
Abstract
Exome and whole-genome sequencing studies are becoming increasingly common, but little is known about the accuracy of the genotype calls made by the commonly used platforms. Here we use replicate high-coverage sequencing of blood and saliva DNA samples from four European-American individuals to estimate lower bounds on the error rates of Complete Genomics and Illumina HiSeq whole-genome and whole-exome sequencing. Error rates for nonreference genotype calls range from 0.1% to 0.6%, depending on the platform and the depth of coverage. Additionally, we found (1) no difference in the error profiles or rates between blood and saliva samples; (2) Complete Genomics sequences had substantially higher error rates than Illumina sequences had; (3) error rates were higher (up to 6%) for rare or unique variants; (4) error rates generally declined with genotype quality (GQ) score, but in a nonlinear fashion for the Illumina data, likely due to loss of specificity of GQ scores greater than 60; and (5) error rates increased with increasing depth of coverage for the Illumina data. These findings, especially (3)–(5), suggest that caution should be taken in interpreting the results of next-generation sequencing-based association studies, and even more so in clinical application of this technology in the absence of validation by other more robust sequencing or genotyping methods.
With the recent development of next-generation sequencing technologies, there has been an explosion in the number of whole-exome (Choi et al. 2009; Ng et al. 2009, 2010) and whole-genome (Lupski et al. 2010; Rios et al. 2010; Roach et al. 2010) biomedical sequencing studies, which have grown to include up to thousands of samples (The 1000 Genomes Project Consortium 2012; Tennessen et al. 2012; Fu et al. 2013). While several different sequencing platforms are available, the vast majority of studies utilize either Illumina (IL) (Bentley et al. 2008) or Complete Genomics (CG) (Drmanac et al. 2010) sequencing technologies. In the only published comparison between the two platforms (Lam et al. 2012), the investigators estimated that 12% of the called genotypes were discordant between IL and CG whole-genome sequences obtained from the same sample. This estimated error rate is several orders of magnitude higher than the published error rates of the two technologies (Bentley et al. 2008; Drmanac et al. 2010), and much larger than the error rates estimated using various genotype-calling algorithms. If correct, a 12% error rate would have serious implications for the interpretation and power of sequence-based genetic studies that are attempting to find rare variants affecting complex disease susceptibility, and even more critically in the clinical translation of this technology.
In this study, we conduct a detailed, quantitative comparison of single-nucleotide variant (SNVs, i.e., genotypes different from the reference sequence) calling with IL and CG platforms, using both whole-genome (WGS) and whole-exome sequence (WES) data generated from blood and saliva samples from four different individuals. SNVs were called separately for each sample. Agilent and Nimblegen in-solution capture methods were used to generate the exome data from both DNA sources and all four individuals. The individuals are members of the Kaiser Permanente Medical Care Plan Northern California Region (KPNC) and participated in the Research Program on Genes, Environment, and Health (RPGEH). We focus on estimating genotype discordance rates as a function of the genotype quality (GQ) score, a phred-like measure that base-calling algorithms use to estimate the accuracy of a genotype call. The GQ score is logarithmic and defined by GQ = −10log10(Error Rate) (so that GQ = 10 corresponds to an estimated error rate of 10%, GQ = 20 reflecting an estimated error rate of 1%, and so on). We also estimate genotype discordance rates as a function of the minor allele frequency (MAF) of the putative variant, since sequence-based genetic and clinical studies are generally searching for rare causal variants. Finally, we perform subsampling experiments to determine what effect depth of coverage has on the number and accuracy of SNV genotype calls for the Illumina sequencing. While some comparisons of the effect of depth of coverage on SNV calls have been performed before (Clark et al. 2011; Meynert et al. 2013), none have estimated error rates as a function of sequencing depth.
Results
We tabulated the discordance rates between genotypes from the four replicate WGS data sets generated from each individual (blood and saliva samples sequenced using CG and IL). Observed discordance rates are much higher than expected based on the GQ score, especially at the higher end of the range (i.e., GQ ≥ 60) (Fig. 1A). There is no significant difference in the error profiles between the blood and saliva samples. To compare the two DNA sources more closely, we tabulated the discordance rates between the IL-generated whole-genome and exome sequence data from the same samples. Specifically, we compared the discordance rate between two sequencing runs of the same sample (e.g., WGS blood vs. WES blood, both sequenced from the same DNA sample using IL) to the discordance rate between blood and saliva samples from the same individual (e.g., WGS blood vs. WES saliva, sequenced using IL from the same individual). We found that there was no difference in the two discordance rates (0.149% vs. 0.152%, P > 0.1, Supplemental Table S1), so all further analyses pool the blood and saliva results together (i.e., consider them as independent data sets) for estimating discordance rates.
Expected and actual genotype discordance rates as a function of GQ (genotype quality) score. (A) Illumina (IL) and Complete Genomics (CG) sequences from saliva (sal) and blood (bld) samples. (B) IL discordance, with or without SNP filters, and for different depths of coverage.
