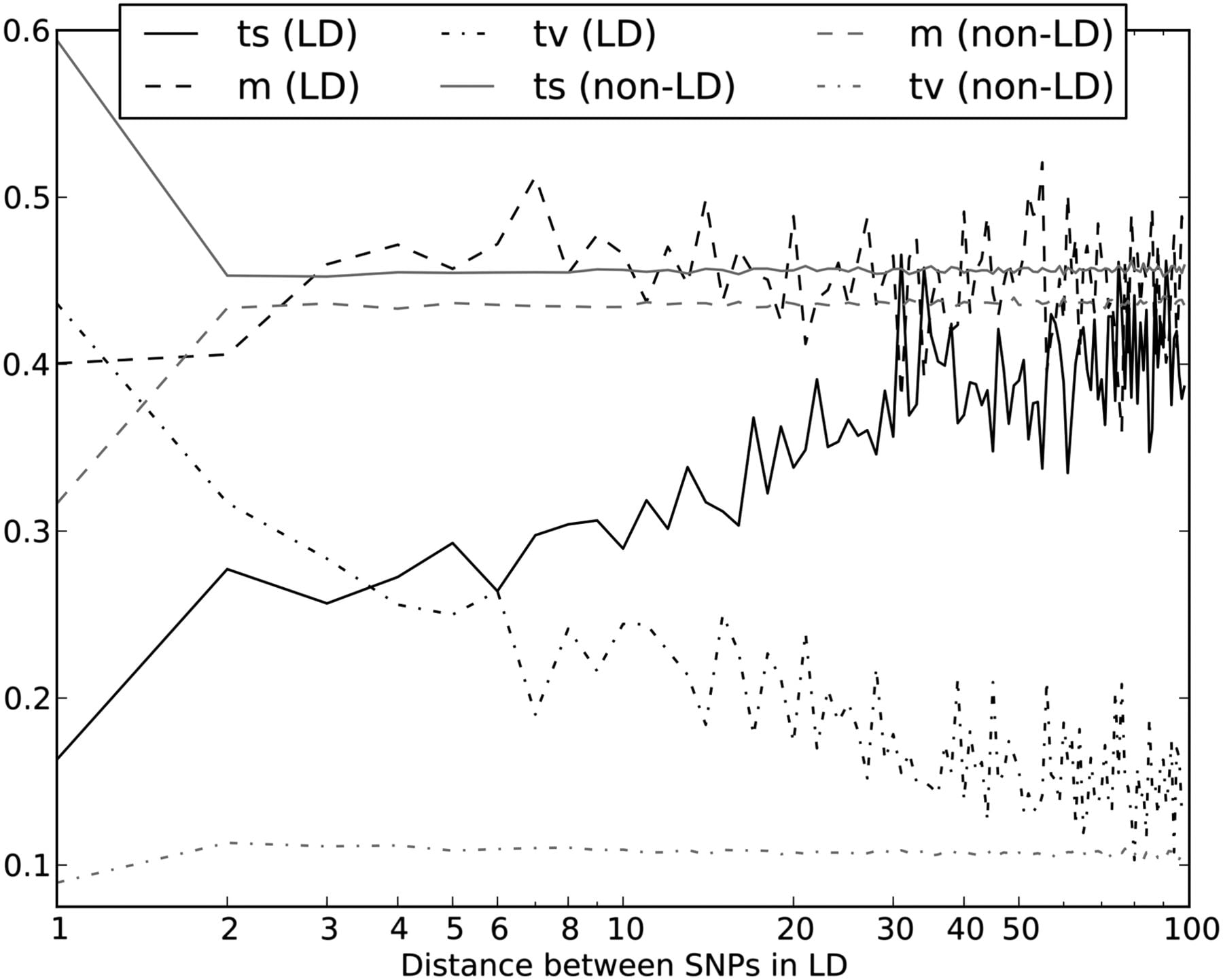
Among mutations in perfect LD, we found that 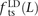 ,
, 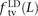 , and
, and 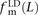 deviate dramatically from the expectation of mutational independence, adding support to the idea that many such SNPs are produced by a nonstandard mutational process. The frequency of transversion pairs declines with L; we found that 36.7% of SNP pairs in LD at adjacent sites consisted of two transversions, compared to 11.1% of SNP pairs in LD at a distance of 100 bp and 10.7% of SNP pairs not in LD. These numbers are not just incompatible with a transition:transversion ratio of 2:1 but are also incompatible with two neighboring SNP types being assigned independently. If the SNP types were assigned independently, it should hold that
deviate dramatically from the expectation of mutational independence, adding support to the idea that many such SNPs are produced by a nonstandard mutational process. The frequency of transversion pairs declines with L; we found that 36.7% of SNP pairs in LD at adjacent sites consisted of two transversions, compared to 11.1% of SNP pairs in LD at a distance of 100 bp and 10.7% of SNP pairs not in LD. These numbers are not just incompatible with a transition:transversion ratio of 2:1 but are also incompatible with two neighboring SNP types being assigned independently. If the SNP types were assigned independently, it should hold that 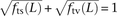 , an assumption that is violated for small values of L. We also found excess close LD transversions in human data sequenced by Complete Genomics (Supplemental Fig. S1), suggesting that this pattern is not an artifact of the Illumina sequencing platform or the 1000 Genomes SNP-calling pipeline.
, an assumption that is violated for small values of L. We also found excess close LD transversions in human data sequenced by Complete Genomics (Supplemental Fig. S1), suggesting that this pattern is not an artifact of the Illumina sequencing platform or the 1000 Genomes SNP-calling pipeline.
Estimating the fraction of perfect LD SNPs that are MNMs
Schrider et al. (2011) previously estimated the abundance of MNMs using the following analysis of a phased diploid genome: For distances L ranging from 1 to 20 bp, they counted heterozygous sites L bp apart where the derived alleles lay on the same haplotype and could potentially have arisen due to MNM. They compared this quantity, S(L), to the number D(L) of heterozygotes L bp apart with the derived alleles on different haplotypes. If all mutations arise independently, S(L) and D(L) are expected to be equal, leading them to propose S(L) − D(L) as an estimate of the number of MNMs spanning L bp. We repeated this analysis on the 1000 Genomes data, subsampling each possible pair H from among the 2184 phased haplotypes. For each L between 1 and 100 bp, we obtained counts 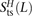 ,
, 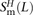 , and
, and 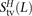 of transitions, mixed pairs, and transversions L bp apart where one haplotype carried the two ancestral alleles and the other haplotype carried the two derived alleles. Similarly, we obtained counts
of transitions, mixed pairs, and transversions L bp apart where one haplotype carried the two ancestral alleles and the other haplotype carried the two derived alleles. Similarly, we obtained counts 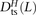 ,
, 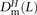 , and
, and 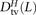 where the derived alleles occurred on opposite haplotypes of H. Adding up these counts over all haplotype pairs subsampled from the 1000 Genomes data, we obtained global counts St(L) and Dt(L) for each pair type t. The quantity (Stv(L) − Dtv(L))/(S(L) − D(L)), a direct estimate of the fraction of MNMs that are transversion pairs, is consistently slightly higher than
where the derived alleles occurred on opposite haplotypes of H. Adding up these counts over all haplotype pairs subsampled from the 1000 Genomes data, we obtained global counts St(L) and Dt(L) for each pair type t. The quantity (Stv(L) − Dtv(L))/(S(L) − D(L)), a direct estimate of the fraction of MNMs that are transversion pairs, is consistently slightly higher than 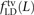 (Supplemental Fig. S2), as expected if close LD SNP pairs are a mixture of MNMs and linked independent mutations.
(Supplemental Fig. S2), as expected if close LD SNP pairs are a mixture of MNMs and linked independent mutations.
We were able to use Sts(L) − Dts(L), Sm(L) − Dm(L), and Stv(L) − Dtv(L) to estimate the abundance of MNMs relative to perfect LD SNPs. Our simulations indicate that fewer than 0.5% of MNMs 100 bp apart should be ultimately broken up by recombination (Supplemental Table S2); guided by this, we assume that MNMs are a subset of perfect LD SNP pairs. To make this assumption robust to phasing and genotyping error, we relax the definition of perfect LD to include site pairs where, at most, 2% of samples carry a discordant genotype (see Methods). For each linked SNP pair, we count the number of subsampled haplotype pairs for which exactly one lineage contains the two derived alleles. Adding up these counts over all perfect LD SNPs, we obtain a count S(LD)(L) that is strictly less than S(L). We estimate that m(L) = (S(L) − D(L))/S(LD)(L) is the fraction of perfect LD SNP pairs created by MNM. Similarly, 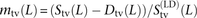 is the fraction of perfect LD transversions created by MNM. The results indicate that >90% of SNPs in perfect LD at adjacent sites are MNMs (Fig. 3). At a distance of 5 bp between sites, 60% of perfect LD transversions are predicted to be MNMs, in contrast to 40% of perfect LD transitions and mixed pairs. At 100 bp between sites, ∼35% of perfect LD pairs appear to be MNMs, a figure that is similar across transitions and transversions. We calculate that MNMs spanning 1–100 bp account for 1.8% of new point mutations (see Methods). Section S5 (Supplemental Material) describes how to simulate data containing 1.8% MNMs with realistic spacings of 1–100 bp.
is the fraction of perfect LD transversions created by MNM. The results indicate that >90% of SNPs in perfect LD at adjacent sites are MNMs (Fig. 3). At a distance of 5 bp between sites, 60% of perfect LD transversions are predicted to be MNMs, in contrast to 40% of perfect LD transitions and mixed pairs. At 100 bp between sites, ∼35% of perfect LD pairs appear to be MNMs, a figure that is similar across transitions and transversions. We calculate that MNMs spanning 1–100 bp account for 1.8% of new point mutations (see Methods). Section S5 (Supplemental Material) describes how to simulate data containing 1.8% MNMs with realistic spacings of 1–100 bp.
