
Understanding the regulatory and transcriptional complexity of the genome through structure
Abstract
An expansive functionality and complexity has been ascribed to the majority of the human genome that was unanticipated at the outset of the draft sequence and assembly a decade ago. We are now faced with the challenge of integrating and interpreting this complexity in order to achieve a coherent view of genome biology. We argue that the linear representation of the genome exacerbates this complexity and an understanding of its three-dimensional structure is central to interpreting the regulatory and transcriptional architecture of the genome. Chromatin conformation capture techniques and high-resolution microscopy have afforded an emergent global view of genome structure within the nucleus. Chromosomes fold into complex, territorialized three-dimensional domains in concert with specialized subnuclear bodies that harbor concentrations of transcription and splicing machinery. The signature of these folds is retained within the layered regulatory landscapes annotated by chromatin immunoprecipitation, and we propose that genome contacts are reflected in the organization and expression of interweaved networks of overlapping coding and noncoding transcripts. This pervasive impact of genome structure favors a preeminent role for the nucleoskeleton and RNA in regulating gene expression by organizing these folds and contacts. Accordingly, we propose that the local and global three-dimensional structure of the genome provides a consistent, integrated, and intuitive framework for interpreting and understanding the regulatory and transcriptional complexity of the human genome.
It is testament to the rapid advances achieved in genome research that our conception of the human genome has changed dramatically since the publication of the first draft assembly over a decade ago (International Human Genome Sequencing Consortium 2001; Venter et al. 2001). At that time, our interpretation of the human genome was largely focused on the ∼1% protein-coding fraction that was interspersed across vast and largely uncharacterized intergenic noncoding regions. Aided by the advent of increasingly cheap high-throughput sequencing technologies, the genome has been rapidly annotated with detailed regulatory landmarks and transcriptional maps, revealing a complex array of overlapping and interlacing transcripts and a layered terrain of open and closed chromatin, diverse histone modifications, nucleotide modifications, and transcription factor occupancies (The ENCODE Project Consortium 2012). These overlapping layers act in concert, and in combination encompass the majority of the genome, comprising a vast landscape whose detail and rich complexity was unanticipated at the outset of the human genome project.
We are now faced with the task of interpreting this huge catalog of data in an integrated and systematic manner. Here, we argue that this interpretation can be achieved by reference to the three-dimensional folding of the genome in the nucleus. We argue that, despite its value, the current one-dimensional representation impairs an intuitive understanding of the genome, and that many current regulatory maps intrinsically reflect, indeed retain, the signatures of its higher order structure, which in turn has an overbearing role in the organization and architecture of genes and in regulating gene expression. Therefore, achieving a detailed and accurate three-dimensional representation of the genome within the nucleus has emerged as one of the major goals currently facing the field of genomic research.
Rendering the regulatory landscape in three dimensions
The human genome sequence exposed vast non-protein-coding regions that are replete with responsive and cell-specific regulatory elements (Thurman et al. 2012). Chromatin immunoprecipitation (ChIP) has been an invaluable technique for surveying these regions and is now widely used to identify transcription factor binding sites and chromatin modifications (Landt et al. 2012). The first genome-wide application of ChIP revealed an intricate landscape containing an unexpectedly large number of transcription factor binding sites across chromosomes 21/22, often in regions distal to gene promoters (Cawley et al. 2004). However, many of these promiscuous sites are cross-linked at low levels, and similar sites fail to drive patterned reporter gene expression when systematically assayed in Drosophila (Fisher et al. 2012). Notably, many of these sites also do not contain corresponding transcription factor sequence recognition motifs, and a further subset, termed “transcription factor hotspots,” exhibit simultaneous overlapping signals to numerous transcription factors (Fig. 1; Moorman et al. 2006; Roy et al. 2010; Neph et al. 2012).

Examples of proximal enrichments resulting from ChIP-seq. (A) ChIP-seq of a transcription factor (green) results in immunoprecipitation of bound DNA sequence (blue) as well as addition of DNA sequence (orange) in close proximity. Only bound sequence shows evidence of DNase I footprint and binding motif. (B) Immunoprecipitation of DNA sequence associated with large multiprotein complex results in artifactual indirect enrichments for a wide range of transcription factors. (C) Active enhancers exhibit a range of ChIP-seq enrichments as a result of a close spatial proximity to histone modification and transcription factors at promoters.