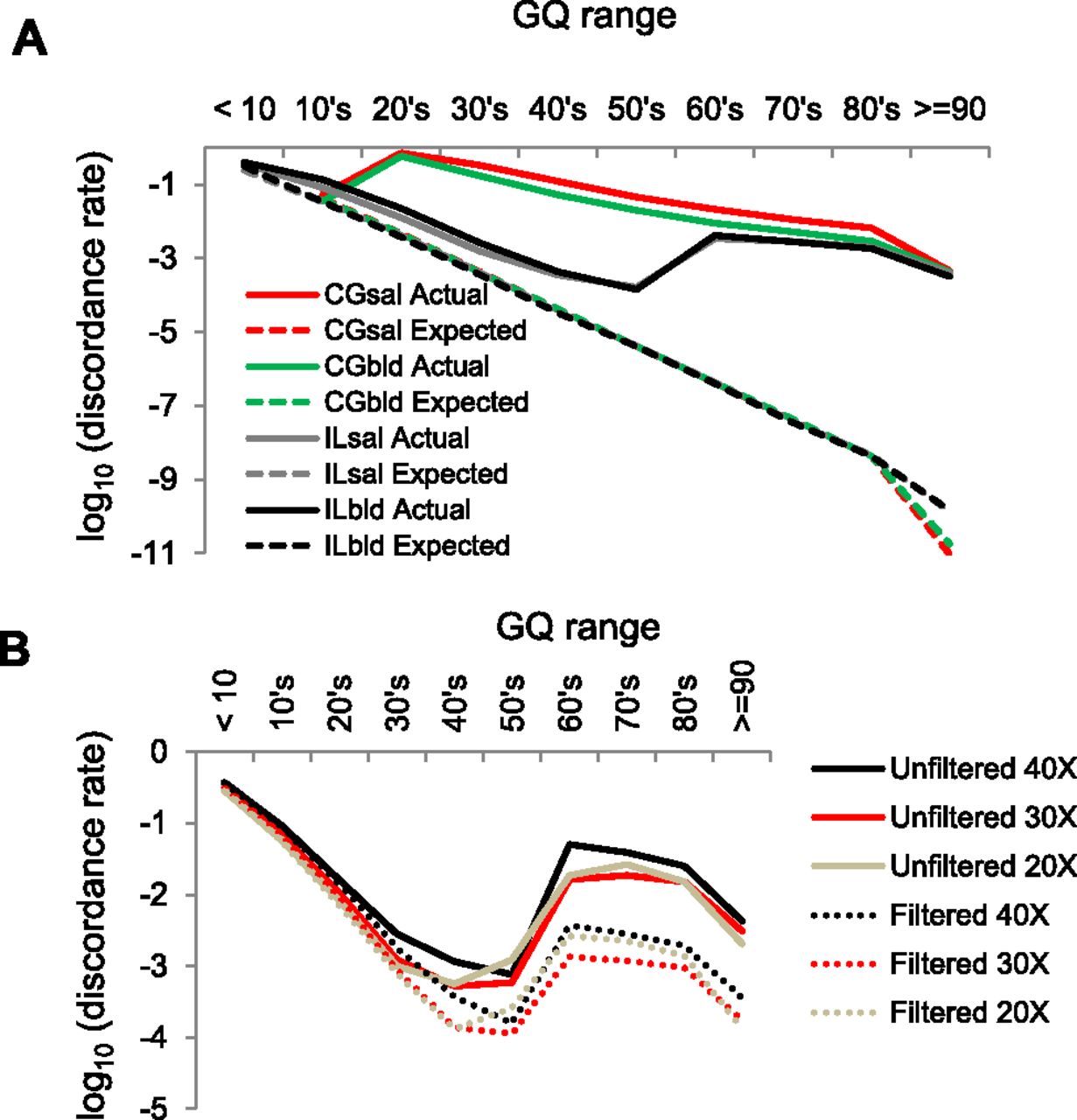
Overall, the sequences generated using IL technology have lower genotype discordance rates than the CG sequences. More troubling, the discordance rates are far higher than expected and do not decrease monotonically with increasing GQ score in either sequencing platform. For the CG data sets, SNVs with a GQ score in the 20’s (corresponding to an estimated error rate of 0.1%–1%) have a staggeringly high 63% discordance rate (i.e., over half of the genotype calls are incorrect). For GQ scores ≥30, there is a monotonic decline in the discordance rate for the CG data, although it is still vastly higher than expected for each GQ score (for example, at a GQ of 50, the expected error rate is 0.00001, but the observed discordance rate is about 0.01). For the IL sequence data, there is a clear monotonic decline in discordance rate for GQ scores between <10 and 60, and the observed discordance rates in this range are only about 10-fold higher than expected. However, there is a discontinuity in discordance rates at GQ = 60, with all samples having a higher discordance rate for SNVs with GQ ≥ 60 than for SNVs with 50 ≤ GQ ≤ 59. This pattern appears to be an intrinsic property of GATK, as it is common to all of the IL-based WGS and WES data sets. We conclude that IL GQ scores greater than 60 are misleading, and from Figure 1B estimate an overall error rate for GQ scores above 60 of about 0.001.
