
Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes
- Leen Abu-Safieh1,15,
- May Alrashed1,2,3,15,
- Shamsa Anazi1,
- Hisham Alkuraya1,4,
- Arif O. Khan5,
- Mohammed Al-Owain6,7,
- Jawahir Al-Zahrani1,
- Lama Al-Abdi1,
- Mais Hashem1,
- Salwa Al-Tarimi1,
- Mohammed-Adeeb Sebai1,
- Ahmed Shamia1,
- Mohamed D. Ray-zack1,
- Malik Nassan1,
- Zuhair N. Al-Hassnan6,7,
- Zuhair Rahbeeni6,
- Saad Waheeb8,9,
- Abdullah Alkharashi10,
- Emad Abboud11,
- Selwa A.F. Al-Hazzaa1,12,13 and
- Fowzan S. Alkuraya1,7,14,16
- 1Department of Genetics, King Faisal Specialist Hospital and Research Center, Riyadh 11211, Saudi Arabia;
- 2UCL Institute of Ophthalmology, Department of Genetics, London EC1V 9EL, United Kingdom;
- 3Department of Clinical Laboratory Sciences, College of Applied Medical Sciences, King Saud University, Riyadh 11573, Saudi Arabia;
- 4Department of Ophthalmology, College of Medicine, Imam Muhammed Bin Saud Islamic University, Riyadh 13317, Saudi Arabia;
- 5Department of Pediatrics, King Khaled Eye Specialist Hospital, Riyadh 11462, Saudi Arabia;
- 6Department of Medical Genetics, King Faisal Specialist Hospital and Research Center, Riyadh 11211, Saudi Arabia;
- 7Department of Anatomy and Cell Biology, College of Medicine, Alfaisal University, Riyadh 11533, Saudi Arabia;
- 8Department of Ophthalmology, King Faisal Specialist Hospital and Research Center, Jeddah 21499, Saudi Arabia;
- 9Department of Ophthalmology, College of Medicine, King Abdulaziz University, Jeddah 21352, Saudi Arabia;
- 10Department of Ophthalmology, College of Medicine, King Saud University, Riyadh 11573, Saudi Arabia;
- 11Department of Retina, King Khaled Eye Specialist Hospital, Riyadh 11462, Saudi Arabia;
- 12Department of Ophthalmology, King Faisal Specialist Hospital and Research Center, Riyadh 11211, Saudi Arabia;
- 13Department of Ophthalmology, College of Medicine, Alfaisal University, Riyadh 11533, Saudi Arabia;
- 14Department of Pediatrics, King Khalid University Hospital and College of Medicine, King Saud University, Riyadh 11573, Saudi Arabia
-
↵15 These authors contributed equally to this work.
Abstract
Retinal dystrophy (RD) is a heterogeneous group of hereditary diseases caused by loss of photoreceptor function and contributes significantly to the etiology of blindness globally but especially in the industrialized world. The extreme locus and allelic heterogeneity of these disorders poses a major diagnostic challenge and often impedes the ability to provide a molecular diagnosis that can inform counseling and gene-specific treatment strategies. In a large cohort of nearly 150 RD families, we used genomic approaches in the form of autozygome-guided mutation analysis and exome sequencing to identify the likely causative genetic lesion in the majority of cases. Additionally, our study revealed six novel candidate disease genes (C21orf2, EMC1, KIAA1549, GPR125, ACBD5, and DTHD1), two of which (ACBD5 and DTHD1) were observed in the context of syndromic forms of RD that are described for the first time.
Deprivation of visual perception is a major form of morbidity worldwide with a wide array of causes that cover the entire spectrum from primarily environmental to primarily genetic. Representing the Mendelian end of the spectrum, retinal dystrophy (RD) is a vast group of blinding diseases that are characterized by loss of photoreceptor function, usually due to mono- or biallelic mutations in an expansive list of genes (Wright et al. 2010). Collectively, RD is a major cause of blindness, particularly in industrialized countries where infectious causes are less common and where treatable blinding diseases such as cataract and glaucoma receive adequate management.