Rather than bona fide sites of transcription factor binding, these promiscuous sites may reflect an artifactual enrichment resulting from proximal nonspecific cross-linking between contacts within a tightly folded genome structure. During the initial step of the ChIP-seq protocol, formaldehyde is used to cross-link occupied DNA and bound proteins, which are then immunoprecipitated by antibodies against the transcription factor of interest and digested to yield the occupied DNA for sequencing. However, the initial formaldehyde cross-linking can also nonspecifically link DNA sequences that are not bound, but rather in close spatial proximity to proteins, resulting in the parallel, collateral precipitation of juxtaposed genomic regions, potentially explaining the lack of binding nucleotide motifs within many ChIP-seq enrichments (Fig. 1A). Similarly, targeted proteins may be constituents of larger multiprotein complexes. As a result, fixation with formaldehyde would immunoprecipitate the entire multiprotein complex, including any sequences bound by intermediate protein partners, resulting in a single sequence exhibiting a simultaneous enrichment for the full range of transcription factors within the complex, providing a potential interpretation for the existence of transcription factors hotspots (Fig. 1B).
While these scenarios argue for the careful interpretation of signal enrichments within ChIP-seq libraries, they also suggest that ChIP-seq libraries retain information on the three-dimensional folding of the genome and its interaction with protein structures. Indeed, this prospect forms the basis for the chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) approach (Fullwood et al. 2009). ChIA-PET uses the same protocol as ChIP-seq, including the initial formaldehyde cross-linking and immunoprecipitation of targeted protein, but with the addition of a ligation step that joins coprecipitating DNA sequences before sequencing, thereby discerning those regions of the genomic sequences that copurify due to close proximity. For example, utilizing ChIA-PET shows not only the residence of the Ser2-hypophosphorylated form of RNA polymerase II at human gene promoters, but also the aggregation of these gene promoters into higher-order networks of coregulated and cotranscribed genes (Li et al. 2012). Similarly, ChIA-PET targeting H3K4me2 modifications is able to delineate interactions between promoters and distal enhancers (Chepelev et al. 2012). A comparison of ChIP-seq and matched ChIA-PET libraries reveals the extent to which numerous ChIP-seq sites may be parsimoniously resolved as alternative contacts with a common transcription factor.
Immunofluorescent microscopy using matched antibodies directly illustrates the structural information implicit within ChIP-seq libraries, visualizing the subnuclear distribution of transcription factors, histone modifications, and specialized subnuclear structures (Mao et al. 2011). Transcription machinery and factors are not uniformly diffused throughout the nucleus but coalesce as distinct and discrete foci, and histone modification often form broad nuclear domains, such as the aggregation ofH3K9 methylated regions to the nucleus periphery (Bartova et al. 2008). These nuclear domains are not obvious when matched ChIP-seq libraries are aligned to the genome sequence. For example, the H3K27me3 domains and sites of polycomb complex occupancy that occur concurrently at Hox gene clusters that are dispersed across the Drosophila genome, in fact reflect the convergent localization of these distal Hox loci to common Polycomb bodies within the nucleus (Cheutin and Cavalli 2012; Sexton et al. 2012; Towbin et al. 2012). This suggests that the complexity apparent within our current linear representation of the regulatory landscape may be interpreted as the complex folding of the genome around common subnuclear structures, and a more judicious understanding of ChIP-seq libraries could be achieved with reference to three-dimensional genome structure.
Resolving genome folding
Chromatin conformation capture techniques are the main current approach by which to infer three-dimensional genome structure (de Wit and de Laat 2012). These techniques also use formaldehyde-mediated cross-linking to resolve contact between genomic loci, followed by restriction enzyme digestion to extract cross-linked fragments from the chromatin. Digested termini undergo proximal ligation to form intramolecular fragments that can be used to measure the population-averaged frequency of interactions between two genomic regions (Dekker et al. 2002). This technique has been instrumental in determining significant and stable interactions between two genomic loci, such as the close physical interaction between the locus control regions and active globin genes that loop out ∼40–60 kb of intervening sequence (Tolhuis et al. 2002).
