
Abstract
Prokaryotes, due to their moderate complexity, are particularly amenable to the comprehensive identification of the protein repertoire expressed under different conditions. We applied a generic strategy to identify a complete expressed prokaryotic proteome, which is based on the analysis of RNA and proteins extracted from matched samples. Saturated transcriptome profiling by RNA-seq provided an endpoint estimate of the protein-coding genes expressed under two conditions which mimic the interaction of Bartonella henselae with its mammalian host. Directed shotgun proteomics experiments were carried out on four subcellular fractions. By specifically targeting proteins which are short, basic, low abundant, and membrane localized, we could eliminate their initial underrepresentation compared to the estimated endpoint. A total of 1250 proteins were identified with an estimated false discovery rate below 1%. This represents 85% of all distinct annotated proteins and ∼90% of the expressed protein-coding genes. Genes that were detected at the transcript but not protein level, were found to be highly enriched in several genomic islands. Furthermore, genes that lacked an ortholog and a functional annotation were not detected at the protein level; these may represent examples of overprediction in genome annotations. A dramatic membrane proteome reorganization was observed, including differential regulation of autotransporters, adhesins, and hemin binding proteins. Particularly noteworthy was the complete membrane proteome coverage, which included expression of all members of the VirB/D4 type IV secretion system, a key virulence factor.
A major goal of the post-genome era is to understand how expression of the functional elements encoded by a genome is orchestrated to allow an organism to develop and adapt to life under varying conditions. Transcriptomics and proteomics technologies both provide important and complementary insights: The former allow researchers to generate global quantitative gene expression profiles and to study gene regulatory aspects like the impact of short RNAs. However, due to the varying correlation of transcriptomics and proteomics data reported in the literature (de Godoy et al. 2008; de Sousa Abreu et al. 2009; Maier et al. 2011; Marguerat et al. 2012), the direct measurement of protein expression levels is often desirable. For certain aspects, proteomics data can provide more informative and accurate data, as it reflects the effects of other important regulatory processes like protein translation rates and protein stability (Schwanhausser et al. 2011). Furthermore, proteomics provides unique functional insights including post-translational modifications, subcellular localization information, and identification of interaction partners of proteins.
Due to enormous advances in mass spectrometry instrumentation, biochemical fractionation methods, and computational approaches, proteomics has matured into a state where the description of complete proteomes expressed in a specific condition is within reach. So far, only one study has claimed the identification of a complete proteome expressed in haploid and diploid baker's yeast (de Godoy et al. 2008), while extensive proteome coverage has been reported for several prokaryotes (Jaffe et al. 2004; Becher et al. 2009; Malmstrom et al. 2009) and archaea (Giannone et al. 2011). Describing extensive proteome maps under different conditions with a discovery proteomics approach is an important first step in defining the protein expression landscape for an organism and facilitates a subsequent shift away from the discovery mode to a remeasurement or scoring mode (Kuster et al. 2005; Ahrens et al. 2010).
Due to the lower transcriptome and proteome complexity compared to eukaryotes, an exhaustive discovery proteomics approach is particularly amenable for prokaryotes. We describe here a generic strategy to achieve an essentially complete coverage of a prokaryotic proteome expressed under specific conditions. Key elements of the strategy are the parallel extraction of RNA and protein from matched samples, and a saturated transcriptome analysis by RNA-seq (Wang et al. 2009). This in turn allows the generation of a condition-specific endpoint estimate of the number of actively transcribed protein-coding genes, which is a more appropriate estimate than considering all annotated protein-coding genes. A combination of experimental and computational strategies is then used to dig very deep into the proteome.
We apply the strategy to two conditions that mimic the changing environment encountered by Bartonella henselae upon transfer by its arthropod vector into its mammalian host. The Gram-negative α-proteobacterium B. henselae is a hemotropic, zoonotic pathogen that frequently causes cat scratch disease in immuno-competent humans, as well as bacteraemia, endocarditis, and vasoproliferative lesions in immuno-compromised patients. Members of the genus Bartonella are considered re-emerging pathogens and are primarily being studied as models for host-pathogen interaction (Harms and Dehio 2012). A particular emphasis was put on achieving an extensive coverage of the important membrane proteome (Savas et al. 2011). Membrane proteins carry out essential functions as transporters, enzymes, receptors to sense and transmit signals, and adhesion molecules. In light of the resurgence of infectious diseases, membrane proteins are, furthermore, prime candidates for the development of urgently needed novel anti-infectives (Norrby et al. 2005).
Relying on a very stringent false discovery rate (FDR) cutoff, we were able to identify 1250 of the 1467 annotated distinct B. henselae proteins, i.e., a proteome coverage of 85%. Several lines of evidence indicated that we have exhaustively measured the expressed proteome and can claim to have identified a complete membrane proteome. This included expression evidence―to our knowledge for the first time―for all protein components of a bacterial type IV secretion system (T4SS) which spans the inner and outer bacterial cell membranes.
Results and Discussion
Model system to explore complete proteome coverage
We chose B. henselae as a model system for several reasons: (1) Its relatively small genome (1.93 Mbp) comprises 1488 predicted protein-coding genes (Alsmark et al. 2004); (2) it is a facultative intracellular pathogen that can be grown in pure culture; (3) protocols for subcellular fractionation have been described (Rhomberg et al. 2004); and (4) in vitro conditions that mimic the pH-dependent induction of virulence genes required for the successful interaction with host endothelial cells, the likely primary niche for B. henselae (Harms and Dehio 2012), have been established (Quebatte et al. 2010). The availability of a model system that eliminates the need for coculture with human endothelial cells is critical to achieve complete coverage of an expressed proteome.
Our in vitro model system relies on the induction of the transcription factor BatR (BH00620) that is essential for the pathogenicity of B. henselae (Quebatte et al. 2010) (for details, see Supplemental Methods; Supplemental Tables S1, S2). In the absence of IPTG (uninduced condition), the batR regulon is not induced, resembling the situation encountered in the arthropod midgut. In contrast, batR expression is up-regulated in the induced condition, resulting in a marked induction of the batR regulon, including the VirB/D4 type IV secretion system (T4SS), which is required for infection of endothelial cells (Schulein and Dehio 2002). This state mimics the environment encountered by bacteria in the mammalian host.
A generic strategy for complete proteome coverage by discovery proteomics
We rely on our previous definition of complete proteome coverage, i.e., having identified protein expression evidence for the annotated protein-coding genes actively transcribed in a given state (Ahrens et al. 2010). A recent proteogenomics study of 46 prokaryotes indicated that, on average, only 0.4% protein-coding genes were missed in the original genome annotations (Venter et al. 2011), justifying our focus on the reference genome. Our strategy to achieve as complete as possible coverage of the expressed proteome of a prokaryote consists of three stages.
In a first stage, RNA and proteins are extracted from identical samples, and whole transcriptome libraries are sequenced to saturation by RNA-seq (Fig. 1A). Thereby, the number of protein-coding genes actively transcribed in a given state can be estimated, shown here for the sum of protein-coding genes expressed in the uninduced and induced condition (orange dashed line, Fig. 1B). Based on such an optimal endpoint estimate, in a second stage, several pilot experiments are performed on cytoplasmic and total membrane fractions of the respective conditions. Following a statistical comparison of the pilot phase proteome (green line, Fig. 1B) to the predicted endpoint, areas of underrepresentation can be targeted by the analysis-driven experimentation (ADE) feedback-loop strategy (Brunner et al. 2007), which can help to overcome the premature saturation of distinct protein identifications and sequence deeper into the expressed proteome (blue lines, Fig. 1B). In a third stage, evidence is presented that virtually no biases remain when comparing protein parameters of all identified proteins to those called actively expressed, justifying the claim to have identified a complete proteome expressed in a specific condition. Analysis of such a data set is expected to provide novel insights regarding the achievable membrane proteome coverage, differential protein expression, and evolutionary conservation and genome structure (Fig. 1C).
Overview of the complete expressed proteome discovery workflow. (A) Extraction of RNA and proteins from matched samples, transcriptome analysis. Total RNA and proteins were extracted in parallel from bacteria grown either under uninduced or induced conditions (schematically shown by black knobs representing the VirB/D4 T4SS). Protein extracts were subfractionated into cytoplasmic (Cyt), total membrane (TM), inner (IM), and outer membrane (OM) fractions. To estimate an upper bound for the number of actively transcribed protein-coding genes, the transcriptome was sequenced to saturation using RNA-seq. (B) Analysis-driven experimentation (ADE). In a first pilot phase, samples are analyzed by LC-MS/MS. Underrepresented proteome areas are identified based on a statistical analysis comparing experimentally identified proteins to all expressed proteins (the estimated RNA-seq endpoint indicated by the orange dashed line within an error envelope). All distinct annotated proteins are indicated by the black dashed line. Subsequently, these areas are investigated by targeted experiments, aiming to overcome the saturation trend. (C) Integrative data analysis. Data from the expressed proteome are integrated with genomic, transcriptomics, orthology, and other information to enable further analyses.