The fraction of SNPs in perfect LD caused by MNM. The dash-dotted curve plots our estimate of the fraction of transversions in perfect LD at distance L that were caused by simultaneous mutation. It is uniformly higher than our corresponding estimates for mixed pairs and transitions, plotted with solid and dashed lines.
By construction, m(L) should accurately estimate the fraction of MNMs among the close LD SNPs that are polymorphic in a single diploid genome. This might be different from the absolute fraction of 1000 Genomes close LD SNPs that are MNMs, because these contain a higher proportion of rare alleles. However, m(L) is the more relevant statistic to the prevalence of MNMs in smaller data sets that many readers will be concerned with.
The 1000 Genomes data contains many SNP pairs that lie in perfect LD at distances of >100 bp apart. Although their transition/transversion ratios are close to the genome-wide average, values of m(L) suggest that >25% of these are MNMs (Supplemental Table S1). Although MNMs spanning 10,000 bp appear to be rare events, 10-fold rarer than MNMs spanning only 100 bp, they appear only about fourfold rarer than independent mutations occurring in perfect LD at 10,000 bp, making it possible to infer their distribution in the genome with high precision.
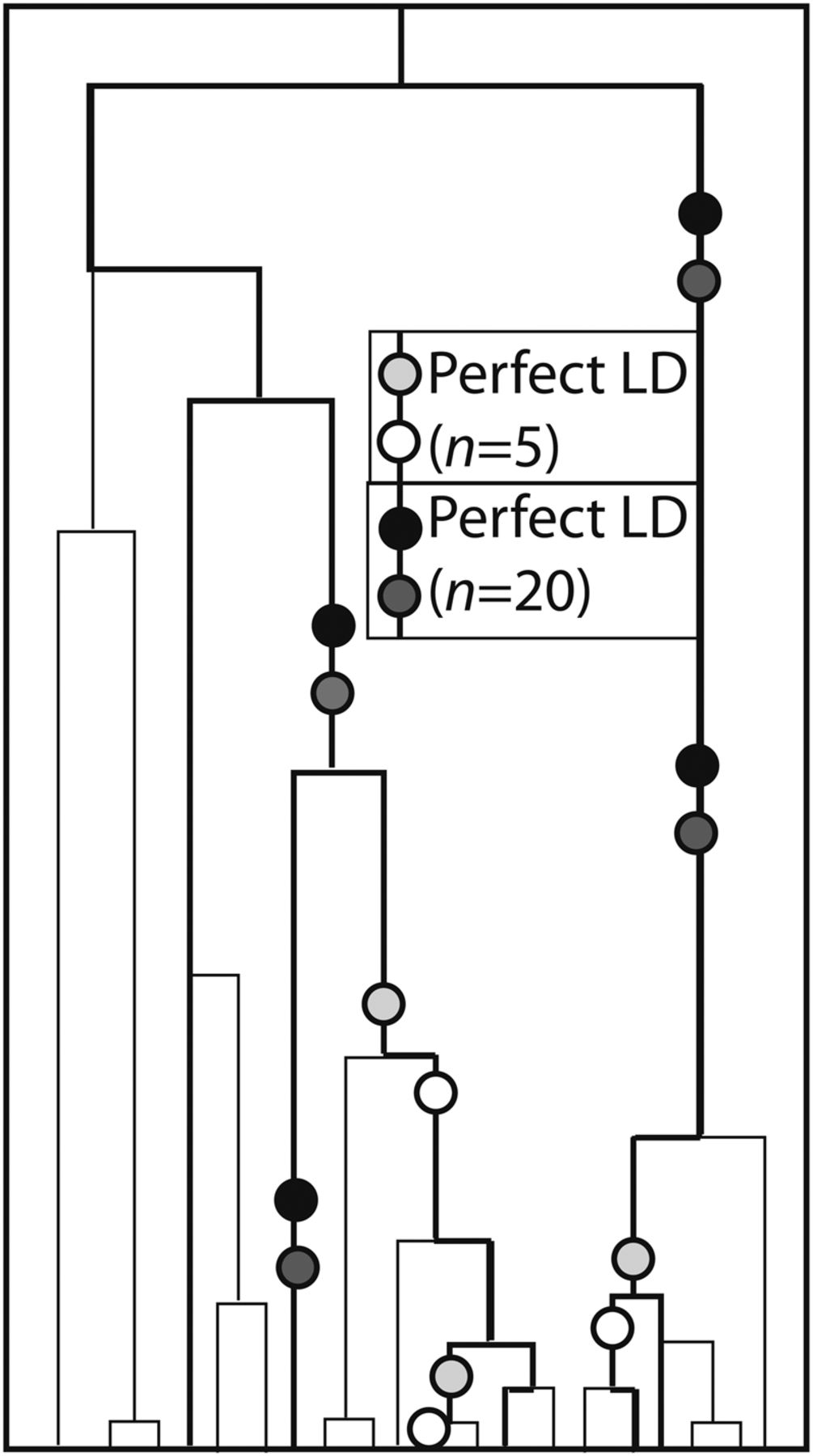
The large sample size of the 1000 Genomes data not only ensures that a huge number of rare mutations can be observed, but also ensures that independent mutations occur in perfect LD much less often than in samples of fewer individuals. The reason for this is illustrated in Figure 4: If two mutations occurred at different time points on the genealogical tree of an entire population, sampling more individuals increases the probability of sampling one who carries the older mutation and not the younger one. To test this prediction, we counted SNP pairs that appear to be in perfect LD in smaller subsets of the 1000 Genomes data. As proved in Section S2 of the Supplemental Material, the genealogies of large samples are dominated by shorter branches, on average, than the genealogies of smaller samples, implying that the percentage of perfected LD SNPs caused by MNM should be an increasing function of the number of sampled lineages. This implies that the abundance of transversions relative to transitions should also increase with the number of sampled lineages.
Independent mutations in perfect LD. This figure depicts a 20-lineage coalescent tree with a five-lineage subsample highlighted in bold. Light circles represent mutation pairs that appear in perfect LD only in the five-lineage sample. In contrast, dark circles represent pairs of independent mutations that occur in perfect LD in the entire 20-lineage sample. These pairs are concentrated on the longest branches of the tree that are often ancestral to many lineages, making their site frequency spectrum enriched for high frequencies.