The global three-dimensional structure of the genome can be inferred from techniques, such as HiC, that combine chromatin conformation capture with sequencing (Lieberman-Aiden et al. 2009). These studies support the adoption of a fractal-globule organization that enables the ready extrication and decondensation of the genome (Bancaud et al. 2012), as well as the organization of chromosomes into distinct radially organized subnuclear territories that were previously visualized by fluorescent in situ hybridization (Bolzer et al. 2005). These territories are further divided into gene-rich domains that extend away from the nuclear periphery and are sites of active gene expression and early replication, with the reciprocal exclusion of gene-poor regions that encompass a compact repressive late-replicating heterochromatin fraction (Simonis et al. 2006; Boyle et al. 2011; Kalhor et al. 2012).
As chromatin conformation capture has achieved higher resolution, smaller structural units, known as topologically associated domains (TADS) have been detected (Dixon et al. 2012; Nora et al. 2012). These mega-base-sized successive domains partition the genome into local, distinct, and introverted folded regions linked by intervening unfolded regions. Although contacts within these domains are dynamic, the borders of these domains are remarkably conserved during differentiation and between cell types, and seem to impose an intrinsic modular architecture to the genome. These topological domains also exhibit a close concordance to transcription factor occupancy and epigenetic domains, including large blocks of H3K27me3 and H3K9me2 repression (Lan et al. 2012; Shen et al. 2012). This correlation may reflect the common measurement by alternative ChIP-seq and HiC approaches of the genome folding around a distinct subnuclear domain, where TAD formation may delimit these segmental chromatin blocks (Nora et al. 2012).
Transcription at factories
The concept of transcription factories was first proposed in response to the clustering transcription factors as distinct and discrete foci within the nucleus (Jackson et al. 1993; Wansink et al. 1993). Transcription factories comprise large subnuclear assemblies that encompass a range of transcription factors and machinery constituents along with additional accessory proteins for RNA processing and splicing (Jackson et al. 1993; Melnik et al. 2011; Edelman and Fraser 2012). A highly specialized example of a transcription factory is provided by the nucleolus, a subnuclear organelle responsible for rDNA transcription that harbors the dedicated machinery required for the ribosomal RNA transcription, elongation, and maturation (Hernandez-Verdun et al. 2010). Over 2000 clustered rRNA copies dispersed over five chromosomes are recruited together to the nucleolus, where they are cotranscribed on the surface of the fibrillar center within the nucleolus (Nemeth and Langst 2011).
RNA polymerase I and II–dependent transcription has also been associated with similar centralized structures, with electron spectroscopic imaging visualizing a porous heterogeneous protein-rich core, with nascent transcription preceding on the surface (Eskiw et al. 2008). Emerging evidence suggests that active RNA polymerase II is commonly bound to the surface of transcription factories (Papantonis and Cook 2011). The use of fluorescent in situ hybridization to register the relative movement of gene loci and nascent transcripts during the transcription cycle shows the DNA sequence tracking through RNA polymerase II complexes that themselves remain immobile with reference to the transcription factory (Papantonis et al. 2010). However, the generality of this model is not yet resolved with, for example, microscopy of the Hsp70 loci in Drosophila polytene chromosomes providing conflicting evidence for a classical model of polymerase II recruitment (Yao et al. 2007).
The immobilization of numerous active RNA polymerase II complexes to a single specialized active compartment affords the coexpression of multiple genes (Zhou et al. 2006). Erythroid genes, located at distal sites across the genome, accrue at common transcription factories when transcriptionally active, with silent genes being excluded (Schoenfelder et al. 2010). These common compartments where the erythroid genes unite also appear specialized, harboring specific transcription factors, such as KLF1, relevant to erythroid gene expression. Similarly, the STAT transcription factor anchors coregulated genes to common compartments during the nuclear reorganization that accompanies T-cell differentiation (Hakim et al. 2013). This aggregation of multiple genes to specialized transcription factories with varying and specific regulatory components may be responsible for the correct and coordinated expression of distinct gene ontologies. Indeed, following transfection, minichromosomes cluster to different transcription factories according to the promoters and introns they contain (Xu and Cook 2008).