Clinically, RD can take various forms, retinitis pigmentosa (RP) being the most common (Buch et al. 2004). RP patients typically present with a predominantly rod dysfunction, which manifests as night blindness, progressively worsening peripheral vision, and typical fundus appearance (Ho 2003; Hamel 2006). In cone dystrophies, it is the cone photoreceptors that are primarily involved, causing a substantial decrease in visual acuity and photophobia (Hamel 2007). In both classes, the other photoreceptor subtype is inevitably affected as the disease progresses, hence the terms rod-cone and cone-rod dystrophy, although the mechanism for this sympathetic cell loss is poorly understood. When severe RD is congenital or early-infantile in onset, it is usually referred to as Leber congenital amaurosis (LCA). Interestingly, the clinical boundaries between these subclasses are blurred by the increasing appreciation of the marked phenotypic variability that is associated with mutations in a large number of RD genes (Daiger et al. 2007).
The remarkable genetic heterogeneity (179 genes as of January 2012; https://sph.uth.tmc.edu/Retnet/sum-dis.htm) and the poor predictive value of the clinical assessment to the specific genetic etiology (at least in nonsyndromic cases) make it extremely challenging to offer a molecular diagnosis to these patients (Koenekoop et al. 2007). Thus, of all Mendelian disorders, this is one disease category where most patients remain unaware of their underlying causative mutation even though such information is critical for informed genetic counseling that aims at prevention and expansion of available reproductive options. This is compounded by estimates that, even if all known RD were to be sequenced in a given patient, the yield is probably 50% (Farrar et al. 2002; Hartong et al. 2006; den Hollander et al. 2008). An additional value in securing a molecular diagnosis lies in the recent progress in gene therapy, which has prompted many RD patients to seek to determine their mutation status in order to know whether they are eligible for these gene-specific treatment protocols (Maguire et al. 2008). In addition, certain classes of mutations have been found to be amenable to treatment in other diseases, e.g., nonsense mutations, which offers hope that RD patients with such mutations could similarly benefit from such innovative strategies, but this will require prior knowledge of the underlying genetic defect (Kerem et al. 2008).
Research in the genetics of RD has greatly improved our understanding of the molecular machinery that enables the retina to play a critical role in the perception of visual stimuli (Inglehearn 1998). While some of the genes were predicted to cause RD based on established physiological roles of the protein they encode, e.g., phototransduction genes, it came as a surprise that almost one in four RD genes plays a role in the photoreceptor cilium (Adams et al. 2007; Wright et al. 2010). Moreover, many genes were completely unsuspected, e.g., pre-mRNA splicing genes, and the function of some remains unknown (Vithana et al. 2001; Faustino and Cooper 2003; Wright et al. 2010). Indeed, the increasing pace of discovery of RD genes over the past few years has widened the gap between our knowledge of the genetic architecture of RD and its functional context.
In this study, we aimed to investigate the utility of genomic approaches in the study of RD genetics. Specifically, we implemented autozygome analysis (Woods et al. 2006; Alkuraya 2010) and exome sequencing in a large cohort of simplex and multiplex patients with different clinical RD subtypes. In addition to providing the most comprehensive analysis to date on the actual contribution of known RD genes to the overall mutation pool, our study reveals six novel RD genes, including two involved in novel syndromic forms of RD, and suggests a framework for mutation identification in these patients.
Results
Clinical characteristics
For the period January 2008–September 2010, 149 eligible families were enrolled representing the three major clinical subtypes of RD, i.e., RP, LCA, and cone-rod dystrophy, but other less common phenotypes such as achromatopsia were also represented. With the exception of cone-dystrophy with supranormal rod response patients in whom KCNV2 was directly sequenced, it was not possible to predict the genetic defect based on the phenotype provided, and they were processed as per the workflow outlined in Figure 1. As expected, RP accounted for the majority of patients (∼55%). The majority of cases were multiplex (60%), and there was remarkable clinical homogeneity among affected members of a given family. Family arRP-FD02 (Supplemental Fig. S1) is worth highlighting in this regard. In this family of four affected members with RP (two siblings on either side of a first cousin relationship), two siblings on one side had additional features of Bardet-Bied syndrome (BBS). Although the two siblings in the other branch lacked any additional BBS feature, one of these two siblings had an infant with BBS in the course of the study. As a result, and based on our experience of rare cases in which BBS presents as nonsyndromic RP (Abu Safieh et al. 2010, 2012), we enrolled this family in this study. However, autozygome analysis and exome sequencing confirmed that the apparent clinical heterogeneity in this family was, in fact, the result of independent segregation of two different diseases: RP secondary to RP1 mutation and BBS due to BBS1 mutation. In Family arRP-F026, which was enrolled as nonsyndromic RP, the finding of a BBS4 mutation prompted us to recall the family for careful phenotyping, and the result indicated that the phenotype should have been labeled as BBS. On the other hand, Family CR-F008, in which we identified a novel MKS1 mutation, was found upon rephenotyping to have no syndromic features. Thus, this appears to be the first report of MKS1 mutation causing nonsyndromic cone-rod dystrophy.