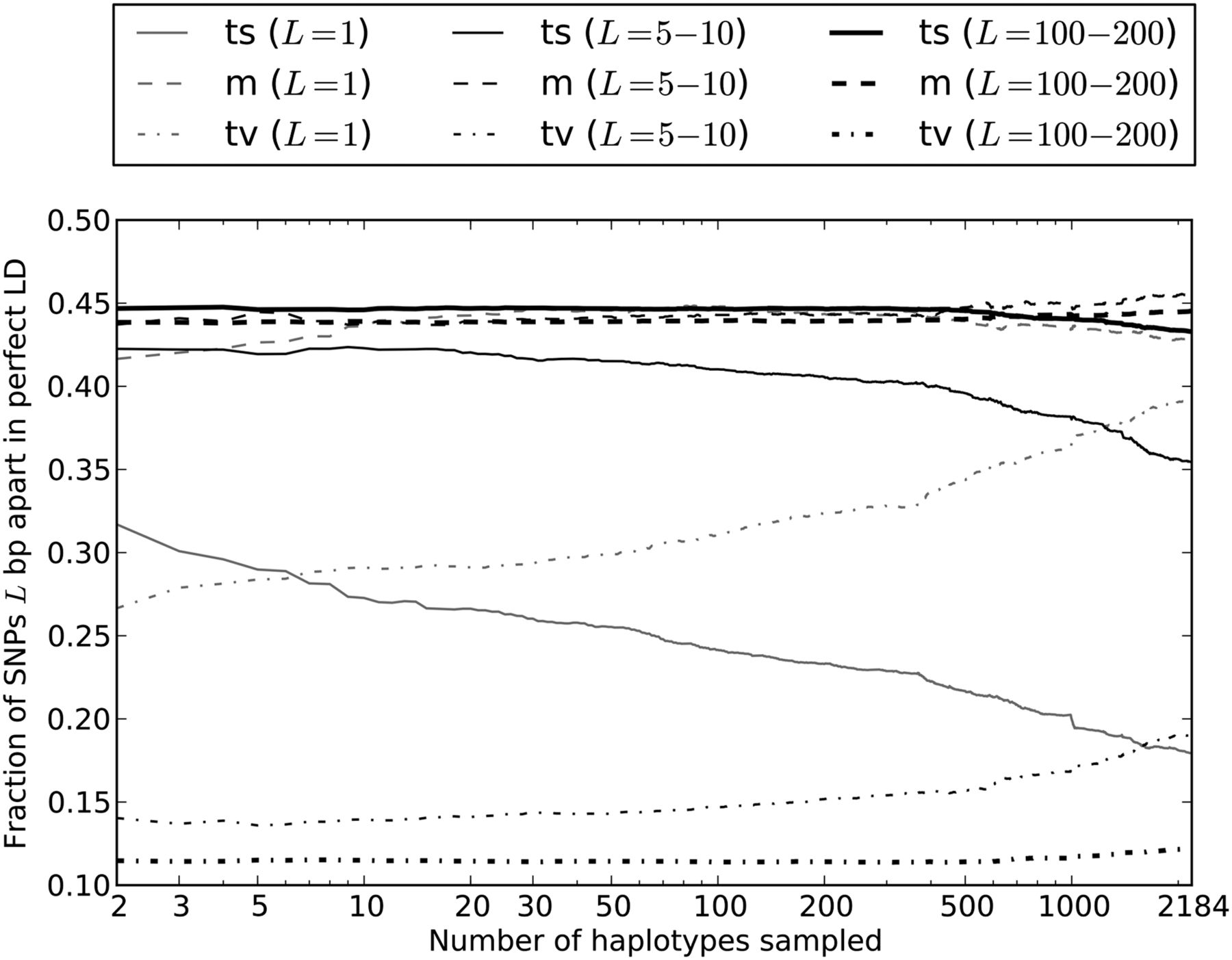
In pairs of adjacent perfect LD SNPs, we find that the percentage of transversion pairs increases very quickly with the number of lineages, making up 27% of the total when only two haplotypes are sampled and nearly 40% of the total when all 2184 haplotypes are sampled (Fig. 5). This result is even more dramatic for transitions at CpG sites, where the rate of nonsimultaneous double mutations is elevated by deamination of methylated cytosine to thymine. When we count the fraction of adjacent transitions in perfect LD that are of the type CG → TA as a function of the number n of lineages sampled, it declines nearly 10-fold as n increases from 2 to 2184 (see Supplemental Fig. S4). For perfect LD SNPs that occur 100–200 bp apart, the percentage of transversion pairs increases much more slowly than for adjacent perfect LD SNPs. However, it is still 10% higher in a sample of 2184 haplotypes than in samples of 2 to 1000 haplotypes (Fig. 5).
Enrichment of transversion pairs and MNMs with increasing sample size. We generated subsamples of the 1000 Genomes data containing 2–2184 haplotypes and computed the percentages of transversion pairs, transition pairs, and mixed pairs for perfect LD SNPs in each data set. As the number of sampled haplotypes increases, the percentage of perfect LD SNPs that are MNMs should increase, leading to an increase in the frequency of transversions and a decrease in the frequency of transitions. This effect is most apparent when the SNPs are adjacent (1 bp apart) or very close (5–10 bp apart). However, perfect LD SNPs that lie 100–200 bp apart display the same pattern, indicating that MNMs spanning 100–200 bp are much less common but are still evident in samples of many lineages.
Clustering of simultaneous mutations
Mutation-accumulation experiments have reported MNMs spanning long genomic distances (Denver et al. 2009; Keightley et al. 2009; Schrider et al. 2011, 2013), and yeast studies have suggested a possible mechanism for their formation. Roberts and colleagues reported that double-strand breakage and subsequent repair can create sparse clusters of mutations spanning a megabase or more, with a mean spacing of 3000 bp between simultaneous mutation events (Roberts et al. 2012). We found evidence for higher-order mutational clustering by counting groups of mutations in perfect LD with fewer than 1000 bp between each adjacent pair and plotting the distribution of cluster size, which ranged from 2 to 31 SNPs. The distribution had a fatter tail than the distribution of perfect LD clusters in an equivalent amount of simulated data, where the largest cluster contained 23 perfect LD SNPs (Supplemental Fig. S3).
The effect of complex mutation on the site frequency spectrum
In addition to showing that large samples contain fewer linked independent mutations than smaller samples, Figure 4 illustrates that linked independent mutations should be enriched for high frequencies relative to the site frequency spectrum (SFS) of ordinary mutations. High-frequency mutations tend to occur on the longest branches of a genealogical tree, whereas low-frequency mutations are scattered across many short branches that are each less likely to be hit with two separate mutations. Simulations confirm that linked independent mutations are biased toward high frequencies, with sixfold fewer singletons and doubletons than the SFS of the data set they come from (Fig. 6F). In contrast, MNMs should have the same SFS as ordinary point mutations as long as they are not affected differently by natural selection.
Site frequency spectra of perfect LD mutations. Each of panels A through E contains site frequency spectra of transitions, mixed pairs, and transversions found in perfect LD in the 1000 Genomes data. Singletons are excluded because they cannot be phased and therefore perfect LD status cannot be determined. For comparison, each panel contains the population-wide SFS of unlinked SNPs as well as the inferred SFS of linked independent mutations. SNP pairs are binned according to the distance between them, showing that close-together SNPs and transversions have spectra closer to the population SFS, while far-apart SNPs and transitions appear more weighted toward linked independent mutations. Dotted lines show the frequency spectra predicted by Equation 1 for each length and pair type category, assuming that the gray dashed line [m (theory)] depicts the correct SFS of linked independent mutations and that Figure 7 shows the correct MNM percentages in each category. For comparison, panel F shows a population SFS and perfect LD frequency spectrum obtained from data simulated under a human demographic model. In the simulated data, there is no difference between the frequency spectra of linked independent mutations that lie 1 bp apart versus 100 bp apart.
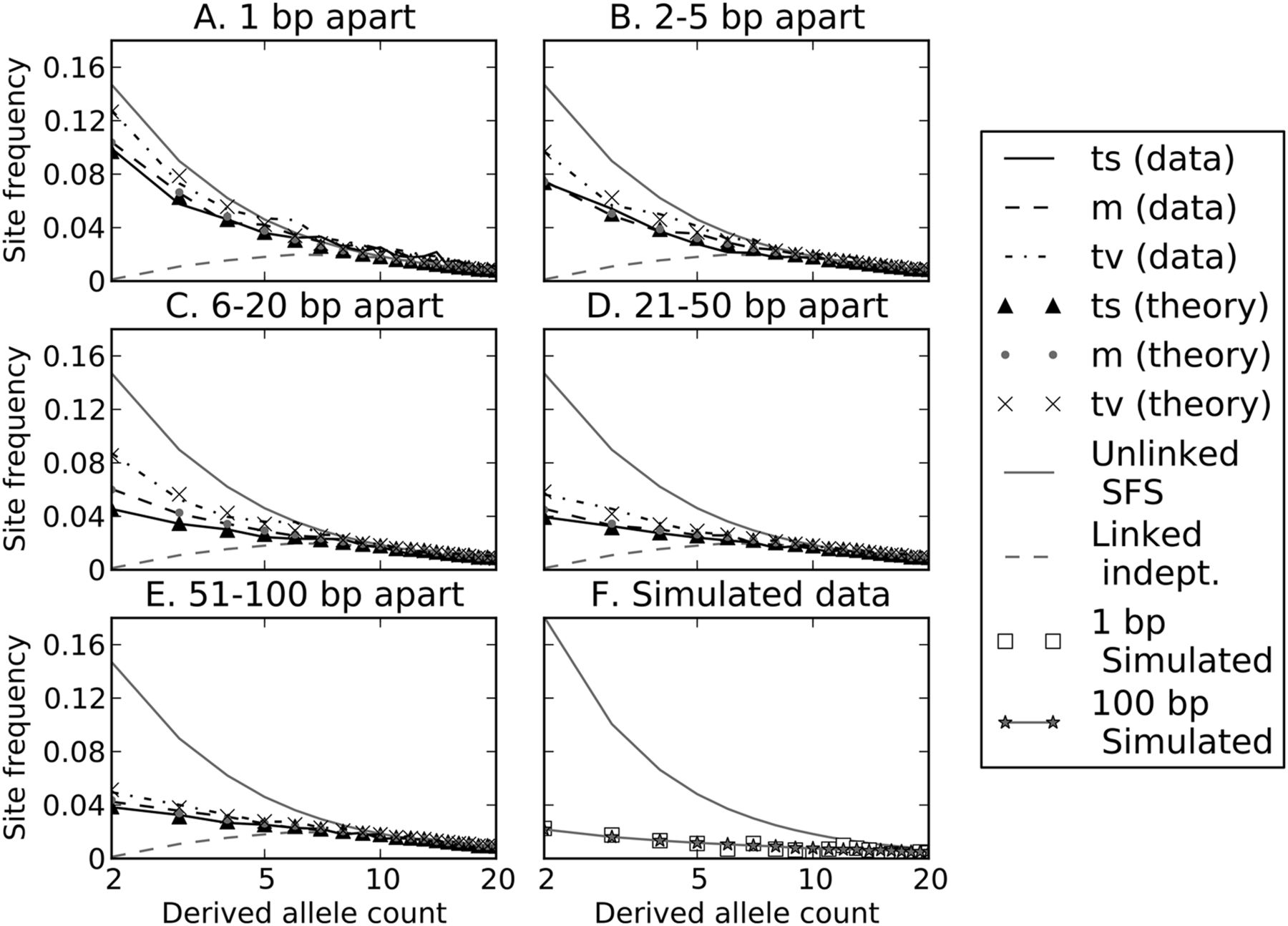
Given a mixture of simultaneous and independent mutations, the SFS should be a linear combination of the site frequency spectra of independent and simultaneous linked mutations. The more heavily the mixture is weighted toward independent mutations, the more the SFS should be skewed toward high frequencies. In agreement with our inference that MNMs contain a high percentage of transversions, we observe that perfect LD transversions have lower frequencies on average than other perfect LD SNP pairs. In addition, far-apart perfect LD SNPs have higher frequencies than close-together pairs on average (Fig. 6).