The activation of a range of genes, including the Myc and globin genes and the collinear activation of Hox genes (Osborne et al. 2004, 2007; Morey et al. 2009; Schoenfelder et al. 2010) is coincident with their nuclear relocation. The potential for this relocation to target genes to pre-assembled transcriptional compartments offers an alternative to the classical model of transcription factor recruitment. Although yet to be realized, this alternative model switches our point of reference from the linear genome being the central structure upon which transcription factors associate de novo to a three-dimensional genome that dynamically traffics genes or promoters to a central scaffold of pre-assembled transcriptional complexes (Cook 2010; Edelman and Fraser 2012). Nevertheless, such movement would be restricted within the confines of the genomes' global architecture. Live cell imaging shows that the movement of gene loci is constrained to a tight volume within the nucleus (Strickfaden et al. 2010), and ligand-induced changes to gene expression that include rapid and global transcriptional changes expand the interactions between genomic regions, but do not incur the major reorganization of chromosomes (Hakim et al. 2011).
An overarching nucleoskeleton
The local folding of enhancers to genes and genes to transcription factories promotes the topography of the genome into an overarching regulatory role. Intranuclear order, including the structure and movement of the genome, is organized by a dense, filamentous nucleoskeleton (Simon and Wilson 2011). Many proteins of the nucleoskeleton, including lamins, titin, actin, myosins, and kinesins associate with DNA, histones, chromatin modifying proteins, transcription factors, and the general transcriptional machinery (de Lanerolle and Serebryannyy 2011). Actin comprises a major component of the nucleoskeleton, of chromatin remodeling complexes, and enhances transcription by interaction with promoter and coding sequences, the RNA polymerase I–III complexes, and other RNA processing proteins (Hofmann et al. 2004). Similarly, specialized nuclear-localized myosins and kinesins are molecular motors that traffic cargo over long ranges along actin or microtubule filaments to transcriptional machinery at active genes (Pestic-Dragovich et al. 2000; Fomproix and Percipalle 2004; Chuang et al. 2006) and, in the case of Myosin 5a, to S35 speckles that harbor splicing factors (Pranchevicius et al. 2008).
Genome folding relies on the nucleoskeleton. Large-scale chromosomal repositioning in response to serum starvation is rapid, requires energy, and is dependent on active nuclear motor complexes (Mehta et al. 2010). The nucleoskeleton may also direct the traction of genes to nuclear bodies such as transcription factories. Following induction by a transcriptional activator, migration of chromosomal loci from the nuclear periphery is perturbed in actin and myosin mutants (Chuang et al. 2006). Furthermore, the collinear induction of HOXB gene expression is actin dependent (Ferrai et al. 2009), and the recruitment of snRNA genes to Cajal bodies, spherical subnuclear organelles that specialize in snRNP biogenesis, requires actin and myosin (Dundr et al. 2007). Similarly, both actin and myosin play a primary role in recruiting rDNA clusters to the nucleolus in response to the requirements of cellular growth and differentiation (Philimonenko et al. 2004). This range of transcription factories that are dependent on actin and myosin anticipates a broad and preeminent role for the nucleoskeleton in organizing genome folding and gene expression.
Complex networks of transcription
The complexity and sheer size of the transcriptional landscape is surely one of the most significant findings to emerge since the publication of the human genome. Given that the signature of genome structure is written into the regulatory landscape, we argue it is likely that this signature is similarly written into the transcriptional landscape. Initial cDNA sequencing and tiling array projects revealed that the transcription of protein-coding genes is accompanied by noncoding RNAs (Carninci et al. 2005; Kapranov et al. 2007a). Vast swaths of noncoding DNA are transcribed into short and long noncoding RNAs that are commensurate in diversity and abundance with protein-coding genes, and have been increasingly accepted as legitimate gene products (Mercer et al. 2009). Indeed, we have still yet to reach the frontiers of the transcriptome, with targeted RNA sequencing revealing further range and complexity of noncoding transcription in intergenic regions not otherwise detected by conventional RNA sequencing (Mercer et al. 2012). The profiling of additional tissues, developmental stages, and cell types continues to expand these limits and collectively ascribe a massive depth and breadth to the human transcriptome.