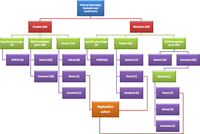
Workflow of the study.
Autozygome-guided gene sequencing
This was pursued in both simplex and multiplex cases because of our past experience of the very high rate of homozygous mutations even in the absence of consanguinity or positive family history (Aldahmesh et al. 2009). The yield was only slightly lower in simplex compared with multiplex cases (42% vs. 52%) (Fig. 2). On average, four genes were sequenced per case (range 1–11). The average number of amplicons per case was 200, with an average cost of $3000. The results of autozygome-guided targeted RD gene sequencing are summarized in Table 1 and Supplemental Table S1 (solved cases) and Supplemental Table S4 (unsolved cases).
Summary of the results of autozygome-guided sequencing among simplex and multiplex cases
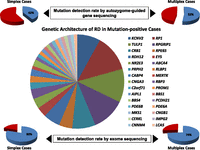
Central pie chart summarizes the contribution of various genes to the overall mutational pool among RD patients in the current study. Pie charts in the upper panel show the percentage of mutation-positive cases among simplex and multiplex cases using the autozygome-guided gene sequencing approach. Pie charts in the lower panel show the percentage of mutation-positive cases among simplex and multiplex cases using the exome sequencing approach. Please note the percentages in these charts do not take into account the novel candidate genes identified in this study.
Exome sequencing for mutation detection in RD
The first group of exomes comprised randomly selected 10 simplex and 23 multiplex cases to investigate the yield of this method in sporadic and familial cases of this extremely heterogeneous disorder. Of the 10 simplex cases, eight (80%) were found to harbor pathogenic mutations in known RD genes. A similar ratio (17/23, 74%) was observed in multiplex cases (Fig. 2). In all these cases, the pathogenic mutation was always homozygous (Table 2; Supplemental Table S2). By checking these mutations against the autozygome data we had prepared for this purpose, it was clear that all these changes could have been identified by autozygome-guided targeted RD gene sequencing because they either resided within one of the largest four runs of homozygosity (ROH) (in simplex cases) (Supplemental Table S5) or an ROH that was exclusively shared by the affected members of a given family (in multiplex cases). However, there is a significant time and money difference in favor of exome sequencing. Indeed, a typical turnaround time for identifying the causative mutation by exome was 8 wk compared to 15 wk for autozygome-guided analysis. The cost of $1500 per exome was also lower than the $3000 per case solved using the autozygome-guided approach. Furthermore, exome sequencing revealed mutations in two multiplex cases that were missed by the autozygome-guided approach because these mutations were in homozygous regions which were shared with an unaffected individual, yet the apparently shared ROH was clearly IBS (identical-by-state) (Supplemental Fig. S2).
Summary of the mutations identified on exome sequencing in known RD genes
Exome as a discovery tool for novel RD genes
The first group of exomes (see above) also revealed two novel candidate genes for RD (Tables 3, 4). In Family sRP-001 (multiplex), a truncating mutation in LGALS9B encoding Galectin-9B protein A2 and a missense mutation in EMC1 were the only variants that survived filtration (Table 3; Supplemental Table S3; Supplemental Fig. S4). However, direct full sequencing of both genes in the 11 patients, out of 210 total RD patients in the replication cohort, whose autozygome overlapped with at least one of the two genes revealed one patient who is homozygous for the same mutation in EMC1 but no additional alleles of LGALS9B. Importantly, the same LGALS9B truncation was later found at high frequency on direct sequencing of additional ethnically matched controls. On the other hand, the novel EMC1 variant was absent from 380 Saudi controls by direct sequencing and in the Exome Variant Server and is highly conserved across species. Taken together, these data strongly support the candidacy of EMC1 in the pathogenicity of RP in the two individuals who are homozygous for that variant, although very little is known about this gene. In Family CR-F024 (simplex), a truncating mutation in the hypothetical protein coding gene C21orf2 was the only variant that survived the various filters. As with the EMC1, this was absent in 160 Saudis by exome sequencing and in 190 Saudi controls by direct sequencing and in the Exome Variant Server (EVS). More importantly, by direct sequencing in the seven individuals in the replication cohort whose autozygome pattern overlapped with this gene, one patient was found to be homozygous for a splicing mutation (NM_004928.2:c.545 + 1G>A) that fully abolishes the donor site in silico, which was absent in the panels of controls described above. Thus, C21orf2 is a compelling candidate in the pathogenesis of cone-rod dystrophy in these two individuals.