Using the empirical spectra of linked versus unlinked mutations, we devised a second method for estimating the fraction of perfect LD SNPs that are MNMs. For each mutation pair type (ts/m/tv), we compute the site frequency spectrum S(L) of perfect LD SNPs L bp apart. We also computed a SFS Sglobal from the entire set of SNPs in the sample. It is not possible to measure the spectrum Sindept−LD of linked independent mutations directly, so we numerically optimized the entries of this spectrum jointly with mixture model coefficients cts(L), cm(L), and ctv(L) between 0 and 1, one for each distance L and mutation pair type t. We treated all entries of Sindept−LD as unknown free parameters and used the BFGS algorithm to minimize the following squared error residual D:
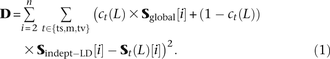
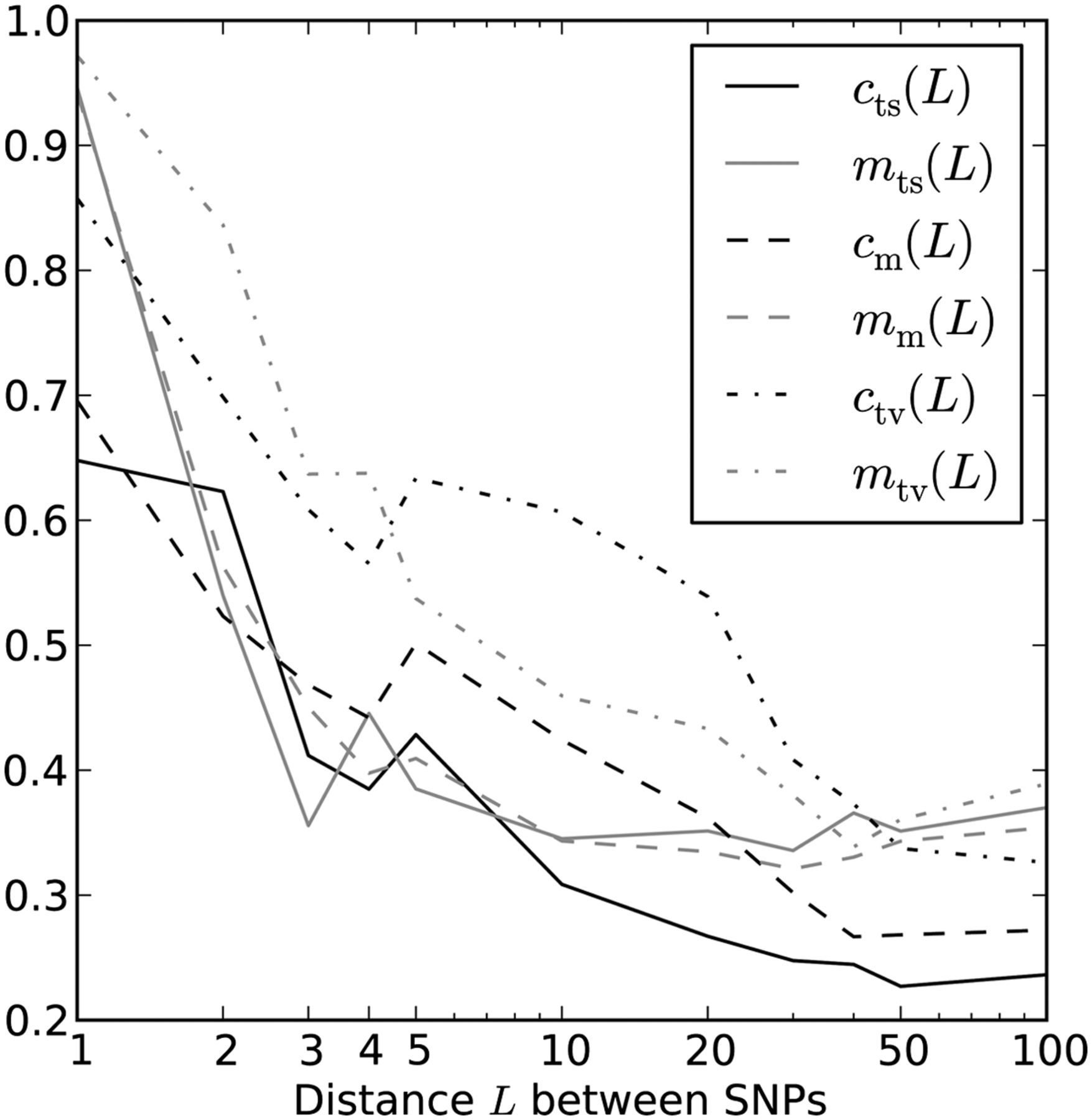
Two estimates of MNM prevalence. Here, the black lines plot ct(L), our SFS-based estimate of the fraction of perfect LD mutations caused by MNM. For comparison, gray lines plot the estimate mt(L) that is based on the excess of same-lineage derived alleles over different-lineage derived alleles in subsampled haplotype pairs.
Evidence for error-prone synthesis by Polymerase ζ
One mechanism that is known to generate MNMs in vivo is error-prone lesion bypass by Polymerase ζ, an enzyme found in all eukaryotes with the unique ability to extend primers with terminal mismatches (Gan et al. 2008; Waters et al. 2009). At a replication fork that has been stalled by a lesion, Pol ζ is responsible for adding bases to the strand containing the lesion and then extending replication for a few base pairs before detaching and allowing a high-fidelity enzyme to resume synthesis. During this extension phase, it has the potential to create clustered errors. Experimental work in yeast has confirmed that Pol ζ generates MNMs (Sakamoto et al. 2007; Stone et al. 2012), and the same enzyme has been linked to somatic hypermutation in the MHC (Daly et al. 2012; Saribasak et al. 2012).
Translesion synthesis by Pol ζ is not the only pathway that has the potential to create MNMs. Eukaryotes utilize at least seven different DNA replication enzymes that are considered “error-prone” (Goodman 2002; Waters et al. 2009) and have mutation spectra with low transition/transversion ratios (McDonald et al. 2011). However, we specifically analyzed human MNMs for signatures of Pol ζ activity because a unique data set was available to make this possible. Specifically, we were able to compare linked adjacent mutations in the 1000 Genomes data to tandem (adjacent) mutations recorded from a yeast strain bred by Stone and colleagues to be deficient in nucleotide excision-repair machinery and rely heavily on Pol ζ to bypass lesions that stall replication forks (Stone et al. 2012). Stone and coworkers recorded a total of 61 spontaneous tandem mutations; these were even more heavily weighted toward transversions than linked SNPs in the 1000 Genomes data, with 52.5% transversion pairs, 37.7% mixed pairs, and only 9.8% transition pairs.