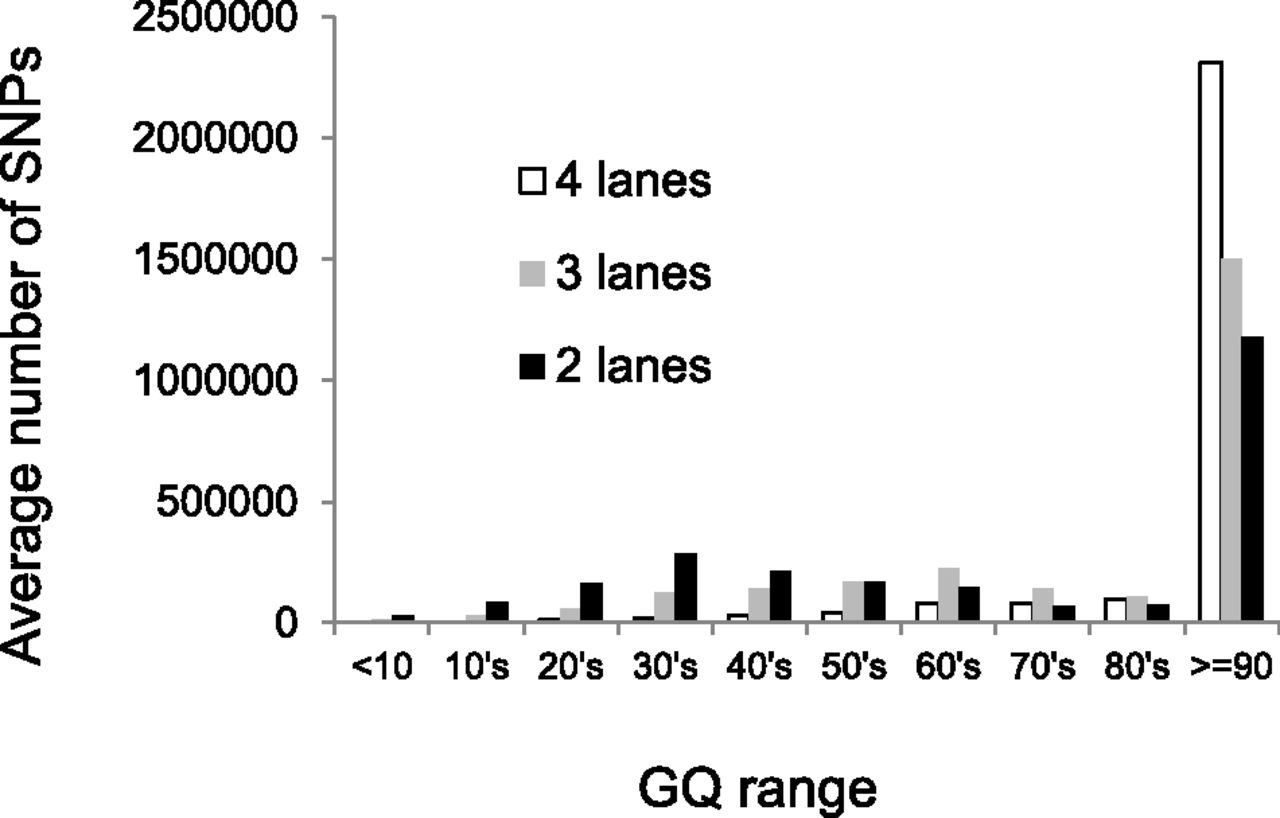
To explore the effect that depth of coverage has on discordance rates, we subsampled the Illumina WGS data to obtain ∼20× and ∼30× coverage levels for each sample, corresponding to two lanes (2L) or three lanes (3L) of HiSeq 2000 sequence data, respectively. For each of these subsampled data sets, we then performed SNV calling using the same pipeline, with or without the filters recommended in the GATK best practices documentation (see Methods for details). We then tabulated discordance rates as a function of GQ score as before (Fig. 1B), summing across individuals and DNA sources (i.e., blood and saliva). As expected, filtered data sets have lower discordance rates than unfiltered data sets, and both show the same discontinuity in discordance rates across the GQ = 60 boundary. Surprisingly, discordance rates for the 2L and 3L data sets are generally lower than for the full data sets when controlling for GQ score. However, this is balanced by the reduced number of SNVs called with high confidence (i.e., high GQ score) in the subsampled data sets (Fig. 2). Decreasing the sequencing depth from four lanes to three or two lanes leads to a 13% or 30% reduction in the number of SNVs called with GQ ≥ 40, respectively. Similar patterns were observed in comparable analyses of the WES data, with lower coverage levels leading to fewer SNVs called and generally lower error rates (Supplemental Fig. S1). Restricting the analysis to variants with GQ ≥ 40 called in the WGS and both WES data sets, the false-positive rate is approximately nine times higher in the exome data compared with the whole-genome data (Supplemental Table S2). This higher discordance rate is directly attributable to the higher coverage levels in the WES data.
Average number of SNVs called per sample, stratified by GQ score (x-axis) and average level of coverage. Each “lane” corresponds to 32 GB of total sequence.
