
Abstract
There is increasing evidence that the phenotypic effects of genomic sequence variants are best understood in terms of variant haplotypes rather than as isolated polymorphisms. Haplotype analysis is also critically important for uncovering population histories and for the study of evolutionary genetics. Although the sequencing of individual human genomes to reveal personal collections of sequence variants is now well established, there has been slower progress in the phasing of these variants into pairs of haplotypes along each pair of chromosomes. Here, we have developed a distinct approach to haplotyping that can yield chromosome-length haplotypes, including the vast majority of heterozygous single-nucleotide polymorphisms (SNPs) in an individual human genome. This approach exploits the haploid nature of sperm cells and employs a combination of genotyping and low-coverage sequencing on a short-read platform. In addition to generating chromosome-length haplotypes, the approach can directly identify recombination events (averaging 1.1 per chromosome) with a median resolution of <100 kb.
Haplotypes are haploid genotypes, i.e., the set of multiple alleles along each chromosome. Although sequencing of individual human genomes can readily identify most heterozygous loci, it remains a challenge to separate these variant bases into haplotypes that span the entire length of each chromosome. Numerous studies have highlighted the importance of understanding haplotype structure. Specific haplotypes have been reported to improve upon individual SNPs for prediction of autoimmune disease or clinical outcomes in transplantations (de Bakker et al. 2006; Petersdorf et al. 2007) or physiological responses to pharmacological agents (Drysdale et al. 2000). Knowledge of haplotype structure is critical for understanding allele-specific events, such as methylation, that are cis-regulated (Tycko 2010), and it can provide valuable validation data for the study of population genetics (Conrad et al. 2006) and genetic ancestry (Green et al. 2010). The haplotype structure of an individual's genome is also essential for predicting instances of compound heterozygosity (McLaughlin et al. 2010; Ng et al. 2010) or for identifying the parental origins of de novo mutations (Glaser et al. 2000; Aretz et al. 2004).
Many approaches have been proposed for extracting the complete haplotype structure from a sequenced genome. Sequencing of paired-end reads (Levy et al. 2007; McKernan et al. 2009) or sequencing of long DNA fragments (Kitzman et al. 2011; Suk et al. 2011; Peters et al. 2012) have been used to link multiple variant loci into large haplotype blocks (N50 values of up to 1.0 Mb), although none of these blocks span entire chromosomes. Other approaches involve the physical separation of chromosomes and include the use of somatic cell hybrids (Douglas et al. 2001), polony sequencing (Zhang et al. 2006), chromosome microdissection (Ma et al. 2010) or chromosome sorting by fluorescence-activated cell sorting (FACS) or microfluidic manipulation (Fan et al. 2011; Yang et al. 2011). Each of these yields chromosome-length haplotypes, although none has yet been implemented to achieve dense maps that include the majority of known variants in a genome. A distinct approach involves sequencing the genomes of both parents and sibling offspring (Roach et al. 2010). Although accurate and comprehensive, it cannot resolve all sites, and it is not always feasible to recruit the required participants for such a study. Consequently, the development of comprehensive haplotyping approaches that can be applied to an individual genome sequence remains a desirable goal.
Here, we describe a distinct approach for full genome haplotyping that involves sequencing the haploid genome content of isolated sperm cells after whole-genome amplification by the multiple displacement amplification method (MDA) (Dean et al. 2001, 2002). PCR-based methodology has been used frequently to study haplotypes and recombination events in single sperm cells (e.g., Hubert et al. 1994), and MDA has enabled extensive genotyping (Jiang et al. 2005). Recently, genome-wide analysis of individual sperm cells after MDA was used to assess recombination activity and de novo mutation rates (Wang et al. 2012). Here, despite amplification bias and allele drop-out, sequencing of a small number of sperm cells at low coverage after MDA was used to phase the vast majority of SNPs in an individual human genome.
Results
Sperm cells (n = 96) from the donor of the HuRef diploid genome sequence (Levy et al. 2007) were isolated by micromanipulation, and the genomic DNA was amplified by MDA. Amplification bias was assessed by qPCR at 12 genomic loci, including loci on chromosomes X and Y. Human DNA was detected in 69 amplifications, and the number of detectable loci ranged from four to 11 per preparation. Sperm cells were rinsed extensively prior to MDA to remove contaminating free DNA, and none of 32 control MDA reactions containing the final rinse buffer were positive for any of the qPCR loci. Positive reactions (n = 57) contained markers for either chromosomes X or Y, but never both, consistent with amplification of single sperm and the absence of contaminating DNA. It was concluded that although each sperm genome undergoes biased amplification, resulting in lack of detection of certain loci by qPCR, the content of contaminating DNA is likely to be minimal. Sixteen of the 57 positive reactions that contained the highest number of detectable qPCR loci were selected for genotyping.
The HuRef genome has been sequenced using multiple technologies, and 1.95 million heterozygous SNPs have been identified by independent analyses of data from at least two of these platforms (Levy et al. 2007; EF Kirkness and JC Venter, unpubl.; Supplemental Table S1). We aimed to phase these SNPs across the entire lengths of all HuRef autosomes using a combination of genome-wide SNP genotyping and low-coverage whole-genome sequencing (WGS). The SNP genotyping was used to identify recombination crossover events for each chromosome of sperm cells and for construction of a low-resolution haplotype map. The low-coverage WGS data could then be used to define the high-resolution haplotype structure.