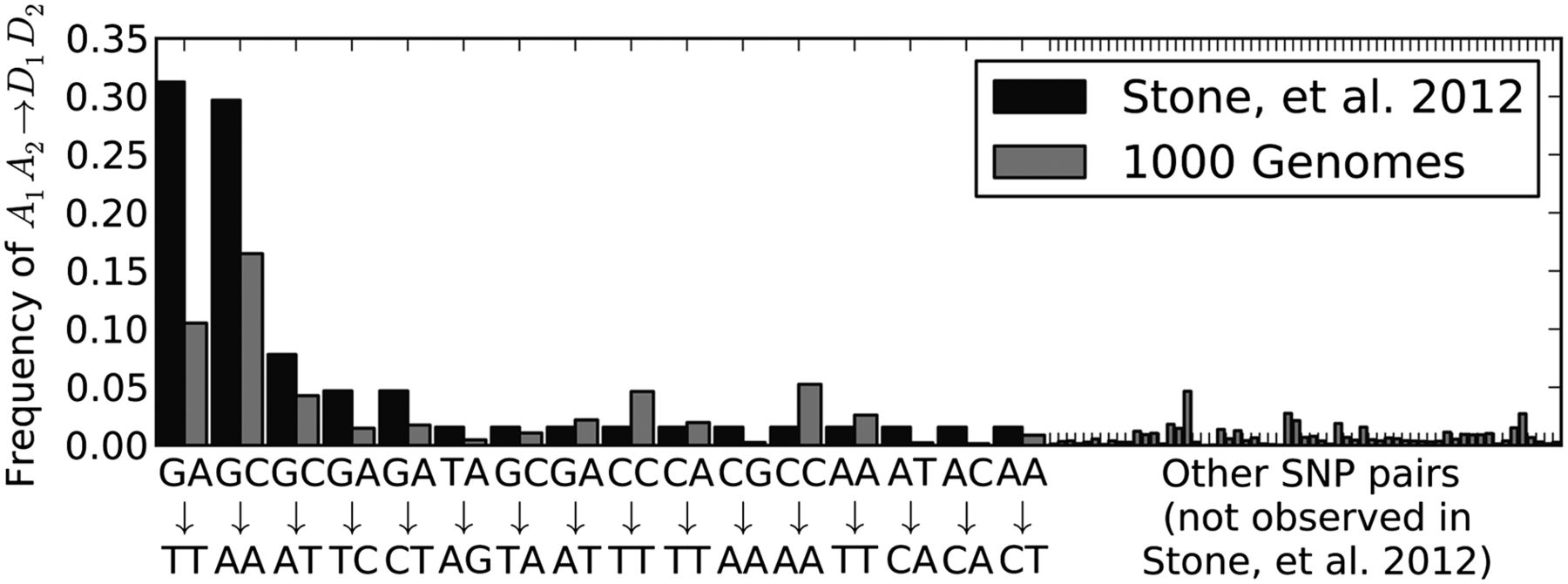
Two particular tandem mutations comprised >60% of the tandem mutations in the Stone et al. (2012) yeast. One of them, GA → TT, is a transversion pair that made up 31% of the total. The other, GC → AA, is a mixed pair that made up 30% of the total. We found that these were also by far the most common adjacent linked SNPs in the 1000 Genomes data, with GC → AA comprising 16% of the total and GA → TT comprising 11%. No other single mutation type accounts for >5% of the linked adjacent mutations in the 1000 Genomes data, and no other type accounts for >7% of the Stone et al. (2012) tandem mutations (Fig. 8).
Tandem mutations caused by Pol ζ. Black bars plot the frequencies of specific tandem mutations observed by Stone and coworkers in yeast deficient in nucleotide-excision repair machinery. Each mutation type is pooled with its reverse complement because there is no way to know on which DNA strand a mutation occurred. The two mutations GC → AA and GA → TT account for >60% of all tandem mutations observed by Stone and coworkers (Stone et al. 2012). As shown in gray, these are also the two most common types of mutations occurring at adjacent sites of the 1000 Genomes data in perfect LD.
In addition to 61 tandem mutations affecting adjacent base pairs, Stone and colleagues recorded 210 complex mutations where two or more substitutions, insertions, and/or deletions occurred at nonadjacent sites within a single 20-bp window (Stone et al. 2012). From this data set, we extracted 84 pairs of simultaneous substitutions at distances of 2–14 bp apart. These pairs had almost the same transition/transversion makeup as the tandem substitutions, being comprised of 53.6% transversions, 36.9% mixed pairs, and 9.5% transitions.
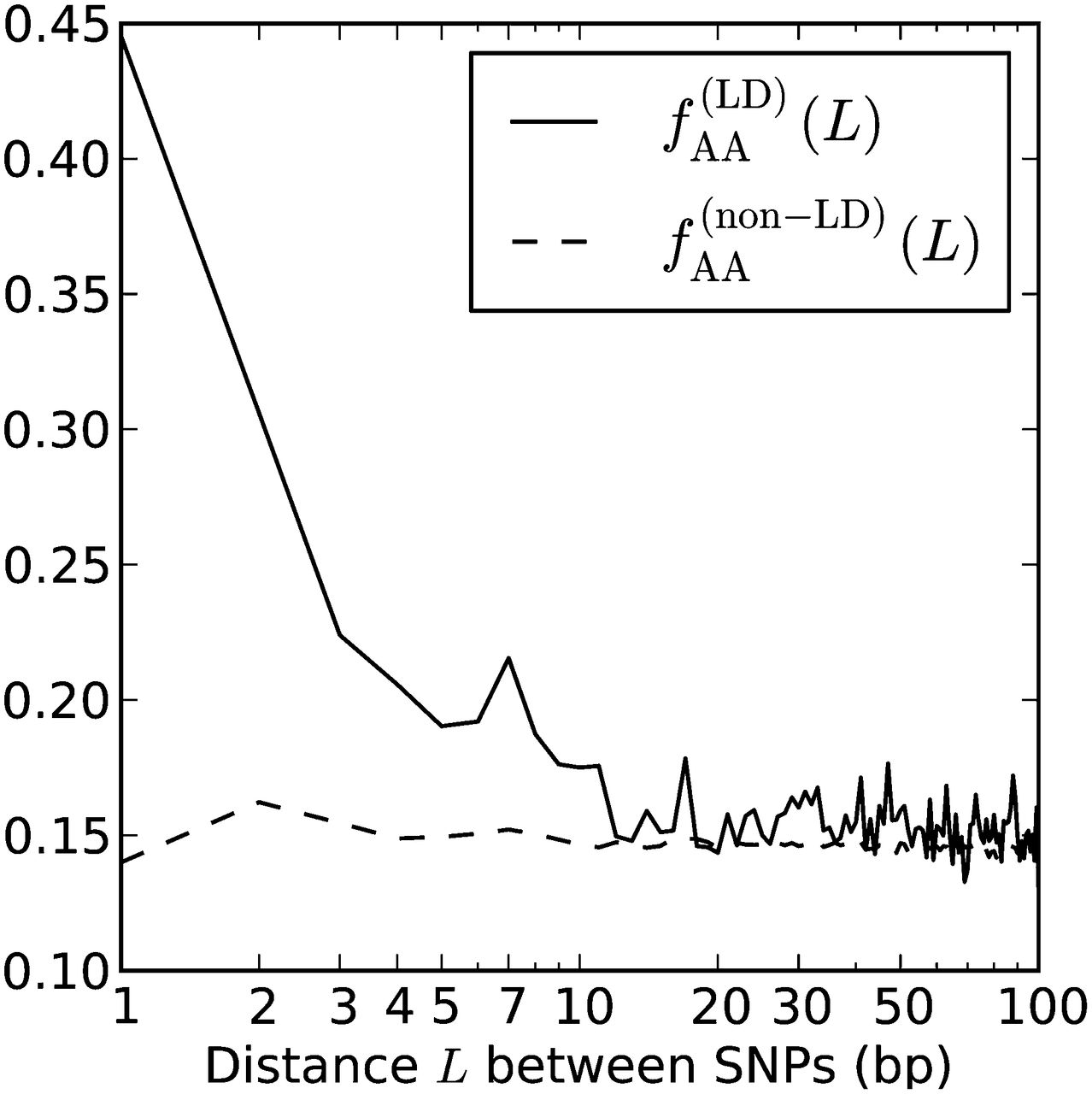
Among the nonadjacent yeast mutation pairs, GA → TT and GC → AA were not particularly common, making up only 4.8% and 1.2% of the total, respectively. However, 44.0% of the derived allele pairs were “AA” or “TT” (compared to 72.1% of adjacent mutation pairs). This percentage is much higher than what we would expect in two mutations that occurred independently. Mutation accumulation studies have shown that 33% of yeast mutations have derived allele A (by A/T symmetry, 33% also have derived allele T) (Lynch et al. 2008). From this, we expect the fraction of AA/TT derived allele pairs to be only 2 × 0.332 = 0.22. We found that AA and TT were similarly overrepresented among the derived allele pairs in linked human SNPs. In Figure 9, we plot the fraction fAA(L) of derived AA/TT allele pairs as a function of the distance L between perfect LD SNPs, charting its decline from fAA(1) = 0.445 through fAA(100) = 0.144.
Linked derived AA/TT allele pairs in the 1000 Genomes data. After observing that a high fraction of yeast MNMs had homogeneous AA/TT derived allele pairs, we tabulated the frequencies 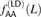 of AA/TT derived allele pairs among perfect LD SNPs L bp apart in the 1000 Genomes data. For comparison, we also plot
of AA/TT derived allele pairs among perfect LD SNPs L bp apart in the 1000 Genomes data. For comparison, we also plot 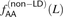 , the frequency of AA/TT derived allele pairs among SNPs not in perfect LD. This fraction is consistently lower than
, the frequency of AA/TT derived allele pairs among SNPs not in perfect LD. This fraction is consistently lower than 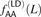 and does not decrease with the distance between SNPs.
and does not decrease with the distance between SNPs.