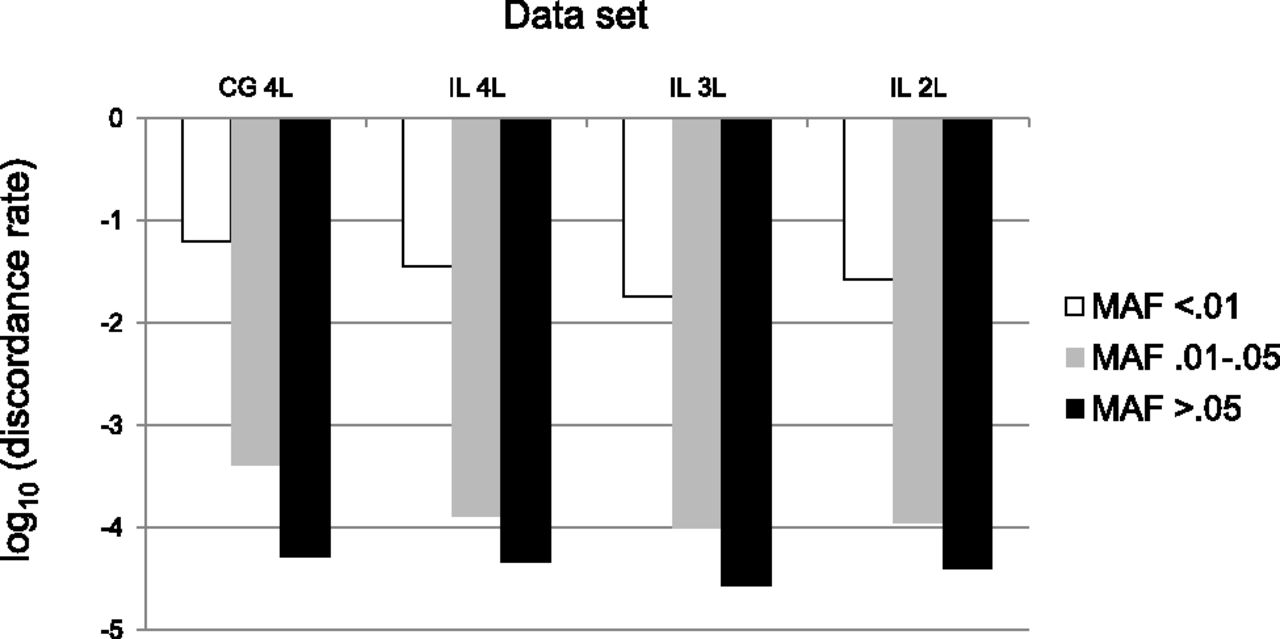
Next, we stratified discordance rates based on the MAF at each putative SNV with GQ ≥ 40 (see Methods), with allele frequencies estimated from the 1000 Genomes Pilot Project European data (The 1000 Genomes Project Consortium 2010). The motivation for this analysis is that genotypes at common SNVs (i.e., SNPs) can generally be ascertained accurately and cheaply using genotyping arrays, whereas large-scale sequencing studies are implicitly concentrating on rare variants. Rare variants (MAF < 0.01) have discordance rates that are more than 100 times higher than discordance rates for common variants (MAF ≥ 0.05), ranging from 6.3% for the CG WGS data to 3.6% for the IL WGS data (Fig. 3). As before, these discordance rates are lower for the subsampled WGS data (1.8% and 2.7% for the comparable 3L and 2L IL WGS data sets). Analyses of the WES data sets from the same samples produced the same general patterns (Supplemental Fig. S2).
Genotype discordance rate for whole-genome data stratified by minor allele frequency (MAF) for different sequencing platforms (IL, Illumina; CG, Complete Genomics) and levels of coverage (4L, four lanes; 3L, three lanes; 2L, 2 lanes).
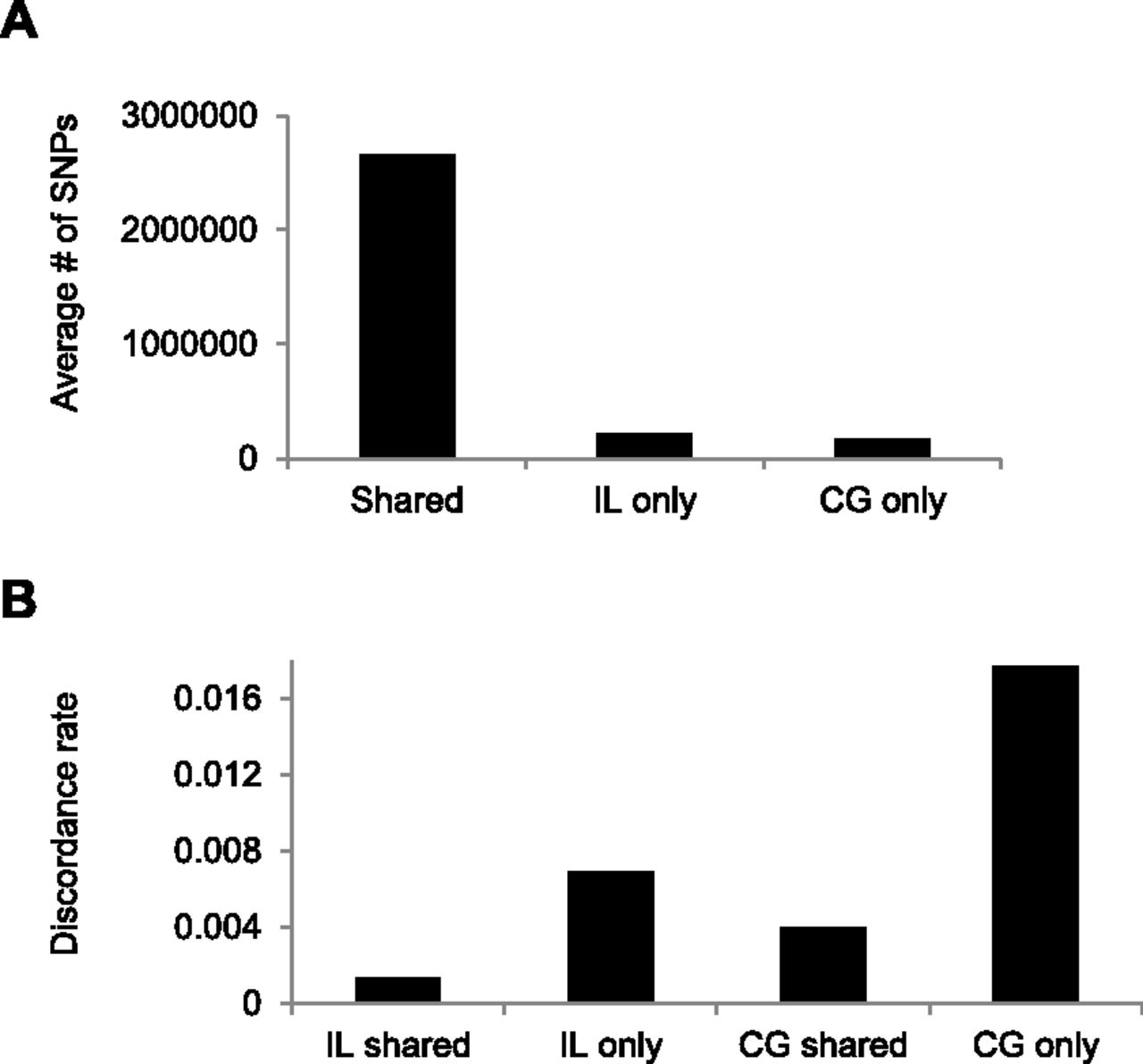
Next, we examined the number and discordance rates for SNVs that were called by both platforms (i.e., all four replicate samples) compared with SNVs that were called by a single platform (e.g., called in both IL samples but not both CG samples), using a genotype quality score cutoff of GQ ≥ 40 (Fig. 4). A priori we might expect that SNVs called by both platforms would tend to have a lower error rate than do platform-specific SNVs. This is exactly the pattern observed, with platform-specific SNVs having a several-fold higher discordance rate compared to shared SNVs. Of note, the IL data sets have both a greater number of called SNVs (Fig. 4A) and a lower discordance rate (Fig. 4B) than do comparable CG data sets from the same individuals. The former might be a result of the greater variance in depth of coverage across the genome for CG data compared with IL WGS data (Ross et al. 2013).