Summary of the filtration strategy used in patients in whom exome sequencing failed to identify a pathogenic mutation in any of the known RD genes
Summary of the novel candidate RD genes based on exome sequencing
The second group of exomes (n = 12) was enriched for novel gene discovery because all known RD genes had been excluded in these multiplex families by the autozygome-guided sequencing approach. As mentioned above, despite this enrichment, 3/12 harbored mutations in known RD genes that were missed for various reasons (two because of IBS [identical by state] being confused with IBD [identical by descent], and one because of a highly unusual pedigree structure; see Supplemental Figs. S1, S2). A novel candidate gene was identified in each of four additional families (Tables 3, 4). In Family sRP-022 (multiplex), a novel missense mutation was identified in an absolutely conserved residue of the sixth transmembrane helix of G protein-coupled receptor 125 (GPR125). Of note, mutations in several other G protein-coupled receptors are known to cause RD (Dryja et al. 1990; Morimura et al. 1999; Ebermann et al. 2009; Hilgert et al. 2009). In Family sRP-004 (multiplex), a novel truncating variant was identified in KIAA1549 as the only variant that remained after applying the various filters. Virtually nothing is known about this hypothetical protein-coding gene. However, it is among the top 4% of genes enriched for CRX-binding sites in a data set used to identify MAK as a novel RD gene (Ozgul et al. 2011; Tucker et al. 2011). Additionally, while the reduction of MAK representation in retina of mice with loss of photoreceptors was ∼26%, that of KIAA1549 was ∼88%, suggesting specific loss of this gene in photoreceptor degeneration (Ozgul et al. 2011). As with the GPR125 variant, this truncating variant was not encountered in any of 160 Saudi exomes or 190 Saudi controls by direct sequencing. Both were also absent in the EVS. These data support the candidacy of these two genes as novel RD genes. However, direct sequencing of both in the 27 patients in the replication cohort whose autozygome overlapped with either of these two genes revealed no additional mutations.
Two families displayed an apparently novel syndromic form of RD. In Family LCA-F045, LCA segregated with a mild-moderate form of nonspecific muscle dystrophy. By only considering the exome variants within the three exclusively shared ROH among the affected members, we uncovered a single nucleotide substitution that abolishes the first methionine residue of DTHD1 encoding death domain-containing protein 1 (Supplemental Fig. S3). Western blot analysis showed a greater than fourfold reduction in the abundance of the mutant protein compared to control (Fig. 3). Virtually nothing is known about this hypothetical protein other than that it contains a death domain. However, the identification of this as the only variant within the shared ROH, its effect on the protein, its full segregation with the phenotype in this extended family, and its absence in a large number of controls strongly support its candidacy as the causal gene for this apparently novel LCA/muscular dystrophy syndrome. In Family CRSPW, an apparently novel association between cone-rod dystrophy and psychomotor delay associated with significant white matter involvement was observed. A single novel variant was identified in the single ROH that is exclusively shared by the three affected siblings (Supplemental Fig. S3). The variant is predicted to abolish a consensus splice donor site in ACBD5. Indeed, RT-PCR confirmed the resulting aberrant transcript that predicts frameshift and premature truncation. However, despite lack of evidence of nonsense-mediated decay (NMD), Western blot analysis showed no evidence of the expected smaller band as a result of the truncation (the normal band was completely absent), even though the antibody targets the N terminus part of the protein (Fig. 3). Thus, it appears that the mutation causes severe instability of the protein and can be considered as a null allele. Reassuringly, as with the DTHD1 variant, this variant was absent in 160 Saudi exomes, 190 Saudi controls by direct sequencing, and EVS. ACBD5 encodes acyl-coenzyme A binding domain-containing protein 5, so it remains to be seen, as is the case with the above-mentioned novel candidates, how deficiency of this protein may have caused this phenotype.