In their 2011 study of MNMs in human trios, Schrider et al. (2011) tabulated frequencies of all possible 144 dinucleotide substitutions but did not report excess AA/TT derived allele pairs or Pol ζ-associated mutations of the types GA → TT or GC → AA. We believe that these results differ because of our theoretical result that sampling more lineages enriches the ratio of true MNMs to linked independent mutations. To verify this, we replicated the Schrider et al. (2011) mutation frequency analysis on the 1000 Genomes data and obtained results that were similar to theirs (Section S3 of the Supplemental Material). We also found that the excess of Pol ζ-associated mutations increases as more lineages are sampled (Supplemental Fig. S5), just as all perfect LD transversions increase in frequency with sample size (Fig. 5). Other AA/TT derived allele pairs increase in frequency as more lineages are sampled when considering SNPs <4 bp apart. Since this effect is not discernible for L > 4, derived AA/TT pairs might only play a significant role in closely spaced MNMs.
Correcting downstream analyses for multinucleotide mutation
As evidenced by Figures 1 and 6, MNMs can have considerable impact on summary statistics like the site frequency spectrum and the prevalence of linkage disequilibrium. These summary statistics provide clues about the genealogical histories of data sets and can be leveraged to infer demographic history, natural selection, population structure, recombination rates, and other quantities of interest. However, accurate inference depends on accurately modeling the process that generates data, and most population genetic models omit MNMs.
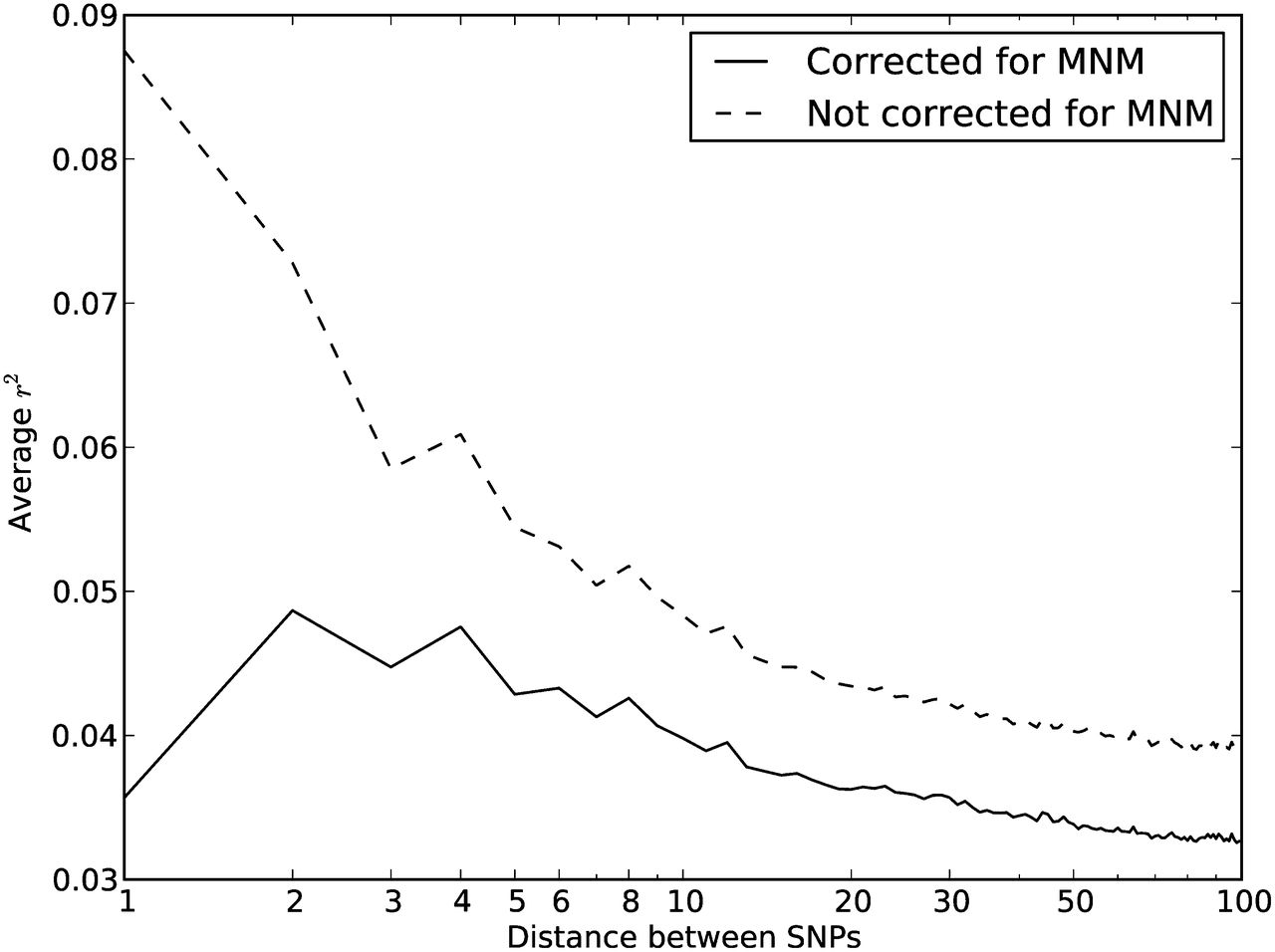
One strategy for improving the accuracy of downstream analyses without adding much to their complexity is to identify MNMs in a probabilistic way and remove them from the data. For each pair of SNPs occurring in perfect LD, we can estimate the probability that they were caused by an MNM as a function of their inter-SNP distance and transition/transversion status, then use this information to correct summary statistics for the presence of MNMs. To illustrate, we devise a method for correcting the correlation coefficient r2(L) that is commonly used to measure linkage disequilibrium as a function of genomic distance L (Hill and Robertson 1968). We computed r2(L) in the 1000 Genomes data as described in the Methods and then devised a corrected statistic 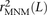 that accounts for MNMs and estimates the average correlation between independent mutations. As shown in Figure 10,
that accounts for MNMs and estimates the average correlation between independent mutations. As shown in Figure 10, 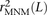 is significantly less than r2(L) at short genomic distances.
is significantly less than r2(L) at short genomic distances.
Average r2 LD correlations between 1000 Genomes SNP pairs. The correlation coefficient r2 between allele frequencies at neighboring sites is often used to measure the decay rate of genealogical correlation with genomic distance. However, we have seen that multinucleotide mutation creates excess LD compared to the expectation under independent mutation. We computed the average r2 across all SNP pairs L bp apart on chromosome 22, then corrected this value for the presence of MNM. 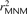 is lower at a distance of 1 bp than a distance of 2 bp because of double deaminations at CpG sites that occur on separate lineages.
is lower at a distance of 1 bp than a distance of 2 bp because of double deaminations at CpG sites that occur on separate lineages.
Discussion
We have uncovered a strong signature of multinucleotide mutation in 1092 genomes sequenced by the 1000 Genomes Consortium, with a large excess of close LD SNPs that cannot be explained by demography or mutational hotspots. This is consistent with earlier reports of MNM in smaller human data sets; however, MNMs are enriched relative to independent linked SNPs as more lineages are sampled and mutations are localized to increasingly short genealogical branches.
By looking at the allelic composition of close LD SNPs containing MNMs, we found several signatures that are consistent with error-prone lesion bypass by Polymerase ζ. One signature is an excess of transversions; the second is an excess of the dinucleotide mutations GA → TT and GC → AA, and the third is a bias toward homogeneous AA/TT derived allele pairs. It remains an open question what percentage of human MNMs is introduced by Pol ζ and how many other DNA damage and repair mechanisms come into play. However, it is interesting that Pol ζ appears to create the same mutation types in the human lineage that it creates in yeast with artificial excision repair deficiencies. We are hopeful that MNM can be understood more completely in the future by comparing perfect LD SNPs to de novo mutations from other sources.
An important alternative hypothesis for the observed patterns is DNA sequencing or assembly errors in the 1000 Genomes data, but there are several different lines of evidence that show that our results cannot be explained by such errors. First, we observed similar patterns in data sequenced by Complete Genomics using non-Illumina technology. Second, the excess close LD SNPs that are enriched for transversions and AA/TT derived alleles are not only singleton mutations but occur at a range of higher allele frequencies. Errors could only cause such patterns if they occurred in an identical fashion in multiple individuals, mimicking the frequency distribution expected for mutations. Third, as already noted, the MNMs we infer are enriched for the same types as MNMs that were observed de novo in yeast. The patterns we observe are consistent with MNM patterns that have been previously found using Sanger sequencing and other high-fidelity variant detection methods (Drake et al. 2005; Levy et al. 2007; Lynch et al. 2008; Chen et al. 2009).