Average number of called SNVs shared between platforms or unique to one platform (A), and the genotype discordance rate (between blood and saliva samples) for shared and platform-specific SNVs (B).
Finally, we performed a related analysis to try to estimate a total error rate for the WGS data from each platform. We set an arbitrary genotype quality score threshold of GQ ≥ 40, and tabulated all genotype calls that were not identical for the different WGS data sets generated for each individual. These sites were then stratified into six different categories:
False positives—Sites called as homozygous reference in three samples and heterozygous in one sample
False negatives—Sites with the same nonreference genotype call in three samples and a homozygous reference call in one sample
Miscalls—Sites called as different from reference in all four samples, with three having one genotype and one having an alternate genotype
Platform-specific fixed differences (Platform FD)—Sites with one genotype in both IL samples (blood and saliva) and a different genotype in both CG samples
Other—Sites where pairs of samples share differing genotypes (e.g., both blood samples share one genotype; both saliva samples share a different genotype)
Errors at platform-specific SNVs—Sites not called in at least one platform sample that differ between the blood and saliva samples from the other platform (e.g., not called in at least one CG sample, different genotype calls in the two IL samples)
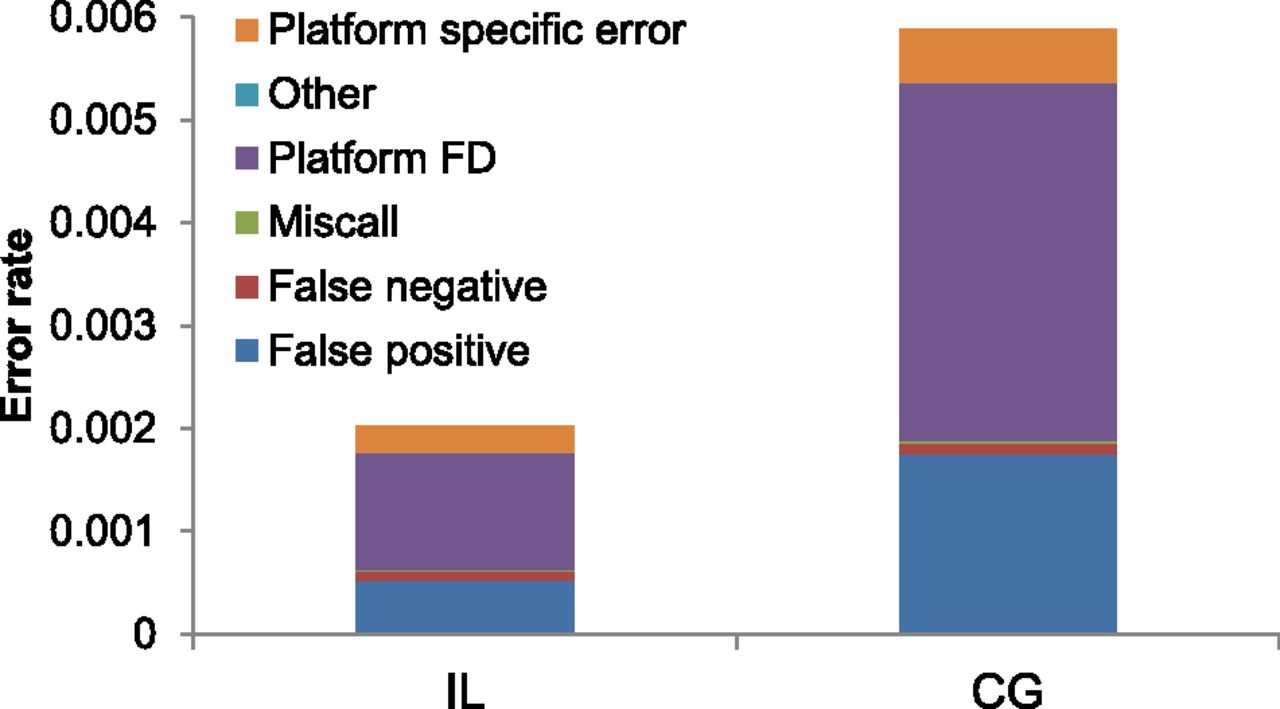
We tabulated the total error rate, stratified by category, for the CG and IL data (Fig. 5). For (1) to (3), we assumed that the outlier genotype is wrong. It is not easy to determine which platform or sample type has the correct genotype in (4) and (5); for platform-specific differences we assumed that the relative error rates for the CG and IL data were proportional to the error rates estimated from (1) to (3). (This assumption is likely to underestimate the ratio of errors attributable to CG versus IL, as described below.) As expected from the previous results, the CG data had a higher total error rate (5.88 × 10−3) than the IL data (2.03 × 10−3). Overall, we found that most errors were category (4), fixed platform differences between the two CG samples and the two IL samples. We also found that false positives were much more common than false negatives, though it is possible that many true positives (i.e., actual nonreference genotypes) were missed simultaneously in all four samples. The results were qualitatively similar for other GQ thresholds (Supplemental Fig. S3).