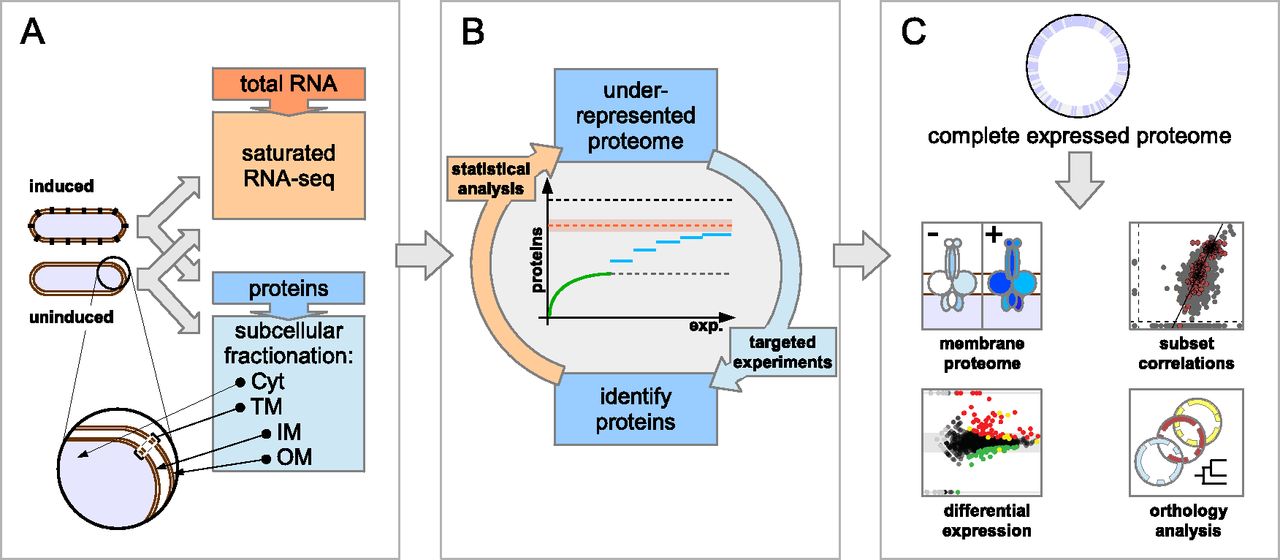
Transcriptome exploration by RNA-seq
We relied on RNA-seq (Wang et al. 2009) primarily to generate an endpoint estimate for the number of expressed protein-coding genes. Whole transcriptome libraries of two biological replicates per condition were generated using a protocol that enriches for mRNA transcripts (see Methods). We sequenced very deep into the transcriptome and obtained 55–87 million single end 50-mer reads per sample. Of these, 10.7–26.7 million reads mapped unambiguously, while the vast majority of remaining reads originated from multiple-copy rRNA genes (see Methods; Supplemental Table S3). Reads per kilobase per million (RPKM) values (Mortazavi et al. 2008) showed very high concordance of the biological replicates (r > 0.97) (Supplemental Fig. S1).
To estimate how many protein-coding genes are actively expressed in the two conditions, we plotted the number of distinct expressed protein-coding ORFs as a function of the sum of uniquely mapping reads. We required at least five distinct reads within a 50-nt window of the 5′ end to deem a protein-coding gene actively expressed (Supplemental Fig. S2), a cutoff similar to that used by Wang et al. (2009). Saturation is characterized graphically through flattening of the curves as the number of reads increases. Due to the asymptotic nature of saturation curves, reaching complete coverage is theoretically only possible with infinite effort. Therefore, we define saturation as the number of discoveries from where, based on nonlinear modeling and extrapolation, a doubling of effort is expected to increase the number of discoveries only marginally. Figure 2A indicates that doubling the number of reads would increase the number of detected protein-coding genes by <3.5% for sample uninduced2 and by ∼1% for induced2. Therefore, our analysis indicated that the transcriptome was sequenced to saturation (Fig. 2A). We acknowledge that different library preparations might potentially identify additional genes and that very low abundance transcripts (and proteins) expressed in only a few cells of the population may not be identified with this approach.
Transcriptome coverage by RNA-seq. (A) Saturated coverage of protein-coding genes. An estimate of the number of actively expressed protein-coding genes based on the number of uniquely mapped RNA-seq reads is shown for both conditions and biological replicates. (B) Density distribution of RPKM values. In addition, boxplots representing the expression level of the 11 members of the virB/D4 operon are shown for the uninduced (black), and induced (red) condition. For clarity, we only show data for the sample pair uninduced2/induced2.
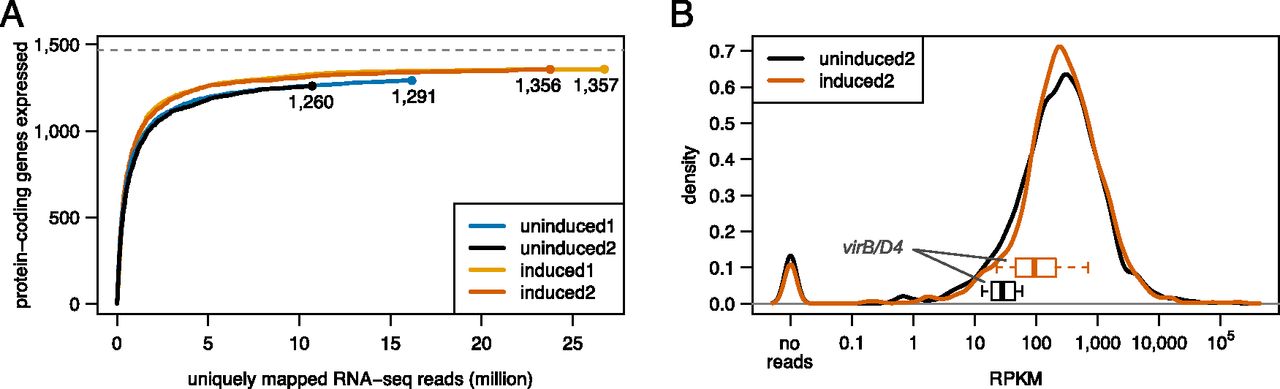
We also plotted the density of the RPKM values in order to assess the distribution of transcription levels for all annotated protein-coding genes: The resulting bimodal graph suggested that, under the conditions studied, not all protein-coding genes are actively expressed; RPKM = 10 might be considered a conservative lower cutoff (Fig. 2B). The average RPKM values for members of the virB/D4 operon in condition uninduced2 (30), where the operon is expected to be expressed at low levels, versus induced2 (160) support this observation.
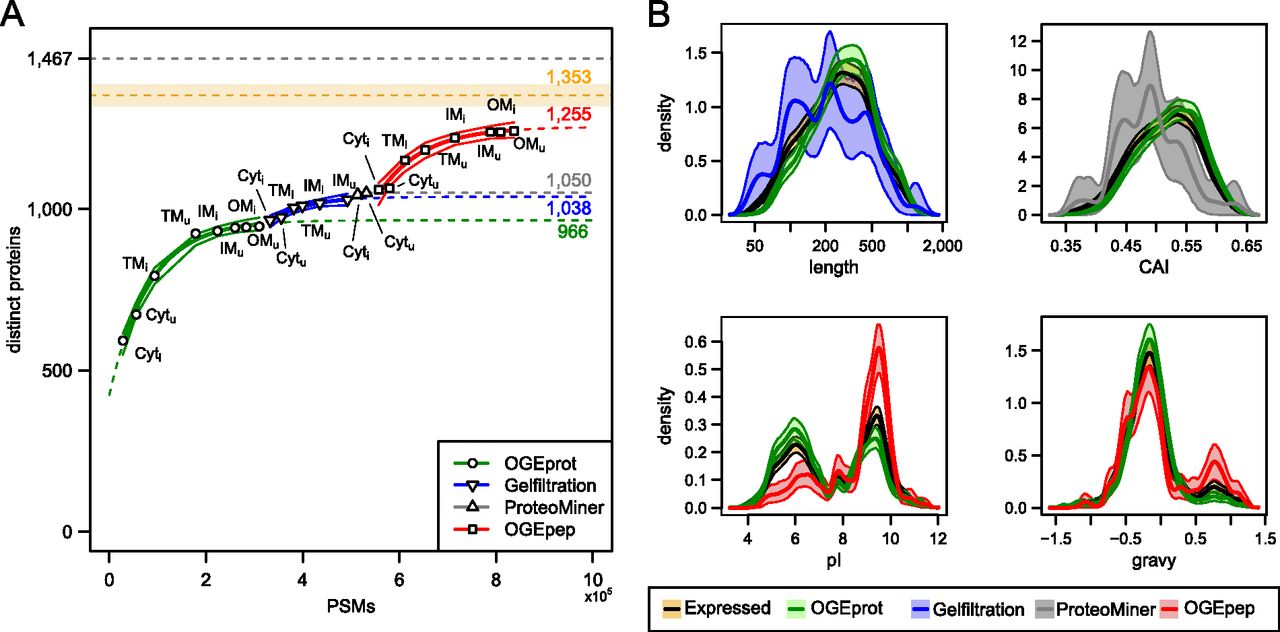
Based on the combined thresholds, 1353 protein-coding genes were expressed in the two conditions (uninduced 1254 and induced 1349). An inter-replicate analysis revealed >95% overlap of the expressed protein-coding genes (Supplemental Table S4). We include an error envelope of ±2.5% to account for uncertainty in the thresholds (Fig. 3A).
Overcoming the saturation of protein identifications using ADE-guided shotgun proteomics. (A) Increase of distinct identified proteins given the number of PSMs observed in different experiments. We fitted an exponential curve (see Methods) to all experiments for a given biochemical fractionation in order to find a saturation limit (see colored numbers on the right-hand side). We also approximated confidence bands for the fitted points (thin lines; see Methods). The black dashed line at the top signifies the total number of distinct B. henselae proteins (1467); the orange dashed line below represents the estimated RNA-seq endpoint of expressed distinct proteins (1353) including a ±2.5%-error envelope (orange shaded area). (B) Density estimates of four physicochemical protein parameters for different protein subsets. The parameter density for proteins newly identified by the ADE approach is contrasted to that of all expressed proteins (orange) and those identified in pilot experiments using OGEprot (green). The most important aspects of over- or underrepresentation can be seen on the abscissa; they indicate that the targeted experiments successfully add new protein identifications in areas of the proteome that were underrepresented in the pilot experiments. For details on the density estimation and the bootstrap confidence bands (shaded areas), see Supplemental Methods.
Extended proteome coverage strategy: Experimental and computational approaches
Our experimental strategy to reach very deep into the proteome relied on four elements: first, we used a combination of subcellular fractionation and additional biochemical fractionation regimens to reduce the overall sample complexity, a measure that had been key to describing the complete expressed proteome of baker's yeast (de Godoy et al. 2008). Second, an exclusion list approach (Kristensen et al. 2004) was applied, which helped to identify a significant amount of low-abundance proteins (Supplemental Fig. S5). Third, we relied on the analysis-driven experimentation feedback-loop strategy (Fig. 1B; Brunner et al. 2007) to target underrepresented areas of the proteome and overcome premature saturation. Finally, for all membrane-derived fractions, we used chymotrypsin in addition to trypsin, thereby maximizing the per-protein sequence coverage and the overall membrane proteome coverage (Fischer et al. 2006).
