
Sumoylation at chromatin governs coordinated repression of a transcriptional program essential for cell growth and proliferation
- Hélène Neyret-Kahn1,2,3,
- Moussa Benhamed1,2,3,6,
- Tao Ye4,
- Stéphanie Le Gras4,
- Jack-Christophe Cossec1,2,3,
- Pierre Lapaquette1,2,3,
- Oliver Bischof1,2,3,
- Maia Ouspenskaia5,
- Mary Dasso5,
- Jacob Seeler1,2,3,
- Irwin Davidson3,4,7 and
- Anne Dejean1,2,3,7
- 1Institut Pasteur, Laboratory of Nuclear Organization and Oncogenesis, Department of Cell Biology and Infection, F-75015 Paris, France;
- 2INSERM, U993, F-75015 Paris, France;
- 3Equipe Labellisée Ligue Contre le Cancer, F-75013 Paris, France;
- 4Department of Functional Genomics and Cancer, Institut de Génétique et de Biologie Moléculaire et Cellulaire, CNRS/INSERM/UDS, 67404 Illkirch Cedex, France;
- 5Laboratory of Gene Regulation and Development, National Institute for Child Health and Human Development, National Institutes of Health, Bethesda, Maryland 20892, USA
Abstract
Despite numerous studies on specific sumoylated transcriptional regulators, the global role of SUMO on chromatin in relation to transcription regulation remains largely unknown. Here, we determined the genome-wide localization of SUMO1 and SUMO2/3, as well as of UBC9 (encoded by UBE2I) and PIASY (encoded by PIAS4), two markers for active sumoylation, along with Pol II and histone marks in proliferating versus senescent human fibroblasts together with gene expression profiling. We found that, whereas SUMO alone is widely distributed over the genome with strong association at active promoters, active sumoylation occurs most prominently at promoters of histone and protein biogenesis genes, as well as Pol I rRNAs and Pol III tRNAs. Remarkably, these four classes of genes are up-regulated by inhibition of sumoylation, indicating that SUMO normally acts to restrain their expression. In line with this finding, sumoylation-deficient cells show an increase in both cell size and global protein levels. Strikingly, we found that in senescent cells, the SUMO machinery is selectively retained at histone and tRNA gene clusters, whereas it is massively released from all other unique chromatin regions. These data, which reveal the highly dynamic nature of the SUMO landscape, suggest that maintenance of a repressive environment at histone and tRNA loci is a hallmark of the senescent state. The approach taken in our study thus permitted the identification of a common biological output and uncovered hitherto unknown functions for active sumoylation at chromatin as a key mechanism that, in dynamically marking chromatin by a simple modifier, orchestrates concerted transcriptional regulation of a network of genes essential for cell growth and proliferation.
The post-translational modification by SUMO is an essential regulatory mechanism of protein function involved in most challenges faced by eukaryotic cells (Hay 2005; Geiss-Friedlander and Melchior 2007; Hochstrasser 2009). Higher eukaryotes have three SUMO paralogs, SUMO1, SUMO2, and SUMO3, with SUMO2 and SUMO3 collectively termed SUMO2/3 because of structural and functional differences from SUMO1. Similarly to ubiquitin, SUMO is covalently conjugated to its targets via a three-step process, including unique E1 (SAE1/UBA2), E2 (UBC9 encoded by UBE2I), and a series of E3 enzymes including the five PIAS members, CBX4, and RANBP2. The SUMO proteases (SENPs) then remove SUMO from its substrates (Yeh 2009).
Investigation of numerous sumoylated transcription factors and chromatin-associated proteins reveals that, in most cases, sumoylation is associated with transcriptional repression (Ouyang and Gill 2009). Moreover, important roles for sumoylation were underscored in heterochromatin configuration (Shin et al. 2005; Maison et al. 2011), and sumoylation of core histones was shown to negatively regulate transcription in yeast and human cells (Shiio and Eisenman 2003; Nathan et al. 2006). However a growing body of evidence also links sumoylation to active transcription (Lyst and Stancheva 2007). In support of these findings, we previously showed that heat shock induces recruitment of the sumoylation machinery at induced promoters (Martin et al. 2009). Moreover, a study performed on a specific set of genes in yeast showed SUMO at promoters of constitutively active genes but without regulating their steady-state expression level, whereas UBC9 is recruited at inducible promoters upon activation, though in this case, to shut off their transcription (Rosonina et al. 2010). These data highlight the high level of complexity and different functions that chromatin-associated SUMO may exert on gene expression.