Total error rate for sites with GQ ≥ 40, partitioned across categories, for different WGS data sets. The six different colors represent the six different error types described in the text.
Discussion
Our results revealed no difference in the sequencing quality or error rate of blood and saliva samples. Since the latter are much easier to gather, we suggest that blood samples do not need to routinely be gathered for future sequence-based biomedical studies. Our analyses also uncovered several other results that have the potential to influence the design and interpretation of future large-scale sequencing studies. First, with current sequencing and SNV-calling technologies, error rates are on the order of one in 200–500 SNVs. This is much lower than a previous estimate (Lam et al. 2012), but several orders of magnitude higher than error rates estimated from the SNV-calling algorithms.
We found that the IL data had a much lower error rate than the CG data, but this is potentially dependent on the assumptions made when calculating a total error rate. Inspection of Figure 5 shows that most errors belong to category 4: fixed differences between platforms. With the data we gathered, it is impossible to unambiguously determine whether these are due to sequencing errors in the two CG samples or due to sequencing errors in the two IL samples. However, there is strong circumstantial evidence suggesting that the vast majority of these sites are false-positive errors in the two CG samples (rather than false-negative errors in the two IL samples). Platform-specific differences can be further divided into IL-variant sites, where the two IL samples have nonreference genotypes and the two CG samples have reference genotypes, and vice versa (CG-variant sites), where the two IL samples have reference genotypes and the CG samples are nonreference (Supplemental Table S3). The vast majority of platform-specific differences (94%) consist of CG-variant sites, which are either CG-specific false positives or IL-specific false negatives. Since analyses of three vs. one sites (i.e., nucleotide sites where three out of four replicate samples share the same genotype and one out of four has an alternative genotype) suggest that false positives are ∼11 times more common than false negatives, it follows that the vast majority of CG-variant sites are likely to be false positives in the CG samples. This conclusion is corroborated by the observation that most of the CG-variants occur at sites that are rare variants (MAF < 0.01 in the 1000 Genomes data, see Supplemental Table S4), which our previous analyses (Fig. 3) showed have an extremely high genotype discordance rate. Since the rate of false negatives does not vary as much across different allele frequency categories, we conclude that the excess of CG-variants at rare SNV sites are due to false-positive genotype calls in the two CG samples. If we conservatively assume that the false-negative rate is the same across the two platforms (even though we observe a higher false-negative rate in the CG samples), then it follows that ∼94% of the platform-specific differences can be attributed to errors in the two CG samples, leading to overall error rate estimates of 6.73 × 10−3 for the CG data and 1.18 × 10−3 for the IL data. False-positive rates for rare (MAF < 0.01) SNVs that include the share of platform-specific fixed differences that can be attributed to false positives range from 4% to 5% in the IL data to 22% to 23% in the CG data.
Our finding of a higher error rate in the CG data compared with the IL data is different from the results of Lam et al. (2012), but similar to what was reported by Ross et al. (2013). We suspect that the difference between our results and those of Lam et al. (2012) is primarily due to (1) the filters used by each study (see, e.g., Reumers et al. 2012), (2) the improvements in GATK over the past 2 yr, and (3) our use of genotype quality (GQ) scores rather than simple quality (QUAL) scores. (QUAL scores reflect the SNP caller’s estimate of how likely there is to be a polymorphism at a given site, while GQ scores are an estimate of how likely the called genotype is to be correct.) If we replace the GQ filter with a QUAL filter (QUAL > 50), then the genotype discordance rate between duplicate IL sequences from the same individual increases approximately fivefold.
We also quantified the relationship between estimated error rate (using GQ scores) and observed discordance rates in both technologies. We found the observed discordance rates to be much higher than the estimated error rates, implying that there might be a systematic source of error unaddressed by the SNV-calling programs as implemented. We also found a nonmonotonic relationship between the observed and expected rates for the IL data. For example, the SNVs with the lowest discordance rate in the IL data had 50 ≤ GQ ≤ 59, while SNVs with GQ scores in the 60’s had 20-fold higher discordance rates. While on the surface this is troubling, it suggests a problem with the GQ estimates above 60 that may be remediable, leading to potential improvement in SNV calling programs.
Error rates also correlate with minor allele frequencies of SNVs, with rare or novel variants much harder to call correctly than common ones. This is not surprising, since GATK and other variant callers use public databases of common SNP calls (e.g., HapMap) as prior data to help calibrate their results. Genotype calls at common SNPs can already be accurately and cheaply obtained from commercial genotyping arrays, so the error rates at common SNPs (Fig. 3) are not as relevant for assessments of the efficacy of sequence-based association studies. The error rates of 4%–6% for rare SNVs implies that an independent source of corroborating evidence (e.g., Sanger sequence or functional data), rather than resequencing to higher depth using the same platform, may be needed to separate false positives from true positives in studies that focus on the role of rare variants in the genetic basis of complex diseases. In fact, if we take false positives identified from IL WGS data and use the high-coverage Nimblegen data from the same sample as a confirmation step, then ∼40% of the false positives will be “confirmed” with the exome data (i.e., 40% of the false positives in the WGS data have the same wrong genotype call in the Nimblegen exome data from the same sample).