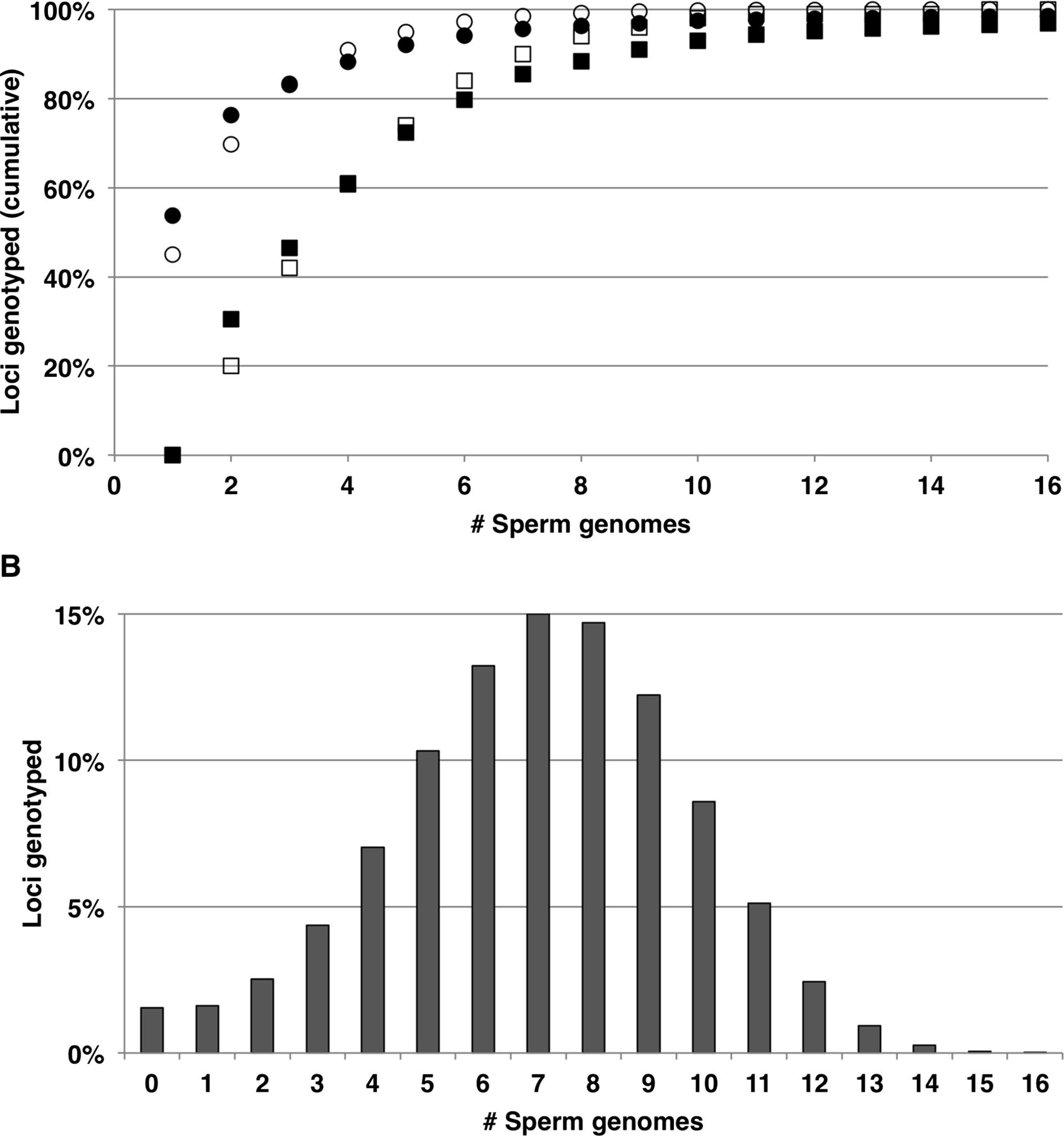
Amplified DNA from 16 independent sperm cells was genotyped at 1 million loci on an Illumina HumanOmni-Quad v1.0 BeadChip. Of these loci, 238,872 were heterozygous autosomal SNPs in the HuRef diploid genome and were therefore informative for haplotype phasing. The yield of genotyping calls at the informative loci ranged from 38.2% to 53.8% (mean 45.4%). Most of the calls (97.4 +/− 0.5%) were homozygous, as expected for a haploid genome. Importantly, although each sperm cell yielded genotypes at only half the informative loci, the missing data were largely random. Consequently, by genotyping multiple sperm cells, it was possible to obtain genotype calls for >98% of informative loci (Fig. 1A). Over 70% of SNP loci were called in six or more cells (Fig. 1B). The ∼2% of loci that failed to yield a genotype were located in 100-bp spans that contained a significantly higher G + C content (0.54 +/− 0.10) than the complete set of 238,872 informative SNPs (0.42 +/− 0.09; P < 0.0001). An underrepresentation of GC-rich sequences after MDA may account for the absence of these loci (and the thicker left tail of the distribution in Fig. 1B). In order to infer the haplotype phase of the HuRef donor (as opposed to individual sperm cells), it was necessary to identify the locations of meiotic crossover events (Supplemental Fig. S1). The genotypes at the informative SNP loci were compared among all pairs of chromosomes. These comparisons identified long haplotype blocks that were either shared or distinct between homologous chromosomal segments of any two sperm genomes. For any given sperm, evidence for a chromosomal crossover was defined by a switch between identity and nonidentity for the paired haplotype comparisons with other sperm (see Methods). The 11 sperm genomes with the largest number of genotype calls were characterized in detail and displayed 260 crossover events, an average of 1.1 events per chromosome (Supplemental Table S2) that is consistent with previous estimates of the average number of recombination events in male gametes (26.2) (Coop et al. 2008).
Genotyping of 238,872 informative loci in the genomes of 16 sperm cells. (A) The cumulative percentage of unique informative loci that was genotyped in at least one (•) or two (■) sperm cells. The calculation of expected values (○,□) assumes a random genotyping dropout rate of 55% per sperm cell. (B) After genotyping all 16 sperm cells, the percentage of informative loci is plotted with respect to the number of independent sperm cells in which they were genotyped.