To comprehensively characterize the SUMO machinery chromatin landscape, its dynamics, and its global role in gene expression, we determined the genome-wide distribution of SUMO1, SUMO2, UBC9, and PIASY (encoded by PIAS4) along with Pol II and histone marks in proliferating versus senescent human fibroblasts coupled to gene expression profiling. We show that the SUMO machinery is prominently enriched at promoters of histone and protein biogenesis genes, as well as Pol I rRNAs and Pol III tRNAs. Remarkably, SUMO-marked genes repressed by sumoylation represent a highly homogeneous set of functionally related genes essential for cell growth and proliferation, notably those histone, ribosomal protein, rRNA, and tRNA genes that figure as the most repressed. Sumoylation therefore appears as a central process governing coordinated transcriptional repression of histone and growth control genes. This latter concept was reinforced by our finding that the SUMO machinery is lost from chromatin and selectively retained on the histone and tRNA genes upon senescence. Together, these data suggest that sumoylated proteins are integral and instructive components of a dynamic chromatin scaffold that actively regulates cell growth/proliferation and the stability of the senescent state.
Results
SUMO1 and SUMO2 are widely distributed over the genome in proliferating human fibroblasts
To profile sumoylated proteins on the genome of proliferating primary cells, we performed chromatin immunoprecipitation coupled to DNA sequencing (ChIP-seq) in WI38 human fibroblasts using antibodies specific for SUMO1 and SUMO2/3 (henceforth referred to as SUMO2). The specificity of these antibodies was confirmed by Western blot analysis both by overexpression and siRNA-mediated knockdown (Supplemental Fig. S1A). Genomic SUMO1 and SUMO2 profiling was performed along with ChIP-seq for RNA polymerase II (Pol II), the active chromatin mark H3K4me3, and the two repressive marks, H3K27me3 and H3K9me3. ChIPed DNA was sequenced, and peak detection and annotation were performed (Supplemental Fig. S1B).
We identified 25,268 peaks for SUMO1 (Fig. 1A, left), using a minimum peak height of 20 as a cutoff in this and all analyses described here. The reproducibility of this profile was verified with a second independent experiment showing a high correlation between the two data sets (Supplemental Fig. S1C). When allocated to defined genomic regions, the greatest enrichment (5.7-fold) is observed at core promoters accounting for 17% of SUMO1 peaks, and takes the form of a major specific peak at the transcription starting site (TSS) (Fig. 1B,C). SUMO1 occupancy at the TSS was further confirmed by ChIP-qPCR experiments (Supplemental Fig. S1D).
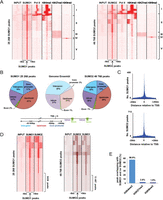
Chromatin profiles of SUMO1 and SUMO2. (A) Association of SUMO1 (left) and SUMO2 (right) with Pol II, H3K4me3, H3K27me3, and H3K9me3 in WI38 cells. Comparison of tag density in the region of ±5 kb around the SUMO-occupied loci. Clustering identifies five classes as indicated. (B) A pie chart representation of the distribution of SUMO sites in five different genomic regions. The definition of each region is described below. (C) Frequency of SUMO1 and SUMO2 site localization with respect to TSS. (D) Comparison of SUMO2 tag density around the SUMO1 peaks (left) and of SUMO1 tag density around the SUMO2 peaks (right). (E) Histogram representing the percentage of the H3K4me3, H3K27me3, or H3K9me3 peaks with an overlapping SUMO1 and/or SUMO2 peak.