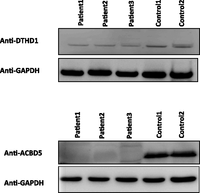
Western blot analysis of DTHD1 and ACBD5 in two families representing novel syndromic forms of RD. Fourfold reduction in the DTHD1 intensity in the patients compared to control and near-absence of the band corresponding to ACBD5 among patients can be seen. GAPDH is used for a loading control.
In the remaining five families, no novel variants were identified after applying the various filters. Interestingly, linkage analysis in four of these families showed one single peak each (Chr17: 3,745,860-7,201,753 in LCA-F037, Chr3: 83,157,375-107,875,119 in arRP-F048, Chr8: 75,000,000-110,000,000 in arRP-F074, and Chr7:105,000,000-147,000,000 in arRP-F077) (Supplemental Table S6). In the remaining one family, we could not narrow the search to a single locus, so several ROHs were used in the filtration of the data.
Discussion
The extreme genetic heterogeneity of RD and the often poor predictive power of clinical assessment in determining the underlying genetic defect have severely hampered the ability of these patients to receive specific genetic diagnosis that can be the basis of informed genetic counseling and gene-specific therapy (Berger et al. 2010; den Hollander et al. 2010). Some attempts have been made to reduce this diagnostic challenge. In one approach, all previously reported mutations in RD genes were captured on a genotyping chip (Koenekoop et al. 2007). Unfortunately, the extreme allelic heterogeneity limits the usefulness of this method. The resequencing chip theoretically circumvents this limitation, but the prerequisite step of amplifying all known RD genes represents a major challenge (Booij et al. 2011). We and others have shown that the autozygome approach can be very effective in guiding the mutation analysis (Aldahmesh et al. 2009; Pomares et al. 2010). Interestingly, this approach was also used successfully in populations where consanguinity is uncommon (Hildebrandt et al. 2009; Collin et al. 2011; Hagiwara et al. 2011; Schuurs-Hoeijmakers et al. 2011). However, this approach has its limitations. Only homoallelic mutations are identified by this method, so compound heterozygosity for recessive RD genes, heterozygosity for dominant RD genes, and hemizygosity for X-linked RD genes are missed. More importantly, novel genes can only be identified in favorable pedigrees, i.e., those in which enough crossing-overs reduce the haplotype sharing to a level that allows a relatively small ROH to be identified that is exclusively shared by the affected members. Indeed, lining up the autozygome pattern of unrelated individuals, which has been used to identify disease loci for autosomal recessive traits in the past as a way to circumvent the limited informativeness of any given family, is largely inapplicable, given the remarkable locus heterogeneity of RD. Finally, as we show in this study, the distinction of IBS and IBD can be challenging (Alkuraya 2012).
Next-generation sequencing allows massively parallel sequencing at an unprecedented scale both in throughput and cost and has recently been used on a smaller scale in the study of retinal dystrophy genetics (Audo et al. 2012; Neveling et al. 2012). Exome sequencing is one of its applications where the protein-coding exons of all known genes can be captured, followed by high-throughput sequencing. Although deep intronic and noncoding regulatory sequence mutations are not covered by this method, we hypothesized that it still lends itself as a powerful genomic tool to at once identify mutations in known RD genes and identify novel RD genes, and we set out to investigate its utility both in isolation and in combination with the autozygome approach.
Our data show autozygome-guided sequence analysis of known RD genes is applicable to both multiplex and simplex cases, which suggests that, even in simplex cases, autosomal recessive RD is the commonest form, at least in our population that is characterized by a high rate of consanguinity. Although a few founder mutations were identified, we find that, similar to our experience with other genetically heterogeneous conditions, there is marked allelic and locus heterogeneity in our population, even within the same tribe. However, we caution against the overinterpretation of this phenomenon as being indicative of high population genetic diversity akin to what is observed in Africa, without empirical population genetic data, which still do not exist for Arabia.