The median resolution with which these crossover events could be located was 82.5 kb. Localization of crossover events for individual sperm cells permitted the reconstruction of the progenitor diploid genome. This was achieved by simply assigning the sequences adjacent to crossover events to distinct haplotypes. Using a consensus of genotype data from the 11 sperm cells, the two chromosome-length haplotypes of a putative diploid progenitor were reconstructed (see Methods). Of the 238,872 informative SNPs, 230,966 (96.7%) were phased along the 22 HuRef autosomes, whereas the remainder either had no genotype data from the 11 sperm (3.0%) or yielded ambiguous genotypes (0.3%). Most of the phased genotypes (91.2%) were supported by data from two or more sperm cells. Discrepancies between the predicted phasing of haplotypes from different cells were generally restricted to isolated loci (see below, this paragraph). However, for one sperm cell, a unique genomic region displayed multiple discrepancies (Y47, chr15:43.1–93.3 Mb, 448 discrepancies). Within this region, it was possible to identify only short haplotype blocks, and this unusual feature was attributed to the existence of both haplotypes within a single sperm preparation, likely due to minor contamination of DNA from lysed sperm cells that was not completely removed during the rinsing and cell isolation procedure. This was consistent with a higher frequency of heterozygous genotype calls in the discrepant regions relative to the flanking regions of the chromosome (5.5% versus 2.0% for sperm Y47, chr15:43.1–93.3 Mb). However, this was exceptional, and within the large blocks of haplotype identity between pairs of sperm genotypes, most discrepancies were isolated instances that conflicted with the overall block pattern (1.3% of genotypes). These may have arisen from gene conversion events, errors that occur early in the MDA reaction, or genotyping errors that were specific to individual sperm cells. Gene conversion tracts are thought to be relatively short (averaging <300 bp), with the relative frequencies of conversions to crossovers at recombination hotspots in the range from 10:1 to <1:12 (Jeffreys and May 2004; Holloway et al. 2006). However, if these ratios are generally true, gene conversion could account for only a small fraction (<20%) of the discrepant calls between individual haplotypes.
The reconstructed diploid haplotypes employed a consensus of genotypes from multiple sperm cells. Consequently, the isolated discrepancies that are unique to individual sperm cells had a minimal effect on the genome-wide haplotype map. In order to validate the haplotype map, we compared it to genotype data from a parent of the sperm donor. All loci for which the parent is homozygous and the HuRef genome is heterozygous should have the parental genotypes phased along the entire length of only one copy from each pair of HuRef chromosomes. Owing to the homozygosity of these parental loci, this feature is unaffected by recombination events in the parental genome. From 576,195 loci that were genotyped in the parental genome, 47,735 were homozygous in the parent, heterozygous in the HuRef genome, and included in the genome-wide haplotype map. Of these, 47,678 (99.9%) were phased on only one of the two HuRef haplotypes for each chromosome. The 47 discrepancies included 22 that were supported by data from only a single sperm cell and 20 for which the underlying sperm genotypes were not unanimous. It was concluded that the genome-wide haplotype map, constructed from genotyping data, is highly accurate; and the low error rate could be reduced further by simply including genotyping data from additional sperm cells.
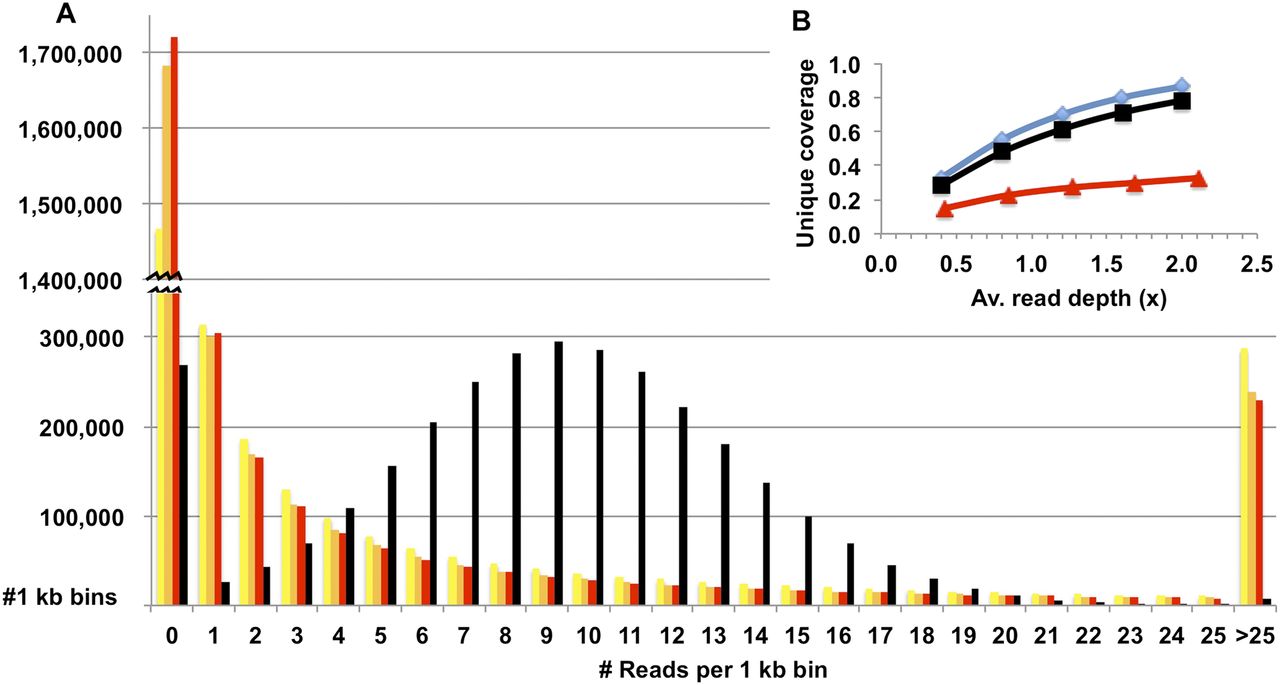
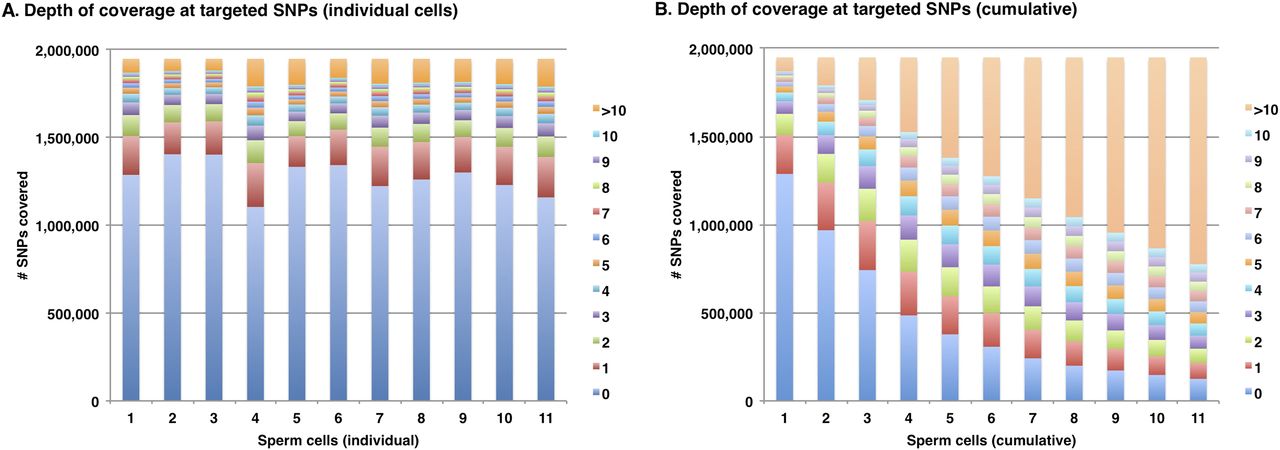