The most commonly used methods for analyzing DNA sequences assume that mutations occur independently of each other. The fact that this assumption is violated in human data, and perhaps most other eukaryotic data, may compromise the accuracy of population genetic inference. Methods based solely on counting mutations, such as SFS-based methods (Gutenkunst et al. 2009), will probably be minimally affected and mostly in their measures of statistical confidence. In contrast, methods that explicitly use the spatial distributions of mutations, in particular the number of mutations in short fragments of DNA (Yang and Rannala 1997; Nielsen and Wakeley 2001; Wang and Hey 2010; Gronau et al. 2011), should be strongly affected. Several recently developed methods analyze genomic data by explicitly modeling the spatial distribution of independent mutations (Hobolth et al. 2007; Li and Durbin 2011; Harris and Nielsen 2013; Sheehan et al. 2013), and these are at risk for bias in regions where SNPs are close together. However, confounding of these methods by MNMs can be minimized by analyzing only a few individuals at a time and by disregarding pairs of SNPs <100 bp apart, which is often coincidentally done for the sake of computational efficiency (Li and Durbin 2011; Harris and Nielsen 2013). MNMs likely have a stronger effect on methods that look at data from many individuals across short, allegedly nonrecombining genomic fragments that are only 1 kb long and contain many SNPs fewer than 100 bp apart (Yang and Rannala 1997; Gronau et al. 2011). However, our results can be used to devise bias-correction strategies because, as illustrated in Figure 3, it is straightforward to estimate the probability that a given pair of linked SNPs is an MNM. This also has the potential to improve the accuracy of phylogenetic tree branch length estimation and molecular-clock-based inferences, as well as dN/dS estimation, and their associated measures of statistical confidence. Our results are also relevant to the interpretation of evidence that genetic variation is being maintained by balancing selection—such evidence typically involves short loci with closely spaced linked SNPs (Charlesworth 2006; Ségurel et al. 2012; Leffler et al. 2013).
A topic worth further investigation is the possibility of local variation in the rate of MNMs. If most MNMs are caused by error-prone polymerase activity, it is likely that high error-prone polymerase traffic should elevate rates of MNMs and simple point mutations in the same genomic regions. Both MNMs and point mutations in these regions might be subject to elevated transversion rates, and it will be important to separate the two classes of mutations to accurately study local variation of the transition/transversion ratio as in Seplyarskiy et al. (2012). Seplyarskiy and coworkers reported that the transition/transversion ratio κ appears depressed in the neighborhood of all human SNPs, even transitions, but we found that the apparent depression of κ in the neighborhood of transitions disappears when SNPs in perfect LD are excluded from the analysis (Supplemental Fig. S7).
MNMs have the potential to accelerate evolution by quickly changing several amino acids within a single gene (Schrider et al. 2011). Our results indicate that they also have the potential to increase both sequence homogeneity and A/T content. There is evidence that repetitive sequences experience more indels and point mutations than sequences of higher complexity (McDonald et al. 2011), possibly due to the recruitment of error-prone polymerases, giving MNM extra potential to speed up local sequence evolution by triggering downstream mutations. We are hopeful that more details about this process can be elucidated by studying the spatial and allelic distribution of MNMs. In this way, population sequencing data could provide new information about the biochemistry of DNA replication, e.g., providing a way to measure the activity of Pol ζ over evolutionary time. Pol ζ is tightly regulated in embryonic and adult cells because over- and underexpression can each be harmful; excess error-prone DNA replication increases the genomic mutation rate, but impaired translesion synthesis ability can lead to replication fork stalling, DNA breakage, and translocations that are more harmful than point mutations (Waters and Walker 2006; Waters et al. 2009; Northam et al. 2010; Ogawara et al. 2010; Lange et al. 2011). An important avenue for future work will be to assess whether different eukaryotes incur different levels of MNM because of changing evolutionary pressures being exerted on error-prone DNA replication activity throughout the tree of life.
Methods
Data summary and accession
We performed all of our analyses on SNP calls that were generated by the 1000 Genomes Project Consortium using joint genotype calling on 2×–6× whole genome coverage of 1092 humans sampled worldwide (The 1000 Genomes Project Consortium 2012). All sequences were mapped to the human reference hg19. To determine ancestral alleles, we downloaded alignments of hg19 to the primate genomes panTro2 (chimpanzee), ponAbe2 (orangutan), and rheMac3 (rhesus macaque) from the UCSC Genome Browser.
Ascertainment of SNP pairs from the 1000 Genomes Phase I data
Let S(L) be a count of SNPs that are polymorphic in a pair of haplotypes and lie L bp apart with their derived alleles on the same haplotype. Similarly, let D(L) be a count of SNPs with derived alleles that lie on opposite haplotypes. To measure S(L) and D(L) precisely from the 1000 Genomes data, we used a stringent procedure for ancestral identification, utilizing only sites that had the same allele present in chimp, orangutan, and rhesus macaque. For each pair p of SNPs L bp apart satisfying this criterion and passing the four-gamete test (to avoid confounding effects of recombination and sequencing error), we counted the number of haplotypes NAA(p) carrying the ancestral allele at both sites, the number NAD(p) carrying the ancestral allele at only the first site, the number NDA(p) carrying the ancestral allele at only the second site, and the number NDD(p) with both derived alleles. Singletons are excluded because they cannot be phased. Combining this information across the set P(L) of SNP pairs L bp apart, we obtain counts
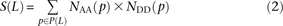
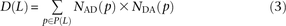
The quantity S(L) − D(L) has been used as an estimate of the number of MNMs lying L bp apart. Since two simultaneous mutations should always lie in perfect LD, S(L) − D(L) should, in theory, always be smaller than the following count of perfect-LD same lineage pairs:
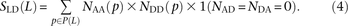
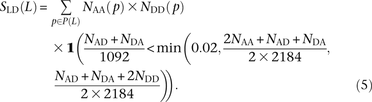
We use a slightly different procedure to obtain the counts 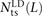 ,
, 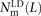 , and
, and 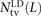 that do not need to be compared to S(L) − D(L). After dividing P(L) into transition pairs, mixed pairs, and transversion pairs to obtain sets Pts(L), Pm(L), and Ptv(L), we simply count the number of pairs with derived alleles that occur in the exact same set of lineages:
that do not need to be compared to S(L) − D(L). After dividing P(L) into transition pairs, mixed pairs, and transversion pairs to obtain sets Pts(L), Pm(L), and Ptv(L), we simply count the number of pairs with derived alleles that occur in the exact same set of lineages:
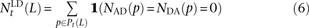
Simulating SNP pairs in LD under the coalescent
The simulated data used to generate Figure 1 were produced using Hudson’s coalescent simulator ms (Hudson 2002). We simulated 2184 human haplotypes (1092 African and 1092 European) under the demographic model published in Harris and Nielsen (2013) that was previously inferred from tracts of identity by state in the 1000 Genomes trios. Because we were only interested in SNP pairs separated by 100 bp or less, we simulated a total of 5.6 × 105 independent “chromosomes” of length 10 kb using the mutation rate 2.5 × 10−8 bp−1gen−1 and the recombination rate 1.0 × 10−8 bp−1gen−1.
Estimating the contribution of MNM to new point mutations
In the 1000 Genomes data, we counted NSNP = 17,140,039 nonsingleton SNPs that met our criterion for ancestral identifiability. For each pair type t, we also counted the number 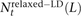 of t-type SNP pairs L bp apart that met the relaxed definition of perfect LD given in Equation 6. We estimate the fraction fMNM = 0.019 produced by MNM using the following equation:
of t-type SNP pairs L bp apart that met the relaxed definition of perfect LD given in Equation 6. We estimate the fraction fMNM = 0.019 produced by MNM using the following equation:
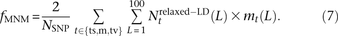
Calculating r2 with a correction for multinucleotide mutation
Given two SNPs sA, sB with major alleles A, B and minor alleles a, b, let pAB, pAb, paB, and pab be population frequencies of each of the four associated haplotypes. Let pA, pa, pB, and pb be the allele frequencies at individual loci. One measure of linkage disequilibrium between the loci is the correlation coefficient
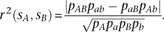
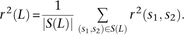
Although it is not possible to classify a SNP pair in perfect LD as an MNM unambiguously, we can correct for MNM by estimating the probability that each observed SNP s was generated as part of a pair of simultaneous mutations. This probability, ℙMNM(s), is calculated as a function of the nearest SNP sLD occurring in perfect LD with s. If s is not in perfect LD with any other SNP within 1000 bp, we assume that s was generated by an ordinary point mutation and let ℙMNM(s) = 0. Otherwise, letting A(s, sLD) denote the allelic state of the pair (s, sLD) (either transitions [ts], transversions [tv], or mixed [m]) and L denote the distance between s and sLD, we estimate that 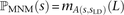 . Note that when s1 and s2 are in perfect LD and mutually closer to one another than to any other SNP in perfect LD,
. Note that when s1 and s2 are in perfect LD and mutually closer to one another than to any other SNP in perfect LD,
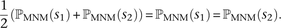
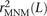 that down-weights each SNP by the probability that it is part of a complex mutation pair:
that down-weights each SNP by the probability that it is part of a complex mutation pair: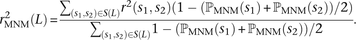
Acknowledgments
We thank Jasper Rine for sharing his deep understanding of replication and mutation in eukaryotes. We also thank members of the Nielsen and Slakin laboratories, particularly Josh Schraiber, Mehmet Somel, and Tyler Linderoth, for helpful discussions and comments. An earlier manuscript was much improved thanks to input from Michael Lynch, Molly Przeworski, and two anonymous reviewers. K.H. was supported by a National Science Foundation Graduate Research Fellowship, and R.N. received funding from NIH grant 2R14003229-07.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.170696.113.