In terms of computational approaches, we combined results from two database search engines, Mascot Percolator (Brosch et al. 2009) and MS-GF+, an updated version of MS-GFDB (Kim et al. 2010; see Methods), which employs the generating function approach (Kim et al. 2008) to compute statistical significance of peptide identifications (spectral probabilities). Based on these spectral probabilities or the target-decoy option, one can estimate and stringently control the FDR rate, a critical step for a complete proteome discovery project. Otherwise, lower quality peptide spectrum matches (PSMs) will start to accumulate false-positive peptide evidence for proteins in a random fashion (Reiter et al. 2009). In addition, the error propagates and increases from spectra to peptides and proteins (Nesvizhskii 2010); a PSM level FDR of 1% can correspond to a protein level FDR of 8%–11% (Balgley et al. 2007). We, therefore, chose a very stringent PSM FDR cutoff of 0.01%, allowing us to report protein identifications with an FDR below 1% (see below).
Identification of the complete expressed B. henselae proteome
The induction of batR and virB/D4 T4SS expression was more pronounced for the sample pair uninduced2/induced2 than for its biological replicate based on the RNA-seq data. Subcellular fractions from this sample pair (i.e., cytoplasmic [Cyt], total membrane [TM], inner [IM] and outer membrane [OM] fractions) were thus analyzed in detail using different biochemical fractionations (see Methods; Fig. 1A).
We first measured the Cyt and TM fractions of both conditions using OFFGEL electrophoresis at the protein level (OGEprot). When requiring at least two independent PSMs to identify a protein, 924 distinct proteins were identified in four experiments, i.e., 63% of all 1467 distinct annotated proteins or 68% ± 2% compared to the RNA-seq endpoint estimate of 1353 ± 34 expressed proteins (Fig. 3A). Analysis of the IM fractions from uninduced and induced condition (IMu/i) and the OMu/i fractions contributed 130,000 additional PSMs (72% more PSMs) but only added 22 previously not identified proteins (Fig. 3A), indicating that we were already in the saturation phase. We fitted a saturation curve to the eight OGEprot experiments, which shows the anticipated trend of further protein identifications assuming no change in the experimental approach, and also calculated confidence intervals (see Methods; Fig. 3A). Carrying out further OGEprot experiments is predicted to lead only to a handful of new protein identifications.
Instead, we relied on the ADE strategy to break the saturation trend. We computed several physicochemical parameters for all distinct B. henselae proteins (see Supplemental Methods). The statistical comparison of the parameters of 946 proteins identified by OGEprot in the pilot phase versus the RNA-seq endpoint estimate of 1353 expressed proteins in both conditions provided evidence for a significant underrepresentation of short, low-abundance, basic, and hydrophobic proteins. These areas of the proteome were subsequently targeted by specific experimental approaches (see Supplemental Methods). Underrepresentation with respect to length was targeted using size exclusion chromatography (gel filtration) (Brunner et al. 2007). These experiments added 83 new protein identifications compared to the OGEprot pilot phase (Fig. 3A, blue color). The enrichment for shorter proteins can be appreciated in the upper left panel of Figure 3B. Low-abundance proteins were targeted using ProteoMiner (Guerrier et al. 2008; Fonslow et al. 2011). These experiments (Fig. 3A, gray) helped to identify 42 additional proteins, which were preferentially lower-abundance proteins as evidenced from the density distribution of their Codon Adaptation Index (CAI) values (Fig. 3B, upper right panel; Sharp and Li 1987). Basic and membrane-localized proteins were targeted using OFFGEL electrophoresis at the peptide level (OGEpep). The 285 proteins newly added by the OGEpep experiments (Fig. 3A, red) were highly enriched for basic proteins (Fig. 3B, lower left panel) and membrane proteins (with a high grand-average hydropathicity [gravy] value) (Fig. 3B, lower right panel).
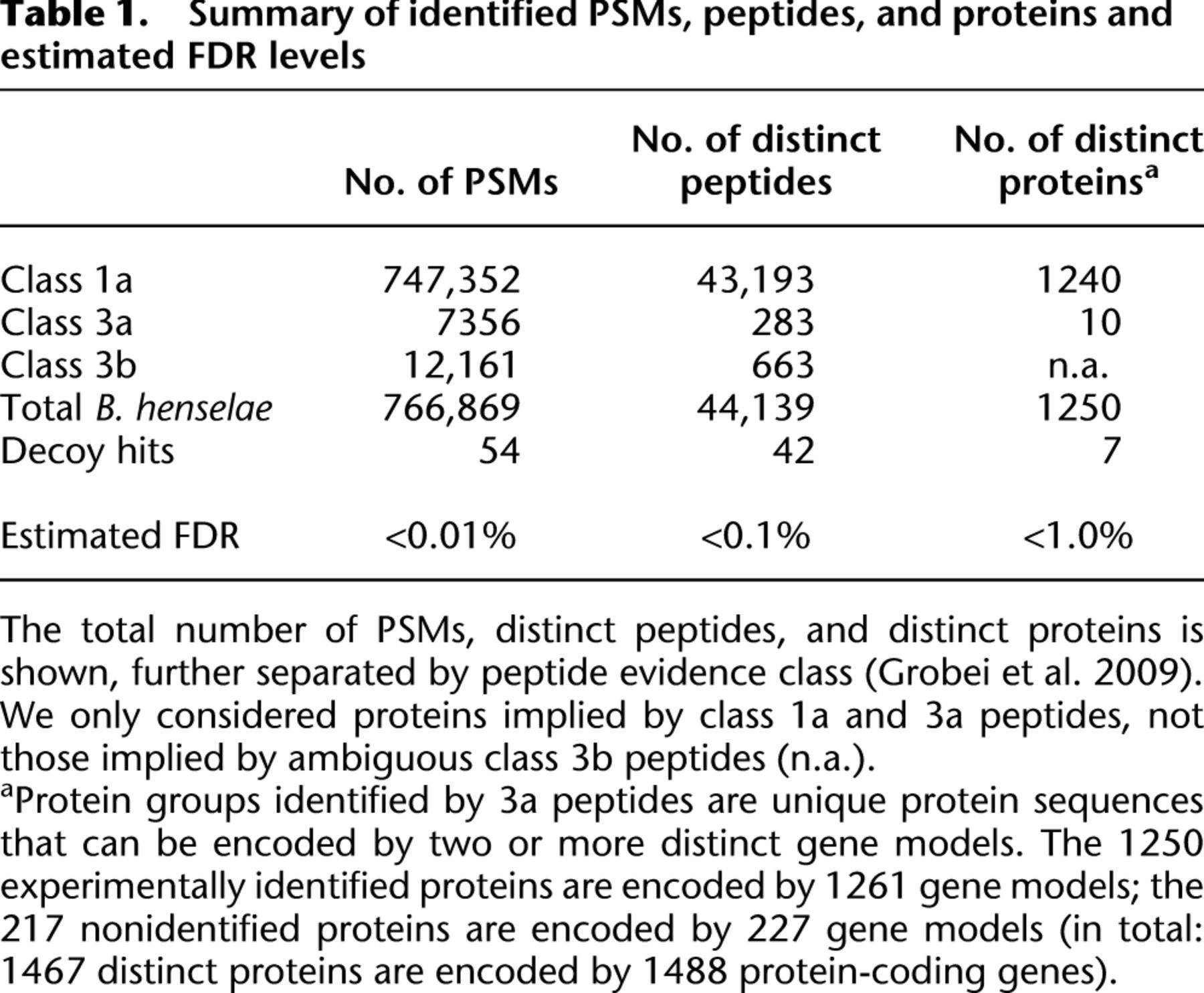
Overall, we identified 1250 distinct proteins requiring at least two PSMs per protein (Supplemental Fig. S3) and only considering peptides that unambiguously identify one bacterial protein (Table 1; Qeli and Ahrens 2010), i.e., 85% of the 1467 distinct protein sequences. The FDRs at the PSM, peptide, and protein level are below 0.01%, 0.1%, and 1%, respectively (Table 1). Only a few among the 1228 proteins identified in the uninduced and the 1231 in the induced condition were selectively expressed (Supplemental Fig. S4); these included several members of the VirB/D4 T4SS in the induced condition. Compared to the expressed transcriptome, the proteome coverage reaches 90% for both the uninduced and induced condition.
Summary of identified PSMs, peptides, and proteins and estimated FDR levels
Although each experimental and computational approach contributed unique protein identifications to the final data set (see Supplemental Fig. S5), for similar studies aiming to maximize coverage of an expressed proteome with a minimum number of experiments, we recommend use of subcellular fractionation (Cyt and TM), and performing OGEpep and measuring each fraction twice using the exclusion list approach. This approach would identify 1153 proteins, i.e., 92%, while requiring only 15% of the mass spectrometry runs needed to identify all 1250 proteins.
Evidence for having reached an expressed proteome endpoint
Several lines of evidence indicated that the 1250 distinct protein groups are very close to the complete proteome endpoint that is actively expressed under the investigated conditions.
First, a comparison of the total number of PSM identifications showed that MS-GF+ added 67% more PSMs than Mascot-Percolator (Supplemental Fig. S3A). Yet, at the level of distinct peptides, this increase was smaller (+37%) (Supplemental Fig. S3B) and amounted to a mere 3%, or 33 additional proteins at the protein level (Supplemental Fig. S3C), despite having added several hundred thousand additional PSMs. Using a third search engine, Sequest, would have only added one additional protein for all experimental spectra. This indicates that, similar to the transcriptome, we have also measured the expressed proteome to saturation. The exponential model fitted to the eight OGEpep experiments (Fig. 3A) supports this: Doubling the number of PSMs on OGEpep samples (roughly 305,000 additional PSMs, i.e., ∼36% more PSMs overall) would only identify five new proteins (red number on top of red dashed line, Fig. 3A).
Second, our expressed proteome encompassed all proteins identified in three previous B. henselae proteomics studies (Rhomberg et al. 2004; Eberhardt et al. 2009; Li et al. 2011), while adding many more low-abundance proteins (Supplemental Fig. S6A–C).