The haplotype map that was built from the genotypes of isolated sperm cells shares features with that derived from genotyping of isolated chromosomes (Fan et al. 2011). That is, the haplotype blocks extend across the entire length of each chromosome but have a relatively low resolution (averaging ∼12 kb between phased heterozygous SNPs). The remaining 90% of heterozygous SNPs in the HuRef genome cannot be genotyped using commercial arrays, and incorporation of these variants into a comprehensive haplotype map requires direct sequencing of sperm DNA. Currently, sequencing of whole genomes from single cells can be challenging due to the biased representation of the genome after amplification. For example, Fan et al. (2011) sequenced amplified products from isolated chromosomes 6 to an average depth of 4×–8× but achieved coverage for up to only 50% of the chromosome. Here we sequenced the amplified genomic DNA from 11 independent sperm cells, each to an average depth of 1.5×–3.7× genome coverage on the Illumina platform. An unamplified preparation of HuRef DNA from diploid cells was sequenced alongside for comparison. The amplified libraries displayed significant bias in the read coverage across the haploid genome, and reads from each sperm genome covered only 28%–43% of the 1.95 million heterozygous SNP loci (Fig. 2; Supplemental Table S3). However, the combined reads covered 94% (1.81 million loci) with 67% of these loci covered by 10 or more reads (Fig. 3). Although there was evidence for a slight bias against G + C-enriched sequences (Supplemental Fig. S2), the dropout appeared to be largely random (Supplemental Fig. S3). Despite the relatively low level of read coverage at a minority of loci, it should be noted that the sperm sequence data was not intended to provide for variant discovery but only to distinguish between the two known alternatives at heterozygous loci. There was a high concordance (>99.9%) between genotypes from the BeadChip array and those from direct sequencing of sperm cells (213,822 common loci). The genotypes derived from sperm genome sequencing were then used to reconstruct the phased haplotypes of a diploid progenitor, using the locations of crossover events inferred as described above. As for the low-resolution map, haplotypes were confirmed using homozygous parental genotypes that are heterozygous and phased in the HuRef genome. For these 84,086 loci, >99.9% were phased on only one haplotype. The combination of BeadChip-derived genotypes and sequencing-derived genotypes permitted phasing of 1.82 million heterozygous SNPs, or 94% of the known complement for the HuRef genome, with an average resolution of 1.6 kb (Supplemental Table S4).
Genome coverage of mapped sequence reads from amplified sperm DNA and unamplified blood DNA. (A) Distribution of mapped reads from amplified DNA of three haploid sperm cells (yellow, orange, and red bars) and unamplified DNA from blood cells (black bars) after dividing the reference genome into nonoverlapping bins of 1-kb length. (B) Unique genome coverage with increasing read depth for libraries prepared from a single sperm cell (▴) or diploid blood cells (■). The ideal mapping to a nonrepetitive genome is included for comparison (♦).
Depth of read coverage at 1.95 million heterozygous SNPs after shallow sequencing of amplified DNA from 11 independent sperm cells. (A) The read depth at target loci for each cell is indicated by the color-coded key. (B) The cumulative read coverage at target loci.
Sequence-derived genotypes from different sperm cells displayed a higher rate of discordance (∼3.5%) than for BeadChip-derived genotypes (1.3%). Interestingly, at the higher resolution obtained by sequencing, it was observed that approximately half of these discrepancies (1.7%) fell within clusters (median spans, 60–80 kb; median number of SNPs per cluster, 21–26; Supplemental Table S5). Smaller clusters, comprised of two or more SNPs within 300 bp, account for 6%–7% of the discrepant genotypes and may derive from gene conversion events. However, it was also noted that clusters of discrepant genotypes were enriched approximately threefold within copy number variants (CNVs) and highly enriched (six- to 11-fold) within segmental duplications (Supplemental Table S6). This suggests that structural differences between the HuRef and NCBI reference genomes, such as copy number differences, coupled with the difficulty in mapping reads from these regions, were a major source of the low-level discrepancy. Notably, the complete set of 1.95 million SNPs contained a larger proportion within segmental duplications (3.6%) than the 0.24 million assayed on the bead arrays (1.4%), and this may explain at least some of the increased discrepancy rate observed after phasing the complete collection of SNPs. The potential for variable numbers of segmental copies between the two haplotypes, coupled with the fact that less than half the genome is sequenced in any one sperm cell, clearly reduces the reliability of haplotype construction within these regions.