Coding and noncoding genes are organized as incredibly complex networks of layered, interleaved, antisense, and overlapping transcripts (Kapranov et al. 2005). This transcriptional complexity has revealed the modular design principles of the genome, whereby a single sequence can be incorporated in numerous ways into a range of coding and noncoding, sense and antisense transcripts that overlap to form complex networks (Kapranov et al. 2007b). In response to this recurrent complexity throughout the genome, we now consider the transcript as the basic unit of the transcriptome, with the concept of a gene being revised to a higher-order definition that encompasses a functionally related group of transcripts influencing a given phenotype (Mattick 2003; Gerstein et al. 2007; Gingeras 2007; Djebali et al. 2012).
The folding of transcriptional complexity
The immobilization of RNA polymerase to nuclear structures ties the complexity of transcriptional initiation and elongation to genome structure. Recognition that splicing is a cotranscriptional process also provides an avenue by which genome structure can influence RNA processing. Therefore, we considered whether the modular design of the genome and its transcription and processing reflects and can be understood through the three-dimensional structure of the genome.
Gene expression requires the combinatorial action of alternative transcription initiation, splicing, and termination, with local chromatin loops communicating close coordination between these processes (Fig. 2; Tan-Wong et al. 2008; Moore and Proudfoot 2009). Chromatin conformation capture routinely resolves a loop that forms across the gene body, localizing gene termini to the promoter and affording contact between transcription initiation and termination processes and coassembly of associated machinery (O'Sullivan et al. 2004; O'Reilly and Greaves 2007; Singh and Hampsey 2007; Tan-Wong et al. 2008; Moore and Proudfoot 2009). This interaction also restricts the divergent transcription of ncRNAs and imposes directionality on the gene's promoter (Tan-Wong et al. 2012). Even further interactions between the RNA polymerase II residing at the alternative promoters used by a gene are anticipated by ChIA-PET (Li et al. 2012).
Formation of chromatin loops at gene loci permits coordination between processes of transcription initiation, termination, and splicing. Promoter and terminal regions of genes colocalize during transcription, forming a looped structure that enhances transcriptional directionality. Gene loop formation depends on contacts between both promoter-associated transcription factors, such as TFIIB, within the pre-initiation complex and polyadenylation factors, such as Ssu72 and cleavage factor subunits, within the terminator complex. Extensive contacts between the spliceosome and the initiating and elongating polymerase II complex also facilitate cotranscriptional splicing.
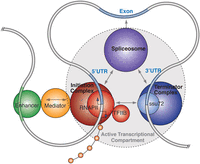
Multiple coding and noncoding transcripts are often interwoven into complex transcriptional networks (Carninci et al. 2005). Genome folding permits these interwoven RNAs to exploit a common regulatory architecture. For example, local intragenic loops permit a single promoter complex to simultaneously drive transcription of both the SPI1 gene promoter and an antisense noncoding RNA that is, counterintuitively, hosted within a downstream intron (Ebralidze et al. 2008). Further loops also bring enhancer elements to bear on the promoter complex, resulting in the assembly of a higher order structure encompassing the loci. The folding of the genome into higher ordered structures that loop out of intervening regions can prevent confusion from overlapping genes and permit compartmentalized transcription for the distinct expression of intronic-hosted genes (Fig. 3). ChIA-PET analysis targeting RNA polymerase II indicates that the genome can fold together multiple overlapping transcripts to share common regulatory features (Li et al. 2012). Such interleaved transcriptional networks, which seem complex in the linear representation of the genome, may be parsimoniously understood in the context of a three-dimensional genome.