Third and most importantly, a comparison of the protein parameter distributions of the data sets expressed protein-coding genes (1353) and final expressed proteome (1250) showed that there is virtually no underrepresentation anymore in those areas of the proteome that we had specifically targeted; i.e., ADE successfully eliminated these differences present in the OGEprot pilot study (Supplemental Fig. S7). Two examples illustrate this point: (1) For the parameter isoelectric point (pI), basic proteins are underrepresented in the OGEprot data set. After carrying out the ADE approach, there is only a small difference between the densities of the data sets “final” and “expressed” (Supplemental Fig. S7, top panels); and (2) for the parameter gravy, membrane proteins with one or more predicted transmembrane domains (gravy values above 0.5) are underrepresented in the OGEprot data set. Again, after the ADE approach, the densities for the data sets “expressed” and “final” are virtually identical (Supplemental Fig. S7, middle panels). This comparison also showed that ADE could add proteins encoded by genes that are expressed at lower levels under the conditions studied (Supplemental Fig. S7, last panels). Two-dimensional density plots of the gene expression level versus the parameters length, pI, and gravy (Supplemental Fig. S8) for the data set final expressed proteome (1250) versus not seen proteins (217) showed that there is still a noticeable tendency for short and basic proteins to be enriched among genes with expression levels close to the threshold whose proteins were not identified (Supplemental Fig. S8A,B). These are not expected to be detectable with the shotgun proteomics approach since short and basic proteins have fewer tryptic peptides in the detectable range of the mass spectrometer. In contrast, for the two-dimensional density plot with the protein parameter gravy (values above 0.5 are found in proteins with transmembrane domains), we observed no bias (Supplemental Fig. S8C), indicative of a complete membrane proteome coverage.
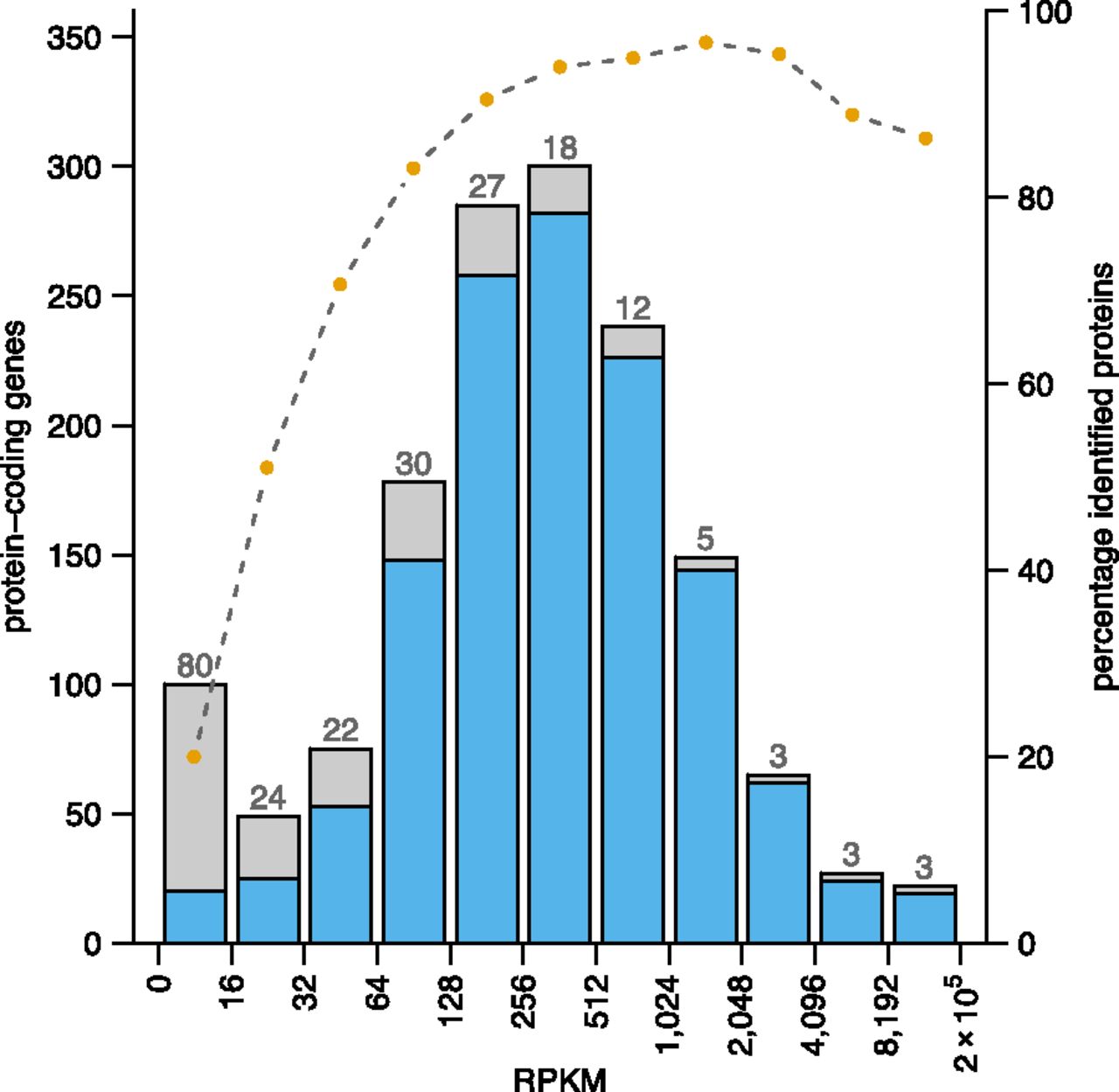
To correlate the gene expression level with the proteome coverage, we binned the protein-coding genes according to gene expression strength (RPKM values) and plotted for each bin the respective percentage of proteins identified (Fig. 4). A clear correlation between higher levels of gene expression with a higher success rate of protein identification can be observed. However, several proteins of highly expressed genes were not identified: among 26 such cases from the five top expression bins, 23 had no conserved ortholog in bartonellae, and 16 were located in a novel, plastic genome region (see next section).
Correlation of gene expression strength and successful protein identification rate. Protein-coding genes are binned according to strength of gene expression (the maximum RPKM value of both states). The success rate in identifying the encoded proteins in each bin is represented by the blue area of the bars; orange dots above the barplot indicate the respective percentage. The numbers above the bars show how many proteins were not identified within a given bin (for a total of 217 distinct proteins).
Integration of genome structure information and evolutionary conservation
We projected transcriptomic and proteomic evidence, ortholog predictions, and repeat regions onto the B. henselae genome sequence (Fig. 5), which contains a large prophage region and three major genomic islands (Alsmark et al. 2004). Genes in such genomic regions are often subject to regulation and become actively expressed only under specific conditions (Juhas et al. 2009). Intriguingly, for 109 of the 198 genes that are located in these four genomic regions, we could not detect any expressed proteins (Fig. 5, fourth ring). This is a significant enrichment, given that only 227 annotated protein-coding genes did not express any protein (P-value < 10−9) (see Fig. 5).
Integration of expression evidence with structural genome information and evolutionary conservation. Genes whose proteins were not identified cluster in specific regions of the B. henselae genome. Outer ring: Genes whose proteins were identified (light blue) or not identified (red). Second ring: Protein-coding genes classified by the RNA-seq analysis as expressed (gray) or not (dark green). Third ring: Genes without a detectable ortholog among species of lineage 4 of the genus Bartonella (Engel et al. 2011) (turquoise) and genes without any functional annotation by the eggNOG classification (black). Fourth ring: Repeat regions identified by RepSeek (orange) (Vallenet et al. 2006) and rRNA repeat regions (light orange). Fifth ring: Location of a prophage region (ochre), three genomic islands (blue), the virB/D4 and trw operons (sky blue), and a novel genomic region enriched in repeats as well as highly expressed genes whose encoded proteins were not identified (yellow). The results of hypergeometric tests for selected data sets are also shown (asterisks indicate statistically significant enrichment; see text). For the hypergeometric test, we used all possible protein-coding genes for the identified “seen” proteins (1250 distinct proteins encoded by 1261 gene models) and “not seen” proteins (217 distinct proteins encoded by 227 gene models). The circular plot was generated using DNAPlotter (Carver et al. 2009).
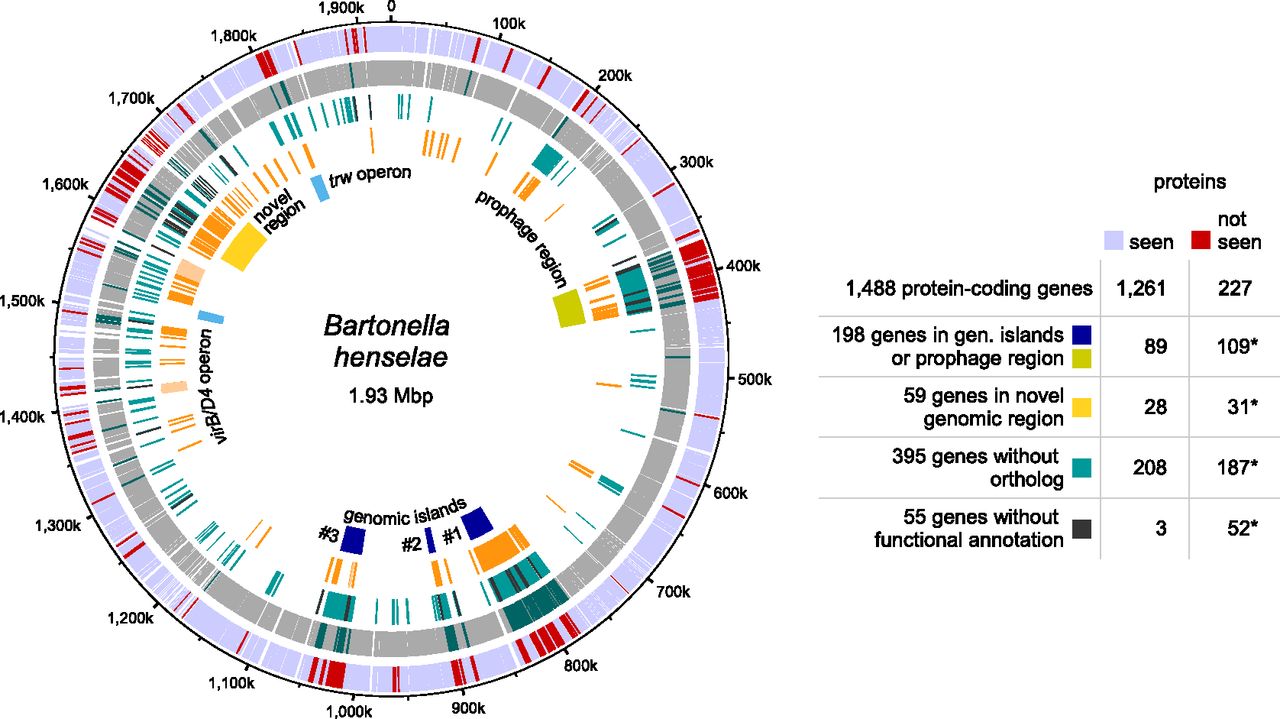
We next investigated whether the products of evolutionarily conserved protein-coding genes were enriched or selected against. In a comparison with B. tribocorum, B. quintana, and B. grahamii, 1093 of the 1488 B. henselae protein-coding genes were predicted to have an ortholog (Engel et al. 2011), while 395 were not (Fig. 5, third ring, turquoise bars). We detected significant overrepresentation of genes lacking an ortholog (187 of 395) among the 227 protein-coding genes whose proteins were not identified (P-value < 10−9) (Fig. 5).