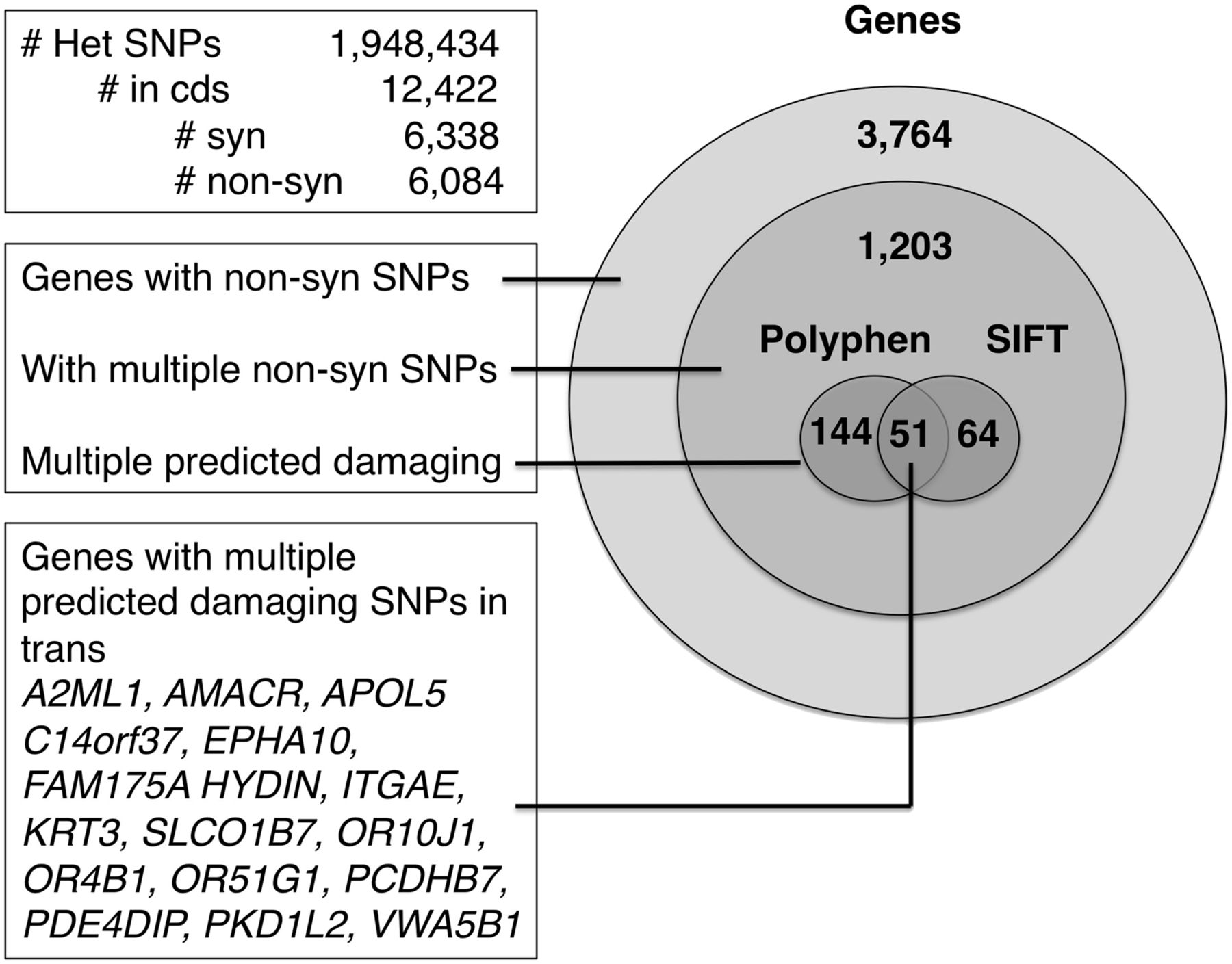
With a majority of HuRef heterozygous SNPs phased into haplotypes, it was possible to identify genes with potentially harmful mutations on both chromosomal copies (compound mutations). Among the 1.95 million heterozygous SNPs, 12,422 fall within protein-coding sequences, and 6084 involve nonsynonymous substitutions in 3764 genes. Polyphen2 and SIFT (sorting intolerant from tolerant) offer predictions of whether or not these nonsynonymous mutations are tolerated or damaging to the encoded gene product. For genes that contain multiple nonsynonymous mutations, Polyphen2 and SIFT each predict multiple damaging mutations in 51 genes (Fig. 4). For 46 of these 51 genes, it is now possible to phase the relevant SNPs, and the ratio of cis:trans mutations is 29:17. The 17 genes for which both chromosomal copies are predicted to have damaging mutations are listed in Supplemental Table S6. Given that phasing can be uncertain in some regions of segmental duplication (see above), it should be noted that 11 SNPs, found within four of the 17 genes (PDE4DIP, HYDIN, PCDHB7, FAM175A) fall within known segmental duplications, and the existence of compound heterozygosity within these genes remains questionable at present. The subject of this study has been generally healthy; and although the current analysis suggests that he may carry 10–20 genes that encode only defective protein products, there is increasing evidence that this could be a common feature of human genomes (Suk et al. 2011; MacArthur et al. 2012).
Heterozygous SNPs and multiple mutations that are predicted to be damaging in the HuRef genome.
Discussion
Low-pass sequencing of genomes from isolated sperm cells is a relatively straightforward and effective means to generate chromosome-length haplotypes without the need for specialized equipment to isolate individual chromosomes (Ma et al. 2010; Fan et al. 2011; Yang et al. 2011). Using genotype data from 16 sperm cells, a simple one-versus-all approach was sufficient to identify recombination breakpoints that were subsequently validated using parental genotypes. In this small sample of sperm cells, the recurrent use of common recombination breakpoints (within the resolution of the genotype data) was observed infrequently (six of 260 breakpoints) and never in more than two sperm cells. Consequently, recombination hotspots did not interfere with the designation of recombination breakpoints, which was based on the comparison of each sperm genome with 15 others. Although the phasing of individual SNPs within segmental duplications is less accurate than in unique regions of the genome, the erroneous assignment of breakpoints to these regions was minimized by using large windows for detection of phase shifts between individual sperm cells. These windows (40 consecutive variant loci; mean length, 453 kb) easily span most segmental duplications in the genome (average length, 19 kb), although the largest segmental duplications remain a potential source of error in this respect. When a genuine breakpoint occurred within a segmental duplication, there was a clear change in variant phase across the flanks of the duplication, although identification of the precise location of the breakpoint presented challenges owing to inconsistent variant detection between sperm cells within the duplication. In these cases, the unambiguous designation of a breakpoint region could encompass a large fraction of the duplicated region, and variants within this region remain unphased. However, for each sperm cell, the number of such unphased variants was <0.5% of those that were phased.
In this study, array-hybridization and genome sequence data from shallow sequencing of 11 sperm cells was used to phase 94% of the known SNPs in a genome. Given the combination of data sets that were used, it is pertinent to discuss the optimal combination for near complete phasing of SNPs in a human genome. The first question is whether the sequence data from the sperm cells (totaling 31×) could be sufficient to both identify and phase the SNPs, thereby dispensing with the need to conduct conventional deep sequencing for variant discovery. The data suggest that, at present, our MDA products from single sperm cells are too biased for comprehensive variant discovery across the genome. As illustrated in Figure 3B, the combined data from 11 sperm cells provides more than 2× coverage for only 85% of the known SNPs. Although variant discovery in a haploid genome requires less sequence depth than for a diploid genome, even a threshold of only 3× coverage would miss at least 15% of the HuRef SNPs. It is possible that shallower sequencing of more independent sperm cells could reduce this deficit, but that remains to be tested.