Three-dimensional interpretation (left) of regulatory and transcriptional complexity in one-dimensional genome representation (right). (A) The genome forms large complex clusters and introspective folded clusters with specialized transcription compartments. Each of these clusters correlates to a collection of transcripts and “background” ChIP-seq enrichment. (B) Within each cluster the genome is folded to associate with subnuclear structures containing transcription factors and machinery, splicing, and other accessory proteins. These associations coregulate genes to generate interleaved complex transcriptional networks of coding (blue) and noncoding transcripts (green). Proximal cross-linking with ChIP-seq results in a complex landscape of enrichment across loci that reflect the folded genome structure. (C) Within each gene, local dynamic chromatin folding determines the association of alternative promoters and local noncoding RNAs with a shared regulatory architecture, thereby mediating coregulated gene expression.
Splicing is increasingly recognized as a cotranscriptional process, and splicing machinery and regulators comprise a major component of transcription factories (Fig. 2; Melnik et al. 2011). Like transcription initiation, a number of observations anticipate that local genome topography can be organized with relation to the gene's internal intron and exon structure, with exons being localized to cognate transcriptional machinery with intervening introns looped out (Tan-Wong et al. 2008; Moabbi et al. 2012). CTCF, better known for organizing chromatin loops and structure in conjunction with cohesin, also occupies alternative exons to mediate exon inclusion (Shukla et al. 2011; Lee and Iyer 2012). Similarly, a range of histone modifications demarcate the intron and exon boundaries within an epigenetic landscape that is intimately linked to genome structure (Luco et al. 2011; Kornblihtt et al. 2013). Such structural and epigenetic features could help direct the spliceosome to recognize correct splice sites across often vast intronic distances.
The imprint of genome structure in the transcriptional landscape
A longer, chromosome-wide perspective shows that these complex transcriptional networks cluster to form active transcriptional foci interspersed by quiescent regions (The FANTOM Consortium 2005; Kapranov et al. 2007b). These active transcriptional foci may associate with a corresponding transcription factory, with the complex internal folding of topological domains around common regulatory cores relating to the internal detail of transcriptional networks. Collectively, these transcriptional clusters crowd within the active nuclear compartment, with distinct knots of folded chromatin that comprise topological domains demarcating boundaries between developmentally regulated transcriptional hubs, with intervening regions replete with insulators, RNA polymerase I genes, and repetitive elements (Fig. 3; Dixon et al. 2012; Sexton et al. 2012).
The folding of the genome within successive three-dimensional structures would impose constraints on the organization of encompassed genes. Transcriptional territories may partition adjacent groups of coexpressed genes in the genome (Caron et al. 2001; Spellman and Rubin 2002). Such territories could create both specialized genome property with, for example, the majority of testes-expressed genes being tightly clustered within the Drosophila genome (Boutanaev et al. 2002) and “valuable” genome property, with ubiquitously expressed genes clustering as the most gene-dense regions (Lercher et al. 2002). Clustering of coexpressed genes also inversely shapes the genomic distribution of transposable elements that space out intervening regions (Fontanillas et al. 2007).
This constraint that genome structure imposes on gene evolution is elegantly demonstrated in the collinear organization of Hox genes, critical developmental genes that evolved in the bilateral ancestor to regulate body plan. Hox genes undergo collinear activation in distinct overlapping domains according to the body axis of animal embryos (Mallo et al. 2010). This collinear transcriptional activation involves the sequential relocation of genes to an active structural compartment, while inactive Hox genes remain sequestered within a single repressive structure delimited from flanking regions (Noordermeer et al. 2011). Despite the duplication, fragmentation, reduction, and expansion of Hox loci that has occurred and correlates with major morphological changes, the collinear order of Hox gene expression and the progressive relocation of genes to active transcriptional compartments has been maintained during evolution (Lemons and McGinnis 2006).
RNA can reciprocally shape nuclear structure
The overarching role for the structure and dynamic movement of the genome in regulating transcription may be reciprocated by RNA on genome structure. Mature RNA is stably associated with the genome, comprising a major part of chromatin where it fullfills well-established epigenetic roles (Mondal et al. 2010). The capacity for sequence-specific interactions with protein makes RNA an ideal guide and/or scaffold for the nucleation and assembly of the large regulatory structures to which the genome folds. The lncRNA, NEAT1, is required for interchromatin paraspeckle formation (Clemson et al. 2009), and the MALAT1 lncRNA sequesters serine/arginine splicing factors to nuclear speckles (Tripathi et al. 2010). Additional structures, including histone locus bodies, stress bodies, and other epigenetic bodies also require RNA for assembly (Shevtsov and Dundr 2011), anticipating a broader role for RNA in subnuclear organization. RNA can also mediate the trafficking of gene loci to subnuclear bodies, a key prediction of the alternative model of gene regulation. In response to growth signals, lncRNAs and associated chromatin modifying proteins relocate gene loci from repressive Polycomb bodies to the activating context of interchromatin granules, whereby gene expression is initiated (Yang et al. 2011).