To extend the evolutionary conservation analysis beyond members of the genus Bartonella, we relied on the eggNOG resource, which contains orthology information from 1133 organisms, including B. henselae (Powell et al. 2012). Among the 1488 B. henselae proteins, only 55 proteins lack any functional annotation; they are a subset of the 395 without ortholog (black bars, third ring, Fig. 5). Strikingly, 52 of these 55 were not detected, again a significant enrichment (P-value < 10−9). A significant number of the genes (16) encoding these 55 proteins clustered in a region from 1612–1674 kbp that harbors 59 predicted ORFs (P-value < 10−9) (yellow box, Fig. 5). Location in this plastic, repeat-rich genome region (orange bars, fourth ring) may lead to strong transcription of genes that do not represent a bona fide protein-coding ORF.
The evolutionary conservation information provided by eggNOG, together with high-quality experimental proteomics data, represents a particular useful combination to identify candidates for overpredicted protein-coding genes in genome annotations: The densities of the protein length distribution of the proteins not identified (217) were clearly separated from that of the proteins seen (1250) (Supplemental Fig. S9A). Among the proteins not seen, those that lack any functional annotation are considerably shorter than those with a functional annotation (Supplemental Fig. S9B). Since we can detect short proteins with our set-up (see density of the 150 shortest proteins detected compared to all, Supplemental Fig. S9C), the proteins that lack an ortholog and any functional annotation may either only be expressed under different conditions or are potential overpredicted ORFs.
Coverage of the membrane proteome and the VirB/D4 T4SS
The membrane proteome serves many essential roles in cellular communication, transport, adhesion to host cells, and evasion of the host immune system. While accounting for up to one third of the gene products, >50% of the druggable targets fall into this category (Hopkins and Groom 2002). However, due to the amphipathic nature and low abundance of membrane proteins, they are notoriously underrepresented in proteomics studies (Poetsch and Wolters 2008; Tan et al. 2008; Helbig et al. 2010).
To reach a high protein sequence coverage for membrane proteins, we used a combination of trypsin and chymotrypsin in all membrane samples and, furthermore, applied proteolytic digestion in 60% (v/v) methanol to improve cleavability of hydrophobic proteins (Fischer et al. 2006; Supplemental Methods). Among 924 proteins identified in the first four pilot phase experiments (63% of all distinct proteins), 182 contained predicted transmembrane domains (54%) (Fig. 6A, left panel). However, the ADE approach was able to eliminate this underrepresentation of membrane proteins: among the final 1250 identified proteins (85% of all distinct annotated proteins), 289 of the 338 distinct proteins with one or more predicted transmembrane regions were found, i.e., 86% (Fig. 6A, right panel; Supplemental Fig. S10A). Notably, the OGEpep fractionation regimen was particularly successful in identifying membrane proteins. We also identified 54 of the 58 predicted secreted proteins (95%). These include many proteins for which PSORTb (Yu et al. 2010) predicts localization in the membrane space and where other studies could confirm their localization in inner or outer membrane, periplasm, or the extracellular space (Supplemental Fig. S10B). Together with the striking result that transmembrane proteins with high gravy values are not overrepresented among the 217 nonidentified proteins compared to 1250 seen proteins (see Supplemental Fig. S8C), the data suggested that we have identified a complete membrane proteome expressed under two specific conditions.
Membrane proteome coverage and dynamics. (A) Comparison of the membrane proteome coverage achieved in four pilot experiments (left panel) and the final data set (right panel). Membrane proteins are binned according to the number of predicted transmembrane domains; the percentage of proteins identified per bin is shown above each bar. The legends summarize the respective coverage achieved comparing the respective data set (pilot phase/final) against all distinct proteins and for the subset of proteins with transmembrane domains. Membrane proteins are underrepresented in the pilot phase but not in the final data set. (B) Transcript and protein expression changes of the virB/D4 T4SS and downstream bep operon. Operon structures (upper panel) are drawn to scale. The lower left panel shows the log2 fold changes at the transcript and protein level for the induced versus uninduced state (the ∞ indicates that the protein was only identified in the induced condition). Fold changes and significance were calculated with DESeq. Regulation at the protein level appears to be more pronounced compared to the transcript level. The lower right panel visualizes the protein expression changes upon induction onto a schematic representation of the assembled VirB/D4 T4SS using different shades of blue. (C) Comparison of expression changes at transcript and protein level. The respective log2 fold changes based on the RPKM values and normalized spectral counts are shown. Members of the VirB/D4 T4SS are shown in blue (BH13360 in light blue), Bartonella effector proteins (Beps) in dark blue. Three proteins that exhibited the most significant differential expression (Supplemental Table S6; Fig. 7) are also shown with their identifiers.
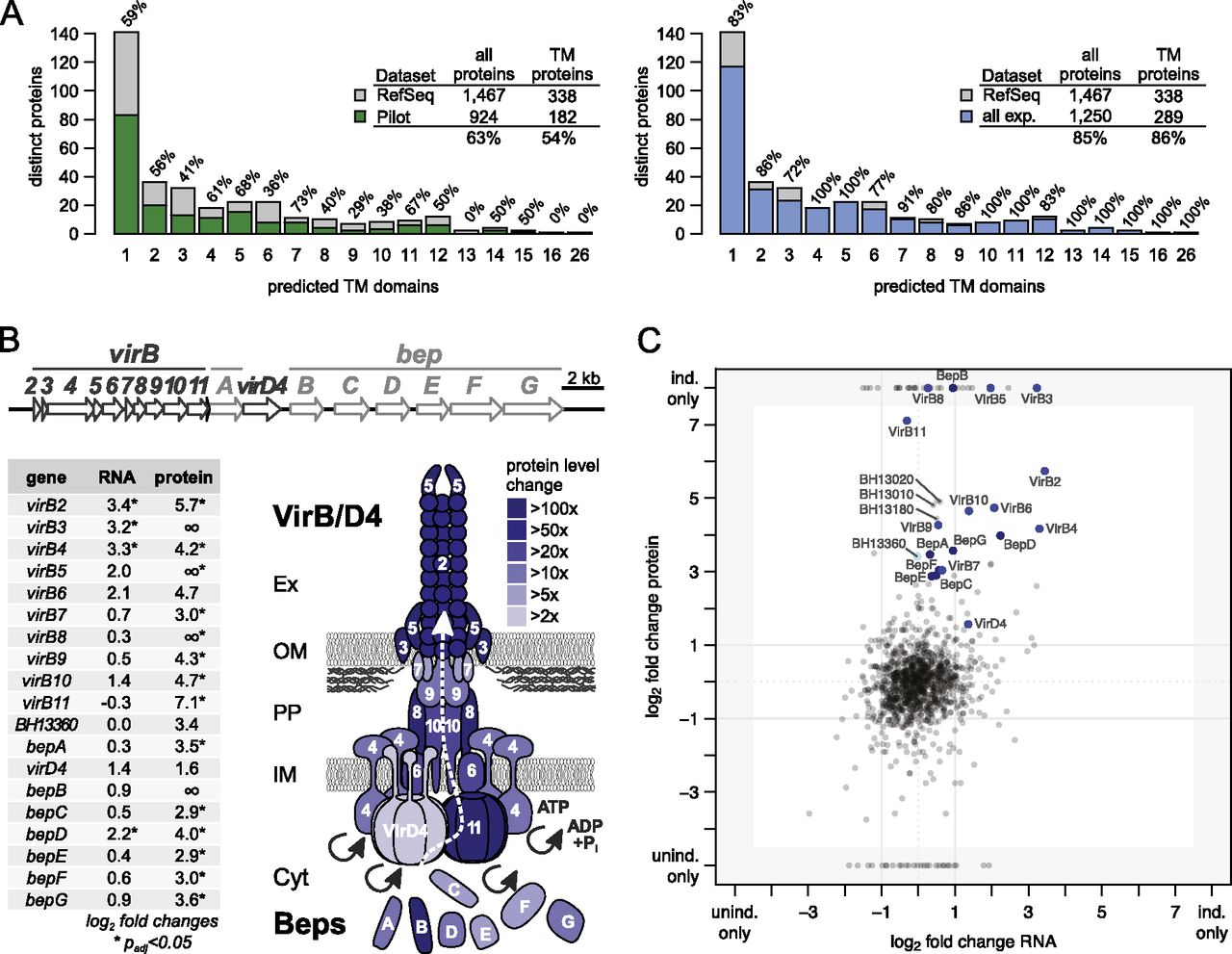
This includes all 11 protein members encoded by the virB/D4 operon in the induced condition (Fig. 6B). To our knowledge, this is the first complete coverage of this important molecular machinery spanning both inner and outer membrane by a shotgun proteomics approach. We also detected all seven Bartonella effector proteins (Beps), which are secreted by the VirB/D4 T4SS into eukaryotic host cells (Fig. 6B). In contrast, many proteins of the Trw complex, a second B. henselae T4SS that is essential for the infection of erythrocytes (Vayssier-Taussat et al. 2010) but dispensable under the conditions studied, were not detected (nine of 24, 38%) (Fig. 5, first and fifth ring), nor was their expression regulated (Supplemental Fig. S11).
When we assessed the level of induction at the RNA and protein level, we observed that the induction of virB/D4 and bep operons, which are direct targets of the transcriptional regulator BatR, seemed to be more prominent at the protein level. They also included more cases with statistical significance of the up-regulation (Fig. 6B, log2 fold changes, left panel). A comparison of the log2 fold changes at the RNA level versus those at the protein level indicated that several of the virB/D4 and bep genes appear to be regulated preferentially at the post-transcriptional level, indicated in Figure 6C by their position close to the vertical axis.