Based on the same cut off criteria, analysis of SUMO2 genomic distribution identified 46,768 peaks (Fig. 1A, right) allocated to genomic regions in similar proportions to SUMO1 (Fig. 1B). Notably, the strongest enrichment (fivefold) corresponding to 15% of the SUMO2 peaks was again found as a specific discrete peak at the TSS (Fig. 1C; Supplemental Fig. S1D). Comparison of the SUMO1 and SUMO2 profiles indicated co-occupancy of about two thirds of the marked loci by SUMO1 and SUMO2 (Fig. 1D). However, a significant fraction (33%) of SUMO1 sites are virtually free of SUMO2. Reciprocally, 29% of SUMO2 sites show almost no SUMO1. These results point to the existence of a significant number of genomic sites that display high specificity for each SUMO paralog.
A de novo motif analysis did not reveal a restricted set of consensus sequences at sites marked by SUMO1 or SUMO2, but rather a complex repertoire of motifs corresponding to multiple mammalian transcription factor binding sites. At SUMO2 sites, a large collection of binding motifs are detected, ∼75% of which are also found at SUMO1 sites (Supplemental Table S1). In contrast, no sites were specific for SUMO1 in this setting. Although many of the transcription factors identified in this way are known to be sumoylated, there was so far very little evidence that their sumoylated form is indeed chromatin-associated. Our analysis now provides evidence that many transcription factors are sumoylated when bound to their cognate sites on DNA, thus identifying SUMO as an integral component of the chromatin.
Integration of the SUMO1 and SUMO2 data sets with those of Pol II and chromatin marks defined five classes of SUMO-labeled genomic sites. In agreement with the observed SUMO1/2 peaks at the TSS, we found a subset of SUMO1 and SUMO2 sites to be tightly associated with high levels of Pol II and H3K4me3 (Fig. 1A, cluster I). A second cluster (II) shows only low levels of H3K4me3 and Pol II. SUMO1 and SUMO2 are also found at repressive chromatin in regions marked by the heterochromatin-type H3K27me3 and H3K9me3 modifications in the absence of Pol II (clusters III and IV, respectively). A further large class of SUMO sites (cluster V) is located in regions that show no high enrichment for any of these chromatin modifications. SUMO1 and/or SUMO2 are present at 56.9% of the H3K4me3 sites, whereas only 2.6% and 1.9% of the H3K27me3 and H3K9me3 sites were occupied by SUMO1/2 (Fig. 1E). SUMO is therefore preferentially and strongly associated with active chromatin regions, yet present at a subset of heterochromatin loci.
We further investigated whether SUMO was also associated with potential active enhancer regions marked by high levels of acetylated H3K27, the EP300 coactivator and DNase I hypersensitivity, but low H3K4me3. To this end, we compared our SUMO1/2 and H3K4me3 data sets with public ENCODE project data sets. This analysis identified 1100 SUMO1 peaks and 2760 SUMO2 peaks that colocalize with high signals for EP300 and H3K27ac, with low H3K4me3, and with DNase I hypersensitivity (Supplemental Fig. S2A,B). These sites are not strongly enriched around the TSS, but dispersed at various distances from the TSS and share the characteristics of active enhancers. SUMO is therefore preferentially and strongly associated with active chromatin regions at both the TSS and enhancers, yet present at a subset of heterochromatin loci.
Sumoylation at the TSS positively correlates with Pol II transcriptional activity
To assess the relationship of the SUMO1 and SUMO2 chromatin landscapes with gene expression, the ChIP-seq data were combined with mRNA-seq profiling of proliferating WI38 cells (Supplemental Table S2). Integration of these data indicates that the majority of the transcriptionally active promoters with high Pol II density and expressed transcripts are strongly marked by SUMO1/2 (Fig. 2A). Of the 3044 and 4718 TSS occupied by SUMO1 and SUMO2, respectively, 67% are associated with significantly expressed transcripts, as defined by an RPKM value greater than two in each of the three biological replicates (Fig. 2B). At active promoters, SUMO1/2 are located immediately upstream of the TSS in the nucleosome-depleted region, where the preinitiation complex (PIC) forms (Choukrallah et al. 2012), while, as previously described (Core and Lis 2009), the major peak of Pol II is found 30–50 nucleotides downstream from the TSS (Fig. 2C). Moreover, we found a global positive correlation of SUMO1/2 and Pol II presence at the TSS with the level of transcript expression (Fig. 2D). Promoters of higher expressed transcripts show generally higher Pol II and SUMO occupancy than those with lower activity. Multiple subunits of the PIC have been shown to be sumoylated in proteomic analysis (Panse et al. 2004; Zhao et al. 2004; Rosas-Acosta et al. 2005; Hornbeck et al. 2012). The specific localization of the SUMO signal immediately upstream of the TSS suggests that sumoylation of one or several component(s) of the PIC is positively associated with promoter activity.