Finally, our finding of higher accuracy for lower coverage IL data sets highlights another weakness in current SNV calling algorithms. As before, we speculate that this is a consequence of systematic sources of error that are unaddressed in the single-sample calling protocol of GATK. As the depth of coverage increases, these errors are compounded, leading to less accurate SNV calls. Indeed, the data set with the highest average coverage level (Nimblegen exome data, with an average of 211× coverage) has the highest error rate. Previous studies have identified several systematic sources of error in IL sequencing data, including higher error rates near the end of reads (Kircher et al. 2009), higher error rates in GC-rich regions (Dohm et al. 2008), and specific sequence motifs associated with high error rates (Nakamura et al. 2011). These are partially addressed in current versions of GATK (DePristo et al. 2011), and they potentially contribute to the high discordance rates that we observed.
A closer examination of the false positives reveals that many of them consist of heterozygous variant calls with a strong allelic imbalance (e.g., most of the reads support the reference allele, while many fewer reads support the alternative allele). If we incorporate an allelic balance filter of 0.25 (i.e., at least 25% of all reads must support each allele for heterozygous variant calls), then this reduces the number of false positives by 62%, while reducing the overall number of heterozygous variant calls by only 7%. For imbalanced sites, depth of coverage influences whether GATK calls a site as variant or reference, and this is one explanation for why higher depths of coverage seem to produce higher genotype discordance rates.
Until the allelic imbalance problem is handled better by GATK, or additional systematic error sources are characterized, or the sequencing technologies improve, our observations should also serve as a particularly important cautionary note regarding the accuracy of current high-coverage next generation sequencing for clinical applications. While false-positive errors can be reduced by subsequent Sanger-based sequencing validation, there is no remedy for false-negative results. As we have shown, there is a clear tradeoff regarding enhanced sequencing depth and error rates. While greater depth does systematically reduce the rate of false negatives, it also increases the rate of false positives. Thus, an optimal strategy would appear to be one involving high-sequencing depth to reduce the rate of false-negative results, but follow up Sanger sequencing (or some other orthogonal genotyping/sequencing platform) for all clinically significant positive results.
Methods
Samples
Blood and saliva samples were obtained for four healthy Caucasian individuals who were participants in the Research Program on Genes, Environment, and Health (RPGEH) for Kaiser Permanente Medical Care Plan Northern California Region members. Oragene kits were used for saliva collection. Protocols for both studies were approved by the KPNC Institutional Review Board.
Sequencing
Blood and saliva samples from all four individuals (eight samples in total) were sent to Complete Genomics for sequencing in the spring of 2012. For each of the four different individuals, we generated WGS data using IL (HiSeq 2000) and CG technologies at an average coverage of 44× and >40×, respectively, from both blood and saliva samples. We also generated WES data from the same eight samples (four individuals × two sample types) using the Agilent SureSelect Human All Exon 50 MB kit and the Nimblegen SeqCap EZ Human Exome Library, with an average on-target depth of coverage of 104× and 211×, respectively. All IL sequencing on the same eight samples was performed on a HiSeq 2000 machine housed at the Genomics Core Facility in the Institute for Human Genetics at UCSF. We first generated short-insert (<400 bp) IL libraries using standard techniques. Then, a total of four lanes of HiSeq 2000 were used for each of the IL WGS data sets, one lane for each Nimblegen exome data set and 0.25 lanes for each Agilent exome data set. We used the Nimblegen SeqCap v3 (64 MB) and the Agilent SureSelect Human All Exon (50 MB) kits for the exome data.
Genotype calling
Complete Genomics uses a proprietary pipeline for aligning reads and calling variants. We extracted SNV calls and GQ scores from the masterVar files, and considered only simple variants where both alleles were called. For the IL data, SNV calling was performed using the bcbio-nextgen pipeline (v0.3) developed by Brad Chapman in the Bioinformatics core at the Harvard School of Public Health (https://github.com/chapmanb/bcbio-nextgen). Briefly, sequencing reads were aligned to the hg19 reference genome using the Burrows-Wheeler Alignment tool (BWA v0.5.9) (Li and Durbin 2010) using the default parameter that allows for two mismatches. Then, indexing, realignment and duplicate removal were performed using Picard (v1.56, from http://picard.sourceforge.net) and SAMtools (v0.1.18) (Li et al. 2009). Variants were called and recalibrated using the Genome Analysis Toolkit (GATK) (McKenna et al. 2010; DePristo et al. 2011), version 1.4-25-g23e7f1b. GATK uses an adaptive error model focusing on known variant sites from HapMap v3.3 and the Omni chip array from the 1000 Genomes Project to differentiate true variants from machine artifacts. Variants were hard-filtered with the recommendations listed in the Best Practice Variant Detection documentation in GATK v3 with “QD < 2.0,” “MQ < 40.0,” “FS > 60.0,” “HaplotypeScore > 13.0,” “MQRankSum < 12.5,” “ReadPosRankSum < −8.0” for SNPs. GQ scores were extracted from the VCF files produced by GATK and analyzed as described in the Results.
GATK does not provide GQ scores for homozygous reference calls. For those analyses calculating discordance rates for all SNVs with a GQ score above a certain threshold (Figs. 3, 4B, 5; Supplemental Figs. S1B, S2, S3), we used a depth of coverage cutoff ≥ 20 in place of GQ ≥ 40 to identify homozygous reference genotype calls that we were confident were correct.