References
- ↵The 1000 Genomes Project Consortium. 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073.
- ↵The 1000 Genomes Project Consortium. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65.
- ↵Bemark M, Khamlichi A, Davies S, Neuberger M. 2000. Disruption of mouse polymerase ζ (rev3) leads to embryonic lethality and impairs blastocyst development in vitro. Curr Biol 10: 1213–1216.
- ↵Charlesworth D. 2006. Balancing selection and its effects on sequences in nearby genome regions. PLoS Genet 2: e64.
- ↵Chen J, Ferec C, Cooper D. 2009. Closely spaced multiple mutations as potential signatures of transient hypermutability in human genes. Hum Mutat 30: 1435–1448.
- ↵Daly J, Bebenek K, Watt D, Richter K, Jiang C, Zhao ML, Ray M, McGregor W, Kunkel T, Diaz M. 2012. Altered Ig hypermutation pattern and frequency in complementary mouse models of DNA polymerase ζ activity. J Immunol 188: 5528–5537.
- ↵Denver D, Morris K, Lynch M, Thomas W. 2004. High mutation rate and predominance of insertions in the Caenorhabditis elegans nuclear genome. Nature 430: 679–682.
- ↵Denver D, Dolan P, Wilhelm L, Sung W, Lucas-Liedo J, Howe D, Lewis S, Okamoto K, Thomas W, Lynch M. 2009. A genome-wide view of Caenorhabditis elegans base-substitution mutation processes. Proc Natl Acad Sci 106: 16310–16314.
- ↵Drake J, Bebenek A, Kissling G, Peddada S. 2005. Clusters of mutations from transient hypermutability. Proc Natl Acad Sci 102: 12849–12854.
- ↵Esposito G, Godin I, Klein U, Yaspo ML, Cumano A, Rajewsky K. 2000. Disruption of the Rev3l-encoded catalytic subunit of polymerase ζ in mice results in early embryonic lethality. Curr Biol 10: 1221–1224.
- ↵Gan G, Wittschieben J, Wittschieben B, Wood R. 2008. DNA polymerase ζ (pol ζ) in higher eukaryotes. Cell Res 18: 174–183.
- ↵Goodman M. 2002. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu Rev Biochem 71: 17–50.
- ↵Gronau I, Hubisz M, Gulko B, Danko C, Siepel A. 2011. Bayesian inference of ancient human demography from individual genome sequences. Nat Genet 43: 1031–1034.
- ↵Gutenkunst R, Hernandez R, Williamson S, Bustamante C. 2009. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet 5: e1000695.
- ↵Harris K, Nielsen R. 2013. Inferring demographic history from a spectrum of shared haplotype lengths. PLoS Genet 9: e1003521.
- ↵Hill W, Robertson A. 1968. Linkage disequilibrium in finite populations. Theor Appl Genet 38: 226–231.
- ↵Hobolth A, Christensen O, Mailund T, Schierup M. 2007. Genomic relationships and speciation times of human, chimpanzee, and gorilla inferred from a coalescent hidden Markov model. PLoS Genet 3: e7.
- ↵Hodgkinson A, Eyre-Walker A. 2010. Human triallelic sites: evidence for a new mutational mechanism? Genetics 184: 233–241.
- ↵Hudson R. 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337–338.
- ↵Keightley P, Trivedi U, Thomson M, Oliver F, Kumar S, Blaxter M. 2009. Analysis of the genome sequences of three Drosophila melanogaster spontaneous mutation accumulation lines. Genet Res 19: 1195–1201.
- ↵Kimura M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16: 111–120.
- ↵Lange S, Takata K, Wood R. 2011. DNA polymerases and cancer. Nat Rev Cancer 11: 96–110.
- ↵Leffler E, Gao Z, Pfeifer S, Ségurel L, Auton A, Venn O, Bowden R, Bontrop R, Wall J, Sella G, . 2013. Multiple instances of ancient balancing selection shared between humans and chimpanzees. Science 29: 1578–1582.
- ↵Levy S, Sutton G, Ng P, Feuk L, Halpern A, Walenz B, Axelrod N, Huang J, Kirkness E, Denisov G. 2007. The diploid genome sequence of an individual human. PLoS Biol 5: e254.
- ↵Li H, Durbin R. 2011. Inference of human population history from individual whole-genome sequences. Nature 475: 493–496.
- ↵Lynch M, Sung W, Morris K, Coffey N, Landry C, Dopman E, Dickinson W, Okamoto K, Kulkarni S, Hartl D, . 2008. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci 105: 9272–9277.
- ↵McDonald M, Wang W, Huang H, Leu J. 2011. Clusters of nucleotide substitutions and insertion/deletion mutations are associated with repeat sequences. PLoS Biol 9: e1000622.
- ↵Nielsen R, Wakeley J. 2001. Distinguishing migration from isolation: a Markov Chain Monte Carlo approach. Genetics 158: 885–896.
- ↵Northam M, Robinson H, Kochenova O, Scherbakova P. 2010. Participation of DNA polymerase ζ in replication of undamaged DNA in Saccharomyces cerevisiae. Genetics 184: 27–42.
- ↵Ogawara D, Muroya T, Yamauchi K, Iwamoto T, Yagi Y, Yamashita Y, Waga S, Akiyama M, Maki H. 2010. Near-full-length REV3L appears to be a scarce maternal factor in Xenopus laevis eggs that changes qualitatively in early embryonic development. DNA Repair (Amst) 9: 90–95.
- ↵Ossowski S, Schneeberger K, Locas-Liedo J, Warthmann N, Clark R, Shaw R, Weigel D, Lynch M. 2010. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327: 92–94.
- ↵Roberts S, Sterling J, Thompson C, Harris S, Mav D, Shah R, Klimczak L, Kryukov G, Malc E, Mieczkowski P, . 2012. Clustered mutations in yeast and in human cancers can arise from damaged long single-stranded DNA regions. Mol Cell 46: 424–435.
- ↵Sakamoto A, Stone J, Kissling G, McCulloch S, Pavlov Y, Kunkel T. 2007. Mutator alleles of yeast DNA polymerase ζ. DNA Repair (Amst) 6: 1829–1838.
- ↵Saribasak H, Maul R, Cao Z, Yang W, Schenten D, Kracker S, Gearhart P. 2012. DNA polymerase ζ generates tandem mutations in immunoglobulin variable regions. J Exp Med 209: 1075–1081.
- ↵Schrider D, Hourmozdi J, Hahn M. 2011. Pervasive multinucleotide mutational events in eukaryotes. Curr Biol 21: 1051–1054.
- ↵Schrider D, Houle D, Lynch M, Hahn M. 2013. Rates and genomic consequences of spontaneous mutational events in Drosophila melanogaster. Genetics 194: 937–954.
- ↵Ségurel L, Thompson E, Flutre T, Lovstad J, Venkat A, Margulis S, Moyse J, Ross S, Gamble K, Sella G, . 2012. The ABO blood group is a trans-species polymorphism in primates. Proc Natl Acad Sci 109: 18493–18498.
- ↵Seplyarskiy V, Kharchenko P, Kondrashov A, Bazykin G. 2012. Heterogeneity of the transition/transversion ratio in Drosophila and Hominidae genomes. Mol Biol Evol 29: 1943–1955.
- ↵Sheehan S, Harris K, Song Y. 2013. Estimating variable effective population sizes from multiple genomes: a sequentially Markov conditional sampling distribution approach. Genetics 194: 647–662.
- ↵Stone J, Lujan S, Kunkel T. 2012. DNA polymerase ζ generates clustered mutations during bypass of endogenous DNA lesions in Saccharomyces cerevisiae. Environ Mol Mutagen 53: 777–786.
- ↵Terekhanova N, Bazykin G, Neverov A, Kondrashov A, Seplyarsky V. 2013. Prevalence of multinucleotide replacements in evolution of primates and Drosophila. Mol Biol Evol 30: 1315–1325.
- ↵Wang Y, Hey J. 2010. Estimating divergence parameters with small samples from a large number of loci. Genetics 184: 363–379.
- ↵Waters L, Walker G. 2006. The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G2/M phase rather than S phase. Proc Natl Acad Sci 103: 8971–8976.
- ↵Waters L, Minesinger B, Wiltrout M, D’Sousa S, Woodruff R, Walker G. 2009. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev 73: 134–154.
- ↵Wittschieben J, Shivji M, Lalani E, Jacobs M, Marini F, Gearhart P, Rosewell I, Stamp G, Wood R. 2000. Disruption of the developmentally regulated Rev3l gene causes embryonic lethality. Curr Biol 10: 1217–1220.
- ↵Yang Z, Rannala B. 1997. Bayesian phylogenetic inference using DNA sequences: a Markov Chain Monte Carlo method. Mol Biol Evol 14: 717–724.