The second question relates to the need for array genotyping. Use of bead arrays is a convenient means to generate a low-resolution map of phased SNPs, as demonstrated here for sperm cells and by Fan et al. (2011), using isolated chromosomes. In order to identify recombination breakpoints by the one-versus-all approach described here, we employed 16 sperm cells. The optimal number of cells has not yet been determined, although it is clearly dependent on the dropout rate for genotypes, which affects the resolution with which recombination breakpoints can be placed on a genome. There is clearly a potential to increase the resolution of a bead-based haplotype map by incorporating more known SNPs into the assay. However, in order to phase the rare and de novo SNPs that are often of greatest interest, it will remain necessary to sequence one or more sperm genomes.
Regarding the additional effort required for phasing a known collection of SNPs, the process (from sample collection to MDA product) can be accomplished in 14–18 h, depending on the number of single cells processed. The preparation of sperm cells, just before micromanipulation, should take <30 min. The time-intensive step is single-cell isolation via micromanipulation. An experienced operator can pick 8–10 cells per hour. After the single cells are picked, they can undergo lysis and an overnight MDA reaction (12 h). The qPCR assays typically take 2.5 h, including the set up and run time. Therefore, cells could be ready for sequencing after ∼20–25 h, depending on the number of cells processed, the operator's level of experience, and the number of qPCR assays needed.
Future efforts to reduce the amplification bias of single-cell MDA reactions will likely reduce the number of independent cells that must be sequenced by this approach. An obvious limitation is that the methodology currently cannot be applied to female genomes without the invasive surgery needed for extraction of egg cells, although recent advances in stem cell research, including cultured ovarian stem cells (White et al. 2012), may circumvent this requirement in the future. The recent development of technology for whole-genome sequencing of isolated cells has found potential applications in diverse fields from bacterial characterization to cancer biology (Chitsaz et al. 2011; Navin et al. 2011). Here, we demonstrate its practical utility for retrieving the complete haplotypes of sequenced genomes—information that is generally lost during conventional approaches to genome sequencing.
Methods
Preparation of sperm cells
Semen (0.2 mL) was diluted 1:100 in a solution of PBS-BSA (ultrapure-grade phosphate buffered saline 1× concentration; 138 mM NaCl, 2.7 mM KCl, pH 7.4 and BSA [5 mg/mL] [Sigma]), and 100-μL aliquots of the diluted sample were frozen and stored at −80oC until use. Individual aliquots were thawed on ice and centrifuged at 400g for 10 min. The supernatant was discarded, and the pellet was rinsed superficially three times with cold PBS-BSA. The pellet was resuspended in 100 μL of cold PBS-BSA solution and spun at 400g for 10 min. The supernatant was aspirated and used as a no-template control (NTC) in downstream assays. The pellet was resuspended in 100 μL of cold PBS-BSA and placed on a glass slide that was kept on ice until ready for micromanipulation.
Single cell micromanipulation
Sperm samples and cells were micromanipulated under bright field conditions with an Olympus IX70 inverted microscope (20× and 40× objectives) and a manual CellTram Oil microinjector (Eppendorf). All consumables were sterilized with shortwave (254 nm) UV crosslinking (CX-2000, UVP). Samples and glass slides were routinely replaced with cold samples and slides after 5 min of micromanipulation. Sperm cells were isolated manually using a sterile glass micropipette (Eppendorf Transfer Tip [ES]) with an inner diameter of 20 μm and deposited onto a glass slide fitted with an adhesive Press-to-Seal silicon isolator well (Invitrogen). Cells were picked from the sample well and bathed in a rinse well containing cold PBS-BSA (50 μL). For rinsing, each cell was drawn into and expelled out of the microcapillary tube approximately 10 times before final capture. Single cells were transferred to a 0.2-mL thin-walled PCR tube (Eppendorf, DNase and RNase free) in a droplet of cold PBS-BSA (3.0 μL). These isolated cells were immediately processed for MDA or flash frozen in liquid nitrogen and stored at −80oC until processing.
Multiple displacement amplification (MDA)
Single cells and controls were subjected to a modified GenomiPhi reaction (GE Healthcare). After addition of 3.5 μL cell lysis solution (400 mM KOH, 100 mM DTT, 10 mM EDTA; pH 8.0), the PCR tubes were incubated for 10 min at 65°C. The mixture then received 3.5 μL neutralization solution (800 mM TrisCl; pH 4.5), and 40 μL GenomiPhi mastermix (22.5 μL GenomiPhi Reaction Buffer, 15 μL GenomiPhi Sample Buffer, and 2.5 μL GenomiPhi Enzyme Mix, lot# 383497). The tubes were centrifuged briefly then incubated at 30°C for 4 h followed by heating at 65°C for 10 min to inactivate the DNA polymerase. The reaction products were stored at 4°C.
TaqMan loci qPCR assay
MDA products were diluted 1:200 in 1× TE buffer, and 5 μL was reacted in a final volume of 20 μL 1× PerfeCTa qPCR FastMix (Quanta Biosciences), 0.3 μM forward and reverse primers, and 0.25 μM TaqMan probe. Real-time thermal cycling conditions were as follows: 2 min at 95°C, 40 cycles (15 sec at 95°C, 60 sec at 60°C). PicoGreen DNA quantification of MDA yield was performed as directed in the GenomiPhi HY kit.