Subsampling
Whole-genome subsampling was based on 320 million (2 × 100 bp) reads or 480 million reads from each sample, roughly corresponding to two or three lanes of HiSeq sequencing, respectively. For the subsampling, reads were grouped into files with 4 million reads, and 80 or 120 files were randomly chosen (without replacement) to generate the 320 million or 480 million reads for analysis. Exome resampling randomly selected 20 or 40 million reads from the data available for each sample; these numbers roughly corresponding to 0.125 or 0.25 lanes of HiSeq sequencing.
Statistical analysis
We first analyzed the WGS data by assuming that a correct genotype call was obtained (i.e., the majority call) if at least three out of four replicate samples (CG blood, CG saliva, IL blood, IL saliva) from the same individual had the same genotype call with GQ ≥ 40 (irrespective of the genotype call or GQ score of the fourth sample). We then measured the (genotype) discordance rate for each sample type-sequencing platform combination by determining the frequency with which that combination’s genotype differed from the other three among all sites with a correct genotype call, as a function of the GQ score, as well as the expected discordance rate if the GQ scores were an accurate reflection of uncertainty. In these calculations, we assume that when three out of four or all four sequences agree, the likelihood of a false call for all four or three out of four (compared to the one discrepant call) is vanishingly small. Because we are not including in this count cases where fewer than three of the four calls agree with each other, these discordance rates can be interpreted as a lower bound on (and underestimate of) the true error rates of the genotype calls.
To quantify the total error rate of genotype calls in each platform, we first partitioned the discordances described above into false positives, false negatives, and miscalls, depending on whether the (homozygous) reference genotype was found in three samples, one sample, or no samples, respectively (Supplemental Table S3). We then examined sites where two samples shared one genotype and the remaining two samples shared a different genotype. If the two CG samples had one genotype and the two IL samples had an alternative genotype, we classified the site as a “platform-specific difference”. Otherwise, we classified the site as “other.” Finally, for those nucleotide sites called in only two or three of the samples, we defined “errors at platform-specific SNVs” as sites where the genotype calls differed between the blood and saliva samples of one platform, but were uncalled in at least one sample from the other platform (see Supplemental Table S3).
Data access
WGS VCF files from this study have been submitted to dbGaP (http://www.ncbi.nlm.nih.gov/dbgap) under accession number phs000786.v1.p1. Workflows are available in the Supplemental Material.
Acknowledgments
This work was supported in part by NIH grants R01 MH085543, U01 GM61390 (to C.S.), and RC2 AG036607 (to C.S. and N.R.).
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.168393.113.
References
- ↵The 1000 Genomes Project Consortium. 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073.
- ↵The 1000 Genomes Project Consortium. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65.
- ↵Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, . 2008. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456: 53–59.
- ↵Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloglu A, Ozen S, Sanjad S, . 2009. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci 106: 19096–19101.
- ↵Clark MJ, Chen R, Lam HY, Karczewski KJ, Euskirchen G, Butte AJ, Snyder M. 2011. Performance comparison of exome DNA sequencing technologies. Nat Biotechnol 29: 908–914.
- ↵DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, . 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498.
- ↵Dohm JC, Lottaz C, Borodina T, Himmelbauer H. 2008. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res 36: e105.
- ↵Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, . 2010. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327: 78–81.
- ↵Fu W, O’Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, Gabriel S, Rieder MJ, Altshuler D, Shendure J, . 2013. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493: 216–220.
- ↵Kircher M, Stenzel U, Kelso J. 2009. Improved base calling for the Illumina Genome Analyzer using machine learning strategies. Genome Biol 10: R83.
- ↵Lam HY, Clark MJ, Chen R, Natsoulis G, O’Huallachain M, Dewey FE, Habegger L, Ashley EA, Gerstein MB, Butte AJ, . 2012. Performance comparison of whole-genome sequencing platforms. Nat Biotechnol 30: 78–82.
- ↵Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler Transform. Bioinformatics 26: 589–595.
- ↵Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genomes Project Data Processing Subgroup. 2009. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25: 2078–2079.
- ↵Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D, Chen DC, Nazareth L, Bainbridge M, Dinh H, Jing C, Wheeler DA, . 2010. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med 362: 1181–1191.
- ↵McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, . 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303.
- ↵Meynert AM, Bicknell LS, Hurles ME, Jackson AP, Taylor MS. 2013. Quantifying single nucleotide variant detection sensitivity in exome sequencing. BMC Bioinformatics 14: 195.
- ↵Nakamura K, Oshima T, Morimoto T, Ikeda S, Yoshikawa H, Shiwa Y, Ishikawa S, Linak MC, Hirai A, Takahashi H, . 2011. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res 39: e90.
- ↵Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler EE, . 2009. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 461: 272–276.
- ↵Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, . 2010. Exome sequencing identifies the cause of a Mendelian disorder. Nat Genet 42: 30–35.
- ↵Reumers J, De Rijk P, Zhao H, Liekens A, Smeets D, Cleary J, Van Loo P, Van Den Bossche M, Catthoor K, Sabbe B, . 2012. Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nat Biotechnol 30: 61–68.
- ↵Rios J, Stein E, Shendure J, Hobbs HH, Cohen JC. 2010. Identification by whole-genome resequencing of gene defect responsible for severe hypercholesterolemia. Hum Mol Genet 19: 4313–4318.
- ↵Roach JC, Glusman G, Smit AF, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M, . 2010. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 328: 636–639.
- ↵Ross MG, Russ C, Costello M, Hollinger A, Lennon NJ, Hegarty R, Nusbaum C, Jaffe DB. 2013. Characterizing and measuring bias in sequence data. Genome Biol 14: R51.
- ↵Tennessen JA, Bigham AW, O’Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, . 2012. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337: 64–69.