The ability to identify complete membrane proteomes of prokaryotes has important implications for studying their expression under different conditions in a quantitative fashion. Ideally, such a task would be performed with the more sensitive targeted proteomics approach (Schmidt et al. 2011), which typically relies on predicted proteotypic peptides (PTPs) using tools like PeptideSieve (Mallick et al. 2007). Our data indicate that a comprehensive discovery proteomics approach adds clear value with respect to experimentally identified PTPs, as we could identify peptides for 145 proteins for which PeptideSieve predicted no PTP (see Supplemental Methods). We provide the proteomics and transcriptomics data with results of several prediction algorithms (Supplemental Table S5A), and all experimentally identified peptides (Supplemental Table S5B), from which the best-suited PTPs can be selected using available guidelines (Picotti and Aebersold 2012).
Identification of differentially expressed proteins
Our in-depth proteome analysis precluded the measurement of biological replicates. We thus relied on DESeq to identify the most significantly differentially regulated proteins between induced and uninduced states (see Methods). The top 10% differentially expressed proteins (Supplemental Table S6), including 68 up-regulated (red dots), and 57 down-regulated proteins (green dots) in the induced condition, are highlighted in Figure 7.
Differential protein expression analysis. The log2 fold change of the expression of all experimentally identified B. henselae proteins in the induced versus uninduced condition is shown against the mean normalized spectral count (MA plot). The 10% most significant differentially expressed proteins are highlighted, including 68 up-regulated proteins (red dots) and 57 down-regulated proteins (green dots). Selected regulated proteins are highlighted in different colors: members of the VirB/D4 T4SS (blue dots), Beps (dark blue dots), and several proteins containing autotransporter beta-domains (yellow dots). Proteins in these categories that rank below the 10% cutoff are shown as open circles.
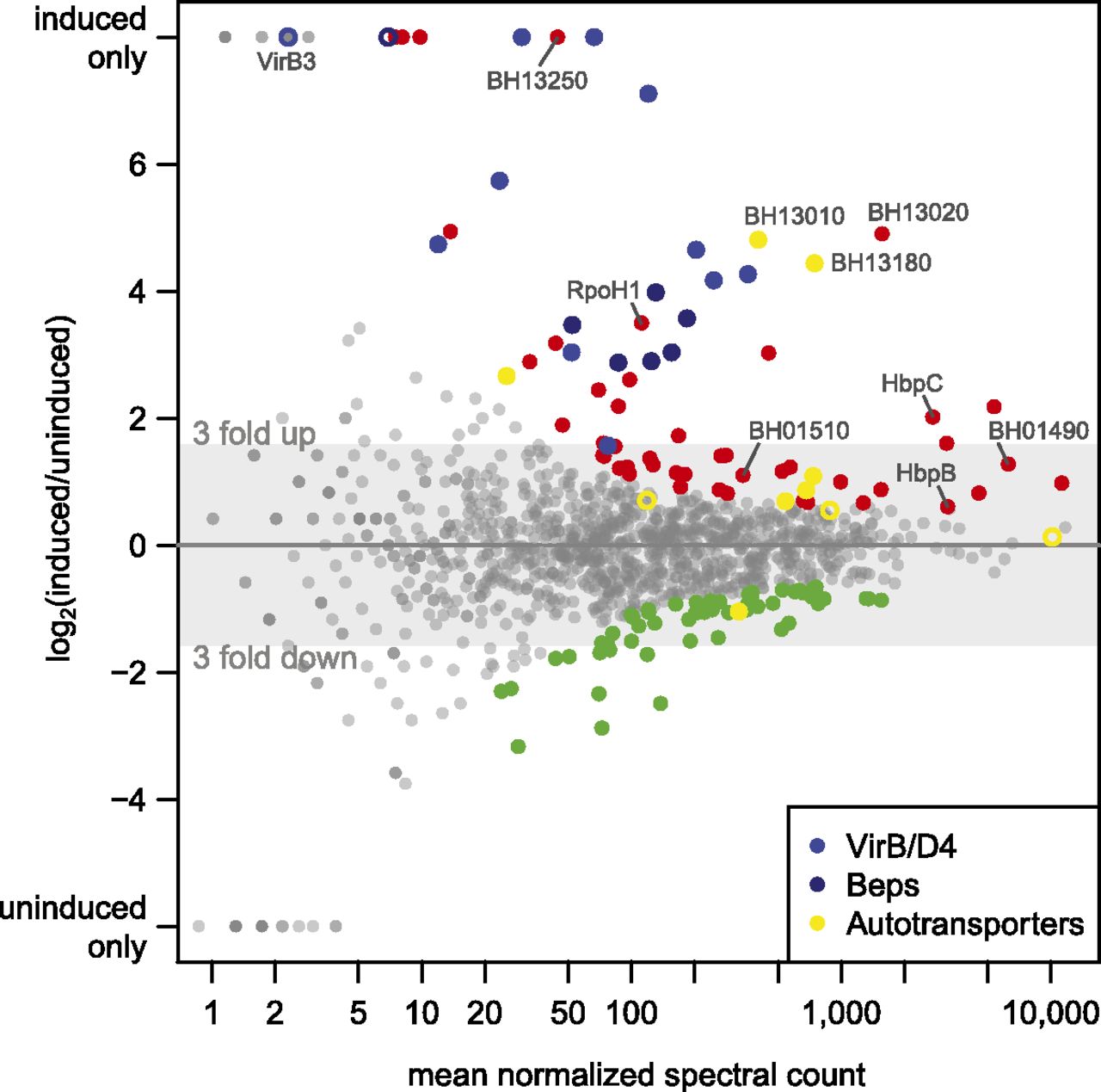
Among these 125, 36 transmembrane and 12 secreted proteins were found, a significant enrichment (P-value < 0.0018) compared to 343 membrane and secreted proteins among the 1250 proteins. A striking feature was the strong regulation of different families of autotransporters, which rely on the type V secretion pathway for their delivery to the surface of Gram-negative bacteria (Leyton et al. 2012). These included two representatives of the trimeric autotransporter adhesins (BH01490, BH01510), a class of virulence factors essential for Bartonella pathogenicity (Franz and Kempf 2011). Furthermore, seven of 10 proteins with an autotransporter beta domain (as predicted by SMART version 7) (Letunic et al. 2012) were among the top 10% differentially regulated proteins (six up-regulated, one down-regulated) (yellow dots, Fig. 7; Supplemental Table S6), i.e., a significant enrichment (P-value < 4 × 10−7). BH13020, BH13180, and BH13010 were the top three up-regulated proteins, which ranked even higher than members of the virB/D4 operon. While less is known about the role of this family of autotransporters in Bartonella, they were found to be up-regulated during infection of endothelial cells (Quebatte et al. 2010) and may be involved in adhesion to host cells (Litwin et al. 2007). Finally, two of the four outer membrane proteins of the hemin binding protein family (HbpC and HbpB) were found. HbpC was shown to protect B. henselae against hemin toxicity and to play a role during host infection (Roden et al. 2012).
The top 10% regulated proteins included six of the seven Beps and all VirB/D4 T4SS proteins except VirB3. For this small protein (103 amino acids) with one predicted transmembrane domain, we only found four spectra, all in the induced condition. This indicates that a large experimental effort is required to detect proteins that combine several parameters which complicate their mass spectrometric identification with shotgun proteomics, i.e., they are short, basic, and hydrophobic. Another protein exclusively identified in the induced condition is BH13250, a hypothetical protein with a transmembrane domain (Supplemental Table S6). Its location just upstream of the virB/D4 operon is conserved in other Bartonella, suggesting that it may potentially carry out a yet to be determined function as a virulence factor. Finally, another interesting up-regulated protein is RpoH1 (BH15210), an alternative RNA-polymerase sigma factor 32. A role in virulence has been documented for its gene in an in vivo mouse infection model for the closely related Brucella (Delory et al. 2006).
Conclusion
Using a discovery proteomics approach, the expressed proteome of B. henselae was exhaustively studied under two conditions that mimic those encountered in different hosts. The saturated transcriptome analysis of RNA extracted from matched samples provided the best possible endpoint estimate for the number of actively transcribed protein-coding genes. ADE was able to virtually eliminate the biases of commonly underrepresented short, basic, and particularly lower-abundance and membrane protein classes, all of which are experimentally tractable. Based on a very stringent FDR at the PSM level, we identified 85% of all distinct, annotated proteins, and ∼90% compared to the expressed protein-coding genes in the two conditions. Several lines of evidence indicated that this is very close to all proteins that can be identified by a discovery proteomics approach with current technology. This is best illustrated by the complete membrane proteome coverage, including evidence for all members of the important VirB/D4 T4SS. The analysis of the genome organization revealed that genes whose transcripts were detected, but not their corresponding protein products, were highly enriched in genomic islands. Information regarding evolutionary conservation provided evidence for preferential expression of genes with a predicted ortholog. In contrast, genes that lacked an ortholog and functional annotation were mostly not observed at the protein level, suggesting possible overprediction in genome annotations.
Our report is the second complete expressed proteome reported (de Godoy et al. 2008). Using a similarly extensive fractionation strategy, our matched transcriptomics and proteomics data correlated quite well (r = 0.57) while identifying the VirB/D4 T4SS as a prominent target of post-transcriptional regulation. The rigorous approach to sequence transcriptome and proteome to saturation and to provide proof for having eliminated observed biases at the protein level is unique. It supports a recent perspective article showing that up to 90% of an expressed proteome (“nearly complete”) can be measured quite quickly (Mann et al. 2013), but the remaining 10% require extensive effort. It also underscores that the difference between “comprehensive” and “complete” can be quite large, in particular with respect to coverage of the membrane proteome (Beck et al. 2011). The higher coverage of distinct annotated proteins (85%) compared to the proteome expressed by haploid and diploid yeast (67%) suggests that prokaryotes express a higher fraction of the encoded proteins, potentially reflecting their need to quickly adapt to changing conditions. This fraction may be lower for more complex prokaryotes.
The data attest to the value of a discovery proteomics approach in providing experimentally identified PTPs beyond those predicted in silico. The sensitive quantitative measurement of such PTPs by SRM holds particular promise to be able to screen entire bacterial surfaceomes and to identify targets for novel anti-infectives. Ideally, such studies would be carried out using in vivo infection models. Enabled by the consideration of organism-specific peptide information (Delmotte et al. 2010), they will bring the analysis of mixed in vivo proteomes within reach and complement the power of dual RNA-seq (Westermann et al. 2012) for this task. We expect that the strategy described here will be useful for some of these exciting applications.