Genotyping
DNA samples (n = 16) were genotyped for 1,140,419 (1,016,423 SNPs) markers using the Illumina HumanOmni-Quad v1.0 BeadChip (Illumina Inc.) according to the manufacturer's protocol with no modifications. Briefly, 200 ng of DNA (4 μL at 50 ng/μL) was independently amplified, labeled, and hybridized to BeadChip microarrays then scanned with default settings using the Illumina iScan System (Illumina Inc.). Analysis and intrachip normalization of the resulting image files was performed using Illumina's Genome Studio (V2010.3) Genotyping Module v1.8.4 software with default parameters. Genotype calls were generated using the Illumina-provided genotype cluster definitions file (HumanOmni-Quad_v1-0_B.egt), generated using HapMap project DNA samples) with a Gencall cutoff of 0.15. The mean call rate was 50.9%. For each sample, base calls among the 238,872 loci of interest (score >0.5) were compared in pairwise combinations with the genotypes at these 238,872 loci for all other 15 samples. These multiple pairwise comparisons indicated identity, nonidentity, or ambiguity (no high-scoring call) at each locus. A custom script then considered the result of this comparison at each locus, together with the preceding 20 loci and the succeeding 20 loci, along each chromosome. This analysis highlighted loci where there was a switch from haplotype identity to nonidentity (or vice versa). When a given sample yielded a switch from haplotype identity to nonidentity in the same chromosomal region for multiple comparisons (at least 13 of 15), this putative crossover event was confirmed and boundaries refined by manual inspection of the pairwise comparisons. The location of each crossover event was therefore bounded by SNPs that represent the termini of haplotype blocks in the test sample. For 11 samples, the identity of the unobserved allele was inferred, and the building of chromosome-wide haplotype blocks employed the locations of putative crossover events. This yielded 11 copies of the two haplotype blocks for each chromosome. These six copies were then compared with each other to build a consensus sequence for each haplotype block (using a simple majority of base calls at each locus or leaving uncalled if there was no majority).
Sequencing
Unamplified genomic DNA from blood cells, or amplified DNA from sperm cells, was fragmented using a Covaris S2 instrument following the recommended conditions for generating a 300-bp peak. For DNA from blood and sperm cells X01, X45, and Y47, the fragmented DNA was end-repaired, tailed with a single ‘A’ base, and ligated to Illumina paired-end adaptors. A 12-cycle PCR was performed, and amplified material was sized using Agencourt AMPure XP beads. The completed libraries were quantified using the Agilent High Sensitivity DNA Kit for the Agilent 2100 Bioanalyzer and sequenced on the Illumina GAIIx platform. One hundred bases were sequenced from each end of the DNA fragments. Image analysis and base calling were performed using Illumina's GA Pipeline version 1.5.1. The other eight sperm cells were treated similarly, except that libraries were quantified using a KAPA Library Quantification Kit on an ABI 7900 and sequenced on the Illumina HiSeq2000 platform using version 3 flow cells and chemistry. Image analysis and base calling were performed using Illumina's Pipeline, RTA version 1.13.48.0. Sequences were aligned to the human reference genome (Build 37.1), using BWA (v. 0.5.9-r16) with q = 20. Alignments were converted to BAM files, duplicate reads identified using picard-tools-1.64, and the consensus call at 1.95 million loci of HuRef heterozygous SNPs was determined using SAMtools (v. 0.1.13) pileup, with a cutoff consensus quality value of 10. For each sample, the identity of the unobserved allele was inferred, and the building of chromosome-wide haplotype blocks employed the locations of crossover events as described for analysis of genotyping data (see above). Details of read coverage and base calling at the 1.95 million heterozygous SNP loci are listed in Supplemental Table S3.
Modeling
Prediction of the cumulative success for genotyping loci in at least one sperm cell used the formula, 1−(0.55)n where 0.55 is the average drop-out rate per sperm cell and n is the number of sperm cells. Similarly, a value of 0.66 was used for the average missing data from sperm sequencing. Prediction of cumulative success for genotyping loci in two or more samples used the binomial distribution probability with the number of successes equal to 2, the number of trials equal to 2−12, the probability of success on each trial was 0.45, and a cumulative probability of X≥2. The relationship between coverage of an ideal (nonrepetitive) genome and the number of sequenced bases used the formula, 1−(e-R) where R is the number of sequenced bases/genome length. Significance values, where reported, were derived from a two-tailed t-test.
Identification of potentially damaging mutations
The potential impact on protein function of 1.95 million heterozygous SNPs in the HuRef genome was assessed using PolyPhen2 v2.2.2 with UniProtKB/UniRef100 Release 2011_12 (14-Dec-2011) (http://genetics.bwh.harvard.edu/pph2/index.shtml) and SIFT Ensembl 63 annotation of NCBI 37 (http://sift.jcvi.org/). Functions and phenotypes that have been associated with specific genes were obtained from GeneCards V3 (http://www.genecards.org).
Data access
The sequencing data used in this study have been submitted to the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/Traces/sra/) under accession numbers SRX209560, SRX209573, SRX209575, SRX209576, SRX209577, SRX209578, SRX209579, SRX209663, SRX209677, SRX209690, SRX209785, and SRX209789.
Acknowledgments
We thank K. Beeson for help with library construction, T. Stockwell for proposing the use of haploid cells for haplophasing, and M. Adams for helpful discussions.
Author contributions: E.F.K. designed the project, analyzed the genotyping and sequencing data, and wrote the manuscript. R.G., J.Y.-G., and R.S.L. designed, conducted, and analyzed data for the whole-genome amplification experiments. C.R.M. conducted the genotyping experiments. S.W.S. and J.C.V. contributed to the design of the project and to manuscript preparation.
References
- ↵S Aretz, S Uhlhaas, R Caspari, E Mangold, C Pagenstecher, P Propping, W Friedl. 2004. Frequency and parental origin of de novo APC mutations in familial adenomatous polyposis. Eur J Hum Genet 12: 52–58.
- ↵H Chitsaz, JL Yee-Greenbaum, G Tesler, MJ Lombardo, CL Dupont, JH Badger, M Novotny, DB Rusch, LJ Fraser, NA Gormley, . 2011. Efficient de novo assembly of single-cell bacterial genomes from short-read data sets. Nat Biotechnol 29: 915–921.
- ↵DF Conrad, M Jakobsson, G Coop, X Wen, JD Wall, NA Rosenberg, JK Pritchard. 2006. A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nat Genet 38: 1251–1260.
- ↵G Coop, X Wen, C Ober, JK Pritchard, M Przeworski. 2008. High-resolution mapping of crossovers reveals extensive variation in fine-scale recombination patterns among humans. Science 319: 1395–1398.
- ↵PI de Bakker, G McVean, PC Sabeti, MM Miretti, T Green, J Marchini, X Ke, AJ Monsuur, P Whittaker, M Delgado, . 2006. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat Genet 38: 1166–1172.