An important yet largely unanswered question is how much the current list of RD genes contributes to the overall genetic architecture of this disease. Only estimates are available because, until recently, the only way to empirically test this was through the PCR amplification of all RD genes, an extremely challenging task. By performing exome sequencing on randomly selected multiplex and simplex cases, we were able to show that the genes identified as of January 2012 account for 74%–80% of the overall mutation pool in our population. Interestingly, all mutations identified by exome sequencing of simplex cases were homoallelic even though hemizygous X-linked, and compound heterozygous mutations in all known RD genes were equally likely to be identified. Indeed, the comparable yield of unselected exomes in simplex and multiplex cases argues against a major contribution of X-linked RD genes in simplex cases in our population. It is unclear how applicable this result is to more outbred populations, although evidence suggests that many sporadic patients in those populations also represent autosomal recessive inheritance (Avila-Fernandez et al. 2010; Iwanami et al. 2012). Another important result from our study is that our exome sequencing data make it unlikely that any additional novel gene will account for a substantial fraction of the remaining cases (see below).
As predicted, in addition to revealing most mutations in known RD genes, exome sequencing was a useful discovery tool as well. We and others have previously demonstrated the power of exome sequencing in revealing novel disease genes based on simplex cases (Gilissen et al. 2010; Aldahmesh et al. 2011; Shaheen et al. 2011). Our data expand the disease phenotypes for which simplex cases can be used to identify novel disease genes to also include RD. Unfortunately, the very low contribution of most RD genes to the overall mutation pool makes it challenging to identify additional pathogenic alleles in the candidate genes we identified in this study, so they remain interesting candidates pending independent verification by future studies (at least in the four for which no additional mutation-positive patients were identified in the replication cohort).
Many syndromes are known to involve the RD phenotype (Ayuso et al. 1995). However, we are not aware of any previously described association between LCA and muscular dystrophy or between cone-dystrophy and severe white matter disease. Thus, we believe these are two novel syndromic forms of RD. In both families, compelling loss of function alleles were identified (DTHD1 and ACBD5), but additional work is needed to explore the presumed causal link mechanistically. These families were part of a collection we tried to enrich for novel RD genes. However, we show how pitfalls in homozygosity scan caused false negative results in three of the 11 families. In fact, the 13.3-Mb IBS that caused confusion in the analysis of Family arRP-F069 is the largest IBS that we are aware of (Supplemental Fig. S2; Alkuraya 2012). Thus, it is possible that the higher yield of exome compared to autozygome-guided analysis can be, at least in part, caused by occasional pitfalls in homozygosity scan. Overall, we show that exome sequencing was superior to the autozygome-guided approach, and although the latter can be very helpful in lending credence to novel disease genes, it does not appear necessary in interpreting exome variants in known RD genes.
In summary, in this largest comprehensive genomic study of RD patients to date, we show that genomic tools are very useful in identifying the underlying genetic lesion. Exome sequencing in particular appears to be an attractive first-line test without prior enrichment for known RD genes, especially with its constantly decreasing cost. The novel disease genes we identified require validation in independent patient cohorts. Similar studies on outbred populations will be needed to explore potential differences in the genetic architecture of RD compared to what we presented in this study.
Methods
Human subjects
Patients with RP, LCA, and cone-rod dystrophy were actively recruited regardless of their age or family history through a wide network of ophthalmologists that covers all regions of Saudi Arabia for the period January 2008 to September 2010. Patients recruited between September 2010 and May 2012 were only used as a “replication cohort” for the purpose of identifying additional mutations in the novel candidate genes we may identify in the main cohort. Assignment to a specific clinical subtype was based on clinical and, in selected cases, electrophysiological assessment. Syndromic patients were only considered further if they did not fit the clinical description of a known syndrome, e.g., Usher, Bardet-Biedl, Alstrom, and Joubert syndrome (these patients were recruited for other projects). Pedigrees were drawn for all recruited patients, and an effort was made to enroll additional affected relatives when present. Whenever possible, we enrolled parents and unaffected siblings for segregation analysis. All subjects signed an IRB-approved written informed consent (RAC# 2070 023), followed by venous blood sampling in EDTA tubes. For selected patients, we also obtained blood samples in sodium heparin tubes followed by establishment of EBV-transformed lymphoblast cell lines for RNA and protein studies.
Workflow
Figure 1 summarizes the algorithm we implemented in the study which is described below in detail.