Methods
Bacterial growth and subcellular fractionation
The B. henselae strain MQB307 harbors a deletion of the response regulator batR (BH00620) and its cognate sensor histidine kinase batS (BH00610) and carries a plasmid-encoded copy of batR under the control of an IPTG-inducible promoter (for details, see Supplemental Methods; Supplemental Tables S1, S2). MQB307 was grown on Columbia blood agar (CBA) plates supplemented with 30 mg/L kanamycin with (induced condition) or without (uninduced condition) 500 μM IPTG at 35°C and 5% CO2 for 60 h. The subcellular fractionation was performed as previously described (Rhomberg et al. 2004; Supplemental Methods). To maximize the recovery of membrane proteins, the total membrane fraction (TM) was further separated into inner membrane (IM) and outer membrane (OM) fraction.
RNA extraction and whole transcriptome sequencing
RNA was isolated from bacterial cells as described (Quebatte et al. 2010). Whole transcriptome libraries were produced using the RiboMinus Bacterial Transcriptome Isolation Kit (Life Technologies), and the SOLiD Total RNA-seq kit (Applied Biosystems). Briefly, cDNA libraries were size-selected and amplified for 18 cycles of PCR. The whole transcriptome library was used for emulsion-PCR based on a concentration of 0.5 pM. Sequencing beads were pooled and loaded on a full SOLiD-4 slide; between 55–87 million 50-base sequencing reads were generated per library (Supplemental Table S3). For details, see Supplemental Methods.
RNA-seq data processing and transcriptome coverage analysis
The sequenced reads were mapped to the genome sequence of the B. henselae Houston-1 strain using the BioScope 1.3.1 mapping pipeline. Among all uniquely mapping reads, those of lower quality were removed (for more detail, see Supplemental Methods; Supplemental Fig. S12). The count data summary for annotated B. henselae ORFs was generated using the HTSeq package. To create Figure 2A, the filtered reads were shuffled and sequentially mapped to the genome; a protein-coding ORF was classified as expressed when accumulating five or more distinct reads in the 5′ end of the ORF. Based on this data, nonlinear regression models were constructed to estimate the effect of doubling the number of reads. For details, see Supplemental Methods.
Protein and peptide fractionation and mass spectrometry
The subcellular fractions (Cytu/i, TMu/i, IMu/i, OMu/i) were further fractionated biochemically, including OFFGEL electrophoresis at the protein (OGEprot) and peptide level (OGEpep), and size exclusion chromatography (SEC, “gel filtration”). To enrich for low-abundance proteins, we used the ProteoMiner approach (Guerrier et al. 2008). More detail on the biochemical fractionations, digest conditions, and the mass spectrometry set-up is given in the Supplemental Methods and in Supplemental Figure S13. Samples were injected into a NanoLC HPLC system (Eksigent Technologies) by an autosampler, separated on a self-made reverse-phase tip column packed with C18 material, and acquired on an LTQ Orbitrap XL or LTQ FT Ultra mass spectrometer (both Thermo Scientific).
Database searching and data processing
To minimize the chance for false positive assignments, spectra were searched against a combined database (1488 B. henselae proteins, 3336 sheep proteins, a positive control [myc-gfp], and sequences of 256 common contaminants [keratins, trypsin, etc.]) either with Mascot (version 2.3.0, Matrix Science) or with MS-GF+ (MS-GFDB v7747). For Mascot, data were further post-processed with Percolator (Brosch et al. 2009). Based on the target-decoy search approach, a Percolator/MS-GF+ score cutoff was determined that resulted in an estimated 0.01% FDR at the PSM level. All PSMs above this cutoff were classified with the PeptideClassifier software (Qeli and Ahrens 2010), and only peptides (tryptic or semitryptic) that unambiguously imply one bacterial protein sequence were considered (Table 1). For details, see Supplemental Methods.
ADE analysis
Exponential curves were fitted to each block of experiments with a shared biochemical fractionation regimen to find a saturation threshold (Fig. 3A). We then used this fit to predict the saturation beyond the point of experimentally observed PSMs for each biochemical fractionation regimen (Fig. 3A, dashed lines). For details on the exponential model, approximating confidence bands, density estimation of physicochemical parameters, and computation of physicochemical parameters and other protein sequence features, see Supplemental Methods.
Statistical analysis
Statistical tests were performed using the statistical software R 2.15.2 (www.R-project.org). All reported P-values are from hypergeometric tests and are adjusted for multiple testing controlling the corresponding FDR (Benjamini and Hochberg 1995). Significance is based on an alpha level of 5%.
Transcript and protein abundance estimation
Transcript abundance was estimated via RPKM values calculated similar to Mortazavi et al. (2008). The sum of mapped and filtered reads per gene was divided by its length (in kilobases) and the sum of reads for all B. henselae protein-coding genes (in million reads). Relative protein abundance (in ppm) (see Supplemental Fig. S6C) was estimated based on spectral counts as described (Schrimpf et al. 2009).
Orthologs, sequence repeats, and functional protein classification
Orthologous genes conserved in B. henselae, B. tribocorum, and B. grahamii were taken from Engel et al. (2011). To find duplicated regions of 50 nt or longer in the B. henselae genome, we used RepSeek (version 6.5) (Achaz et al. 2007). For functional protein classification, we relied on the eggNOG resource (http://eggnog.embl.de). For details, see Supplemental Methods.
Differential expression analysis
Differential transcript and protein expression analysis was carried out with the R package DESeq (version 1.6.1) (Anders and Huber 2010). Our description of condition-specific complete expressed proteomes precluded the analysis of biological replicates. Since DESeq ranks proteins according to statistical significance, i.e., the top-ranked proteins are observed by many spectra, we minimized the potential to erroneously identify differentially expressed proteins by chance. On the other hand, without replicates, we lack the power to detect lower expressed, truly differentially regulated proteins.
Data access
RNA-seq data have been submitted to the NCBI Genome Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under the GEO Series accession number GSE44564. Proteomics data associated with this manuscript can be downloaded from ProteomeXchange (http://proteomecentral.proteomexchange.org/) under accession number PXD000153.
Acknowledgments
We thank Dr. Ermir Qeli for initial work on the project, Sisi Wu and Dr. Ljiljana Pasa-Tolic (PNNL, USA) for helpful discussions, Dr. Sangtae Kim (PNNL, USA) for the MS-GF+ software, and Drs. Bernd Wollscheid (IMSB, ETH Zürich), Aurelien Carlier, and Gabriella Pessi (Institute of Plant Biology, UZH) for critical reading of the manuscript. C.H.A. acknowledges support from the Swiss National Science Foundation (SNSF) under grant 31003A_130723. C.D. acknowledges support from the SNSF under grant 31003A_132979 and from SystemsX.ch under grant 51RT 0_126008 (RTD InfectX). A part of the research was performed using EMSL, a national scientific user facility sponsored by the Department of Energy's Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory.
References
- ↵G Achaz, F Boyer, EP Rocha, A Viari, E Coissac. 2007. Repseek, a tool to retrieve approximate repeats from large DNA sequences. Bioinformatics 23: 119–121.
- ↵CH Ahrens, E Brunner, E Qeli, K Basler, R Aebersold. 2010. Generating and navigating proteome maps using mass spectrometry. Nat Rev Mol Cell Biol 11: 789–801.
- ↵CM Alsmark, AC Frank, EO Karlberg, BA Legault, DH Ardell, B Canback, AS Eriksson, AK Naslund, SA Handley, M Huvet, . 2004. The louse-borne human pathogen Bartonella quintana is a genomic derivative of the zoonotic agent Bartonella henselae . Proc Natl Acad Sci 101: 9716–9721.
- ↵S Anders, W Huber. 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106.
- ↵BM Balgley, T Laudeman, L Yang, T Song, CS Lee. 2007. Comparative evaluation of tandem MS search algorithms using a target-decoy search strategy. Mol Cell Proteomics 6: 1599–1608.
- ↵D Becher, K Hempel, S Sievers, D Zuhlke, J Pane-Farre, A Otto, S Fuchs, D Albrecht, J Bernhardt, S Engelmann, . 2009. A proteomic view of an important human pathogen–towards the quantification of the entire Staphylococcus aureus proteome. PLoS ONE 4: e8176.
- ↵M Beck, M Claassen, R Aebersold. 2011. Comprehensive proteomics. Curr Opin Biotechnol 22: 3–8.
- ↵Y Benjamini, Y Hochberg. 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57: 289–300.
- ↵M Brosch, L Yu, T Hubbard, J Choudhary. 2009. Accurate and sensitive peptide identification with Mascot Percolator. J Proteome Res 8: 3176–3181.
- ↵E Brunner, CH Ahrens, S Mohanty, H Baetschmann, S Loevenich, F Potthast, EW Deutsch, C Panse, U de Lichtenberg, O Rinner, . 2007. A high-quality catalog of the Drosophila melanogaster proteome. Nat Biotechnol 25: 576–583.
- ↵T Carver, N Thomson, A Bleasby, M Berriman, J Parkhill. 2009. DNAPlotter: Circular and linear interactive genome visualization. Bioinformatics 25: 119–120.
- ↵LM de Godoy, JV Olsen, J Cox, ML Nielsen, NC Hubner, F Frohlich, TC Walther, M Mann. 2008. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 455: 1251–1254.
- ↵N Delmotte, CH Ahrens, C Knief, E Qeli, M Koch, HM Fischer, JA Vorholt, H Hennecke, G Pessi. 2010. An integrated proteomics and transcriptomics reference dataset provides new insights into the Bradyrhizobium japonicum bacteroid metabolism in soybean root nodules. Proteomics 10: 1391–1400.
- ↵M Delory, R Hallez, JJ Letesson, X De Bolle. 2006. An RpoH-like heat shock σ factor is involved in stress response and virulence in Brucella melitensis 16M. J Bacteriol 188: 7707–7710.
- ↵R de Sousa Abreu, LO Penalva, EM Marcotte, C Vogel. 2009. Global signatures of protein and mRNA expression levels. Mol Biosyst 5: 1512–1526.
- ↵C Eberhardt, S Engelmann, H Kusch, D Albrecht, M Hecker, IB Autenrieth, VA Kempf. 2009. Proteomic analysis of the bacterial pathogen Bartonella henselae and identification of immunogenic proteins for serodiagnosis. Proteomics 9: 1967–1981.
- ↵P Engel, W Salzburger, M Liesch, CC Chang, S Maruyama, C Lanz, A Calteau, A Lajus, C Medigue, SC Schuster, . 2011. Parallel evolution of a type IV secretion system in radiating lineages of the host-restricted bacterial pathogen Bartonella . PLoS Genet 7: e1001296.