- ↵FB Dean, JR Nelson, TL Giesler, RS Lasken. 2001. Rapid amplification of plasmid and phage DNA using phi29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res 11: 1095–1099.
- ↵FB Dean, S Hosono, L Fang, X Wu, AF Faruqi, P Bray-Ward, Z Sun, Q Zong, Y Du, J Du, . 2002. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci 99: 5261–5266.
- ↵JA Douglas, M Boehnke, E Gillanders, JM Trent, SB Gruber. 2001. Experimentally-derived haplotypes substantially increase the efficiency of linkage disequilibrium studies. Nat Genet 28: 361–364.
- ↵CM Drysdale, DW McGraw, CB Stack, JC Stephens, RS Judson, K Nandabalan, K Arnold, G Ruano, SB Liggett. 2000. Complex promoter and coding region β2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness. Proc Natl Acad Sci 97: 10483–10488.
- ↵HC Fan, J Wang, A Potanina, SR Quake. 2011. Whole-genome molecular haplotyping of single cells. Nat Biotechnol 29: 51–57.
- ↵RL Glaser, W Jiang, SA Boyadjiev, AK Tran, AA Zachary, L Van Maldergem, D Johnson, S Walsh, M Oldridge, SA Wall, . 2000. Paternal origin of FGFR2 mutations in sporadic cases of Crouzon syndrome and Pfeiffer syndrome. Am J Hum Genet 66: 768–777.
- ↵RE Green, J Krause, AW Briggs, T Maricic, U Stenzel, M Kircher, N Patterson, H Li, W Zhai, MH Fritz, . 2010. A draft sequence of the Neandertal genome. Science 328: 710–722.
- ↵K Holloway, VE Lawson, AJ Jeffreys. 2006. Allelic recombination and de novo deletions in sperm in the human β-globin gene region. Hum Mol Genet 15: 1099–1111.
- ↵R Hubert, M MacDonald, J Gusella, N Arnheim. 1994. High resolution localization of recombination hot spots using sperm typing. Nat Genet 7: 420–424.
- ↵AJ Jeffreys, CA May. 2004. Intense and highly localized gene conversion activity in human meiotic crossover hot spots. Nat Genet 36: 151–156.
- ↵Z Jiang, X Zhang, R Deka, L Jin. 2005. Genome amplification of single sperm using multiple displacement amplification. Nucleic Acids Res 33: e91.
- ↵JO Kitzman, AP Mackenzie, A Adey, JB Hiatt, RP Patwardhan, PH Sudmant, SB Ng, C Alkan, R Qiu, EE Eichler, . 2011. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat Biotechnol 29: 59–63.
- ↵S Levy, G Sutton, PC Ng, L Feuk, AL Halpern, BP Walenz, N Axelrod, J Huang, EF Kirkness, G Denisov, . 2007. The diploid genome sequence of an individual human. PLoS Biol 5: e254.
- ↵L Ma, Y Xiao, H Huang, Q Wang, W Rao, Y Feng, K Zhang, Q Song. 2010. Direct determination of molecular haplotypes by chromosome microdissection. Nat Methods 7: 299–301.
- ↵DG MacArthur, S Balasubramanian, A Frankish, N Huang, J Morris, K Walter, L Jostins, L Habegger, JK Pickrell, SB Montgomery, . 2012. A systematic survey of loss-of-function variants in human protein-coding genes. Science 335: 823–828.
- ↵KJ McKernan, HE Peckham, GL Costa, SF McLaughlin, Y Fu, EF Tsung, CR Clouser, C Duncan, JK Ichikawa, CC Lee, . 2009. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res 19: 1527–1541.
- ↵HM McLaughlin, R Sakaguchi, C Liu, T Igarashi, D Pehlivan, K Chu, R Iyer, P Cruz, PF Cherukuri, NF Hansen, . 2010. Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am J Hum Genet 87: 560–566.
- ↵N Navin, J Kendall, J Troge, P Andrews, L Rodgers, J McIndoo, K Cook, A Stepansky, D Levy, D Esposito, . 2011. Tumour evolution inferred by single-cell sequencing. Nature 472: 90–94.
- ↵SB Ng, KJ Buckingham, C Lee, AW Bigham, HK Tabor, KM Dent, CD Huff, PT Shannon, EW Jabs, DA Nickerson, . 2010. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 42: 30–35.
- ↵BA Peters, BG Kermani, AB Sparks, O Alferov, P Hong, A Alexeev, Y Jiang, F Dahl, YT Tang, J Haas, . 2012. Accurate whole-genome sequencing and haplotyping from 10 to 20 human cells. Nature 487: 190–195.
- ↵EW Petersdorf, M Malkki, TA Gooley, PJ Martin, Z Guo. 2007. MHC haplotype matching for unrelated hematopoietic cell transplantation. PLoS Med 4: e8.
- ↵JC Roach, G Glusman, AF Smit, CD Huff, R Hubley, PT Shannon, L Rowen, KP Pant, N Goodman, M Bamshad, . 2010. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 328: 636–639.
- ↵EK Suk, GK McEwen, J Duitama, K Nowick, S Schulz, S Palczewski, S Schreiber, DT Holloway, S McLaughlin, H Peckham, . 2011. A comprehensively molecular haplotype-resolved genome of a European individual. Genome Res 21: 1672–1685.
- ↵B Tycko. 2010. Allele-specific DNA methylation: Beyond imprinting. Hum Mol Genet 19: R210–R220.
- ↵J Wang, HC Fan, B Behr, SR Quake. 2012. Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell 150: 402–412.
- ↵YA White, DC Woods, Y Takai, O Ishihara, H Seki, JL Tilly. 2012. Oocyte formation by mitotically active germ cells purified from ovaries of reproductive-age women. Nat Med 18: 413–421.
- ↵H Yang, X Chen, WH Wong. 2011. Completely phased genome sequencing through chromosome sorting. Proc Natl Acad Sci 108: 12–17.
- ↵K Zhang, J Zhu, J Shendure, GJ Porreca, JD Aach, RD Mitra, GM Church. 2006. Long-range polony haplotyping of individual human chromosome molecules. Nat Genet 38: 382–387.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.144600.112.