Autozygome analysis
Genotyping was performed on an Affymetrix Axiom or Affymetrix 250K SNP chip platform following the manufacturer's instructions on the index only (in simplex cases) and the entire sibship, when possible (in multiplex cases). Autozygome analysis was performed using Genotyping Console (Affymetrix) or autoSNPa as described before (Carr et al. 2006). In simplex cases, we only considered the four largest runs of homozygosity (ROH) initially, but if negative, we expanded our search to all ROHs that are >2 Mb in size. In multiplex cases, we considered all ROHs that are exclusively shared by the affected members of a given sibship. All RD genes within an ROH were sequenced even when they appeared incompatible with the specific phenotype or pattern of inheritance to account for the known phenotypic variability of mutations in RD genes and the dual inheritance pattern for some of them. Twelve out of 30 multiplex cases in which autozygome-guided targeted RD gene sequencing failed to identify the causative mutation were processed for exome sequencing.
Exome sequencing and analysis
Two groups of samples were processed for exome sequencing. The first group represents randomly selected simplex (10) and multiplex (23) cases. The second group represents samples in which autozygome-guided targeted RD gene sequencing failed to identify the causative mutation by the first freeze point (12 out of 30) (Fig. 1). The aim of the first group was to investigate the utility of exome sequencing as a first-pass diagnostic test in RD, whereas the aim was to enrich for novel RD genes in the second group. Exome capture was performed using the TruSeq Exome Enrichment kit (Illumina) following the manufacturer's protocol. Samples were prepared as an Illumina sequencing library, and in the second step, the sequencing libraries were enriched for the desired target using the Illumina Exome Enrichment protocol. The captured libraries were sequenced using an Illumina HiSeq2000 Sequencer. The reads were mapped against UCSC hg19 (http://genome.ucsc.edu/) by BWA (http://bio-bwa.sourceforge.net/). The SNPs and indels were detected by SAMtools (http://samtools.sourceforge.net/). For subsequent analysis, we always started by checking all genes reported to cause RD until January 2012. We considered homozygous, heterozygous, hemizygous, and compound heterozygous changes in these genes that are likely to be pathogenic, i.e., coding (excluding synonymous unless they affect splice site) or splice-site variants that are not present in 160 in-house Saudi exomes. It is important to mention that we manually checked all dbSNP variants in these genes against the Human Genome Mutation Database since a lot of previously reported pathogenic mutations are listed in dbSNP. Only when no such changes are identified did we proceed with the analysis of sequence variants following the filtration scheme outlined in Table 3 and Supplemental Figure S4. The autozygome filter refers to variants present within the four largest blocks of homozygosity in simplex cases and all blocks of shared homozygosity in multiplex cases.
Replication analysis of novel candidate genes
Novel candidate genes were fully sequenced in the “replication cohort” in search of additional alleles using standard PCR and Sanger sequencing. We specifically sequenced patients whose autozygome overlapped with any of these novel candidates.
RT-PCR and immunoblotting
Splice-site mutations were checked for potential effect on splicing in silico. Whenever possible, mutations that are likely to affect splicing were verified on RT-PCR using custom-made primers and lymphoblast-derived RNA as a template. Truncating mutations in novel genes were verified whenever possible by Western blot analysis using commercially available antibodies and lymphoblast-derived protein as the target and following standard protocols.
Data access
All novel sequence variants in known RD genes as well as those in the novel candidate genes that we report in this study have been submitted to the Leiden Open Variation Database (http://grenada.lumc.nl/LOVD2/eye/variants.php) under the following IDs: ABCA4_00014, ACBD5_00001, C21orf2_00001, C2orf71_00006, CDHR1_00001, CERKL_00001, CNNM4_00001, CRB1_00038, CRB1_00037, DTHD1_00001, EMC1_00001, EYS_00006, GPR125_00001, KIAA1549_00001, LCA5_00001, PRPH2_00003, RDH12_00002.
Acknowledgments
We thank the patients and their families for their enthusiastic participation. We thank the Genotyping and Sequencing Core Facilities at KFSHRC for their technical help. This work was supported by KACST Grant 08MED497-20 (F.S.A.), DHFMR Collaborative Research Grant (F.S.A.), and PSCDR Research Grant (F.S.A.).
Footnotes
-
↵16 Corresponding author
E-mail falkuraya{at}kfshrc.edu.sa
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.144105.112.
- Received June 3, 2012.
- Accepted October 4, 2012.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