The looping of long-range regulatory enhancers brings regulatory sequences and complexes into contact with promoters to regulate gene expression. In conjunction with this folding, enhancers themselves are often bidirectionally transcribed as nonpolyadenylated noncoding RNAs that are thought to contribute to the activation of genes targeted by the enhancer (Kim et al. 2010; Melo et al. 2012). Similarly, despite being retained at the site of transcription, the lncRNA HOTTIP recruits the WDR5/KMT2A (previously MLL) complex to impart active modifications to multiple distal sites throughout the HOXA loci via chromatin looping (Wang et al. 2011). The abundance of lncRNAs and eRNAs organized adjacent to developmental genes could similarly facilitate the tightly regulated local folding of these loci and their structural reorganization during development.
A new representation of the human genome
The linear representation of the genome enabled early efforts of gene mapping by classical genetic techniques of pedigree analysis, molecular techniques of physical mapping, and finally the assembly of the human genome sequence. Since this sequence was published, it has formed an invaluable reference to which genome-wide data has been aligned and interpreted. However, the abstraction of the genome to a single dimension ignores the tight folding of the genome within the nucleus, and we are beginning to realize the limits of this linear representation and how it impairs an intuitive conception of the genome. We consider the determination and development of three-dimensional representation of the human genome to be one of the most significant challenges currently facing genome biology.
In recent years the tools and expertise have been developed that make a detailed and global description of genome topology feasible (de Wit and de Laat 2012). The integration of whole-genome and targeted chromatin conformation capture approaches, along with ChIA-PET, ChIP-seq, immunofluorescent microscopy, and fluorescent in situ hybridization are required to construct and refine such a model. However, the size, complexity and dynamism of genome structure represents a major challenge to achieving these ambitions.
In addition to its massive complexity, the genome is a highly dynamic structure. While relatively inert large-scale topodomains and nuclear structures apply constraints, the genome, particularly at a local level, is in continual and stochastic motion. It will be a major technical challenge to reproducibly resolve such dynamic features. Current chromatin conformation capture techniques provide a population-averaged depiction of genome structure, affording the identification of recurrent, stable, and significant genome interactions whereas, in contrast, high-resolution single-cell microscopy can resolve individual chromatin interactions and identify dynamic genome folding. Nevertheless, despite the dynamism, size, complexity, and plasticity of the genome that confounds any easy determination of the genome structure, laudable efforts to tackle this challenge have already been initiated (Asbury et al. 2010; Marti-Renom and Mirny 2011).
These technical challenges will also require accompanying novel visual solutions to render the dynamic genome in three dimensions. A semi-schematic depiction of the genome's internal interaction circuitry may achieve a compromise between clarity and an accurate representation of detail and complexity. This map would have to incorporate and denote dynamic regions that undergo motion and may be recast in a cell-specific manner.
Despite these challenges, achieving such a three-dimensional representation of the genome would provide an invaluable reference for biologists. Aligning and analyzing functional genomic and transcriptional data within this spatial context could provide an integrated, consistent, and judicious basis for understanding the transcriptional and regulatory complexity that has emerged as a hallmark of the human genome.
Acknowledgments
This work was supported by the Australian National Health and Medical Research Council (Australia Fellowship 631668). We also thank Professor John Stamatoyannopoulos (University of Washington) for informative and helpful discussions.
Footnotes
-
↵1 Corresponding author
E-mail j.mattick{at}garvan.org.au
-
Article is online at http://www.genome.org/cgi/doi/10.1101/gr.156612.113.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported), as described at http://creativecommons.org/licenses/by-nc/3.0/.