- ↵F Fischer, D Wolters, M Rogner, A Poetsch. 2006. Toward the complete membrane proteome: High coverage of integral membrane proteins through transmembrane peptide detection. Mol Cell Proteomics 5: 444–453.
- ↵BR Fonslow, PC Carvalho, K Academia, S Freeby, T Xu, A Nakorchevsky, A Paulus, JR Yates 3rd. 2011. Improvements in proteomic metrics of low abundance proteins through proteome equalization using ProteoMiner prior to MudPIT. J Proteome Res 10: 3690–3700.
- ↵B Franz, VA Kempf. 2011. Adhesion and host cell modulation: Critical pathogenicity determinants of Bartonella henselae . Parasit Vectors 4: 54.
- ↵RJ Giannone, H Huber, T Karpinets, T Heimerl, U Kuper, R Rachel, M Keller, RL Hettich, M Podar. 2011. Proteomic characterization of cellular and molecular processes that enable the Nanoarchaeum equitans-Ignicoccus hospitalis relationship. PLoS ONE 6: e22942.
- MA Grobei, E Qeli, E Brunner, H Rehrauer, R Zhang, B Roschitzki, K Basler, CH Ahrens, U Grossniklaus. 2009. Deterministic protein inference for shotgun proteomics data provides new insights into Arabidopsis pollen development and function. Genome Res 19: 1786–1800.
- ↵L Guerrier, PG Righetti, E Boschetti. 2008. Reduction of dynamic protein concentration range of biological extracts for the discovery of low-abundance proteins by means of hexapeptide ligand library. Nat Protoc 3: 883–890.
- ↵A Harms, C Dehio. 2012. Intruders below the radar: Molecular pathogenesis of Bartonella spp. Clin Microbiol Rev 25: 42–78.
- ↵AO Helbig, AJ Heck, M Slijper. 2010. Exploring the membrane proteome–challenges and analytical strategies. J Proteomics 73: 868–878.
- ↵AL Hopkins, CR Groom. 2002. The druggable genome. Nat Rev Drug Discov 1: 727–730.
- ↵JD Jaffe, N Stange-Thomann, C Smith, D DeCaprio, S Fisher, J Butler, S Calvo, T Elkins, MG FitzGerald, N Hafez, . 2004. The complete genome and proteome of Mycoplasma mobile . Genome Res 14: 1447–1461.
- ↵M Juhas, JR van der Meer, M Gaillard, RM Harding, DW Hood, DW Crook. 2009. Genomic islands: Tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol Rev 33: 376–393.
- ↵S Kim, N Gupta, PA Pevzner. 2008. Spectral probabilities and generating functions of tandem mass spectra: A strike against decoy databases. J Proteome Res 7: 3354–3363.
- ↵S Kim, N Mischerikow, N Bandeira, JD Navarro, L Wich, S Mohammed, AJ Heck, PA Pevzner. 2010. The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: Applications to database search. Mol Cell Proteomics 9: 2840–2852.
- ↵DB Kristensen, JC Brond, PA Nielsen, JR Andersen, OT Sorensen, V Jorgensen, K Budin, J Matthiesen, P Veno, HM Jespersen, . 2004. Experimental Peptide Identification Repository (EPIR): An integrated peptide-centric platform for validation and mining of tandem mass spectrometry data. Mol Cell Proteomics 3: 1023–1038.
- ↵B Kuster, M Schirle, P Mallick, R Aebersold. 2005. Scoring proteomes with proteotypic peptide probes. Nat Rev Mol Cell Biol 6: 577–583.
- ↵I Letunic, T Doerks, P Bork. 2012. SMART 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res 40: D302–D305.
- ↵DL Leyton, AE Rossiter, IR Henderson. 2012. From self sufficiency to dependence: Mechanisms and factors important for autotransporter biogenesis. Nat Rev Microbiol 10: 213–225.
- ↵DM Li, QY Liu, F Zhao, Y Hu, D Xiao, YX Gu, XP Song, JZ Zhang. 2011. Proteomic and bioinformatic analysis of outer membrane proteins of the protobacterium Bartonella henselae (Bartonellaceae). Genet Mol Res 10: 1789–1818.
- ↵CM Litwin, ML Rawlins, EM Swenson. 2007. Characterization of an immunogenic outer membrane autotransporter protein, Arp, of Bartonella henselae . Infect Immun 75: 5255–5263.
- ↵T Maier, A Schmidt, M Guell, S Kuhner, AC Gavin, R Aebersold, L Serrano. 2011. Quantification of mRNA and protein and integration with protein turnover in a bacterium. Mol Syst Biol 7: 511.
- ↵P Mallick, M Schirle, SS Chen, MR Flory, H Lee, D Martin, J Ranish, B Raught, R Schmitt, T Werner, . 2007. Computational prediction of proteotypic peptides for quantitative proteomics. Nat Biotechnol 25: 125–131.
- ↵J Malmstrom, M Beck, A Schmidt, V Lange, EW Deutsch, R Aebersold. 2009. Proteome-wide cellular protein concentrations of the human pathogen Leptospira interrogans . Nature 460: 762–765.
- ↵M Mann, NA Kulak, N Nagaraj, J Cox. 2013. The coming age of complete, accurate and ubiquitous proteomes. Mol Cell 49: 583–590.
- ↵S Marguerat, A Schmidt, S Codlin, W Chen, R Aebersold, J Bahler. 2012. Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells. Cell 151: 671–683.
- ↵A Mortazavi, BA Williams, K McCue, L Schaeffer, B Wold. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628.
- ↵AI Nesvizhskii. 2010. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J Proteomics 73: 2092–2123.
- ↵SR Norrby, CE Nord, R Finch. 2005. Lack of development of new antimicrobial drugs: A potential serious threat to public health. Lancet Infect Dis 5: 115–119.
- ↵P Picotti, R Aebersold. 2012. Selected reaction monitoring-based proteomics: Workflows, potential, pitfalls and future directions. Nat Methods 9: 555–566.
- ↵A Poetsch, D Wolters. 2008. Bacterial membrane proteomics. Proteomics 8: 4100–4122.
- ↵S Powell, D Szklarczyk, K Trachana, A Roth, M Kuhn, J Muller, R Arnold, T Rattei, I Letunic, T Doerks, . 2012. eggNOG v3.0: Orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res 40: D284–D289.
- ↵E Qeli, CH Ahrens. 2010. PeptideClassifier for protein inference and targeted quantitative proteomics. Nat Biotechnol 28: 647–650.
- ↵M Quebatte, M Dehio, D Tropel, A Basler, I Toller, G Raddatz, P Engel, S Huser, H Schein, HL Lindroos, . 2010. The BatR/BatS two-component regulatory system controls the adaptive response of Bartonella henselae during human endothelial cell infection. J Bacteriol 192: 3352–3367.
- ↵L Reiter, M Claassen, SP Schrimpf, M Jovanovic, A Schmidt, JM Buhmann, MO Hengartner, R Aebersold. 2009. Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Mol Cell Proteomics 8: 2405–2417.
- ↵TA Rhomberg, O Karlberg, T Mini, U Zimny-Arndt, U Wickenberg, M Rottgen, PR Jungblut, P Jeno, SG Andersson, C Dehio. 2004. Proteomic analysis of the sarcosine-insoluble outer membrane fraction of the bacterial pathogen Bartonella henselae . Proteomics 4: 3021–3033.
- ↵JA Roden, DH Wells, BB Chomel, RW Kasten, JE Koehler. 2012. Hemin binding protein C is found in outer membrane vesicles and protects Bartonella henselae against toxic concentrations of hemin. Infect Immun 80: 929–942.
- ↵JN Savas, BD Stein, CC Wu, JR Yates 3rd. 2011. Mass spectrometry accelerates membrane protein analysis. Trends Biochem Sci 36: 388–396.
- ↵A Schmidt, M Beck, J Malmstrom, H Lam, M Claassen, D Campbell, R Aebersold. 2011. Absolute quantification of microbial proteomes at different states by directed mass spectrometry. Mol Syst Biol 7: 510.
- ↵SP Schrimpf, M Weiss, L Reiter, CH Ahrens, M Jovanovic, J Malmstrom, E Brunner, S Mohanty, MJ Lercher, PE Hunziker, . 2009. Comparative functional analysis of the Caenorhabditis elegans and Drosophila melanogaster proteomes. PLoS Biol 7: e48.
- ↵R Schulein, C Dehio. 2002. The VirB/VirD4 type IV secretion system of Bartonella is essential for establishing intraerythrocytic infection. Mol Microbiol 46: 1053–1067.
- ↵B Schwanhausser, D Busse, N Li, G Dittmar, J Schuchhardt, J Wolf, W Chen, M Selbach. 2011. Global quantification of mammalian gene expression control. Nature 473: 337–342.
- ↵PM Sharp, WH Li. 1987. The codon Adaptation Index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res 15: 1281–1295.
- ↵S Tan, HT Tan, MC Chung. 2008. Membrane proteins and membrane proteomics. Proteomics 8: 3924–3932.
- ↵D Vallenet, L Labarre, Z Rouy, V Barbe, S Bocs, S Cruveiller, A Lajus, G Pascal, C Scarpelli, C Medigue. 2006. MaGe: A microbial genome annotation system supported by synteny results. Nucleic Acids Res 34: 53–65.
- ↵M Vayssier-Taussat, D Le Rhun, HK Deng, F Biville, S Cescau, A Danchin, G Marignac, E Lenaour, HJ Boulouis, M Mavris, . 2010. The Trw type IV secretion system of Bartonella mediates host-specific adhesion to erythrocytes. PLoS Pathog 6: e1000946.
- ↵E Venter, RD Smith, SH Payne. 2011. Proteogenomic analysis of bacteria and archaea: A 46 organism case study. PLoS ONE 6: e27587.
- ↵Z Wang, M Gerstein, M Snyder. 2009. RNA-Seq: A revolutionary tool for transcriptomics. Nat Rev Genet 10: 57–63.
- ↵AJ Westermann, SA Gorski, J Vogel. 2012. Dual RNA-seq of pathogen and host. Nat Rev Microbiol 10: 618–630.
- ↵NY Yu, JR Wagner, MR Laird, G Melli, S Rey, R Lo, P Dao, SC Sahinalp, M Ester, LJ Foster, . 2010. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26: 1608–1615.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.151035.112.