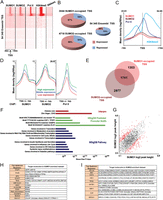
SUMO is highly enriched at promoters of actively transcribed genes. (A) Clustering comparison of SUMO1, SUMO2, Pol II, and RNA-seq reads on TSS. (B) Expression status of SUMO-bound TSS (expression defined by a minimum of two RPKM in three mRNA-seq replicates). (C) Merged profiles of SUMO1, SUMO2, Pol II, and H3K4me3 read density with respect to distance from active TSS. (D) Mean profile of SUMO1, SUMO2, and Pol II read density with respect to distance from TSS of genes showing low, middle, and high levels of expression. (E) Venn diagram representing overlap between SUMO1- and SUMO2-marked TSS. (F) Functional annotation of TSS commonly occupied by SUMO1 and SUMO2 using GREAT. The top overrepresented categories belonging to three different ontologies are shown. (G) Scatterplot comparison of common SUMO1- and SUMO2-marked TSS. In red, TSS with a SUMO1/SUMO2 read ratio ≤0.5 or ≥2. (H) IPA upstream regulator analysis of SUMO1-enriched TSS (≥ twofold). (I) Same as in H but for SUMO2.
SUMO1 and SUMO2 co-occupy the TSS at numerous promoters (Fig. 2E). Ontology analysis using the Genomic Regions Enrichment of Annotations Tool (GREAT) (McLean et al. 2010) revealed that these promoters are enriched for genes associated with fundamental processes such as transcription, translation, protein degradation, and cell cycle as well as for those involved in diabetes and insulin pathway, viral infection, and autoimmune disorders (Fig. 2F). Also, SUMO1/2 are strongly enriched at promoters of genes encoding zinc finger and histone proteins. Analysis of the transcription factor binding motifs at SUMO-marked promoters indicated an enrichment for ELK1, GABPA, E2F1, as well as for another motif that does not match known transcription factor sites but is frequently found in histone gene promoters (Fig. 2F).
In a complementary analysis, we determined the TSS selectively enriched (≥ twofold) in either SUMO1 or 2 (Fig. 2G, indicated in red) and analyzed their associated upstream regulators. Although the genes enriched in SUMO1 are associated with diverse pathways and regulators (Fig. 2H), SUMO2-enriched genes are associated with cell cycle control and the key regulatory factors RB1, E2F, and TP53 (Fig. 2I).
Thus, in apparent contrast with the notion that the prominent effect of sumoylation on transcription is repression, we found here a positive correlation with promoter activity as well as an enrichment of SUMO2 at the TSS of genes involved in cell cycle control.
Distinct sumoylation targets in H3K27me3- and H3K9me3-repressed chromatin
Although active promoters are major sites of SUMO1/2 in the genome, SUMO1/2 are also associated with the repressive H3K27me3 and H3K9me3 chromatin modifications (Fig. 1A, clusters III and IV). In the H3K27me3-marked cluster III, the SUMO1/2 sites are enriched at the TSS of a subset of inactive promoters devoid of Pol II (Supplemental Fig. S3A,B). Ontology analysis of the genes associated with these promoters showed enrichment in terms such as homeobox and in developmentally regulated genes (Supplemental Fig. S3C). These promoters direct expression of highly tissue-specific genes, most of which are silenced in fibroblasts. De novo sequence motif analysis under the SUMO1/2 peaks at these promoters identified a high proportion with the consensus for the CTCF transcription factor (CCCTC-binding factor) (Supplemental Fig. S3D). Although CTCF consensus motifs are present at SUMO sites in all clusters, they are most enriched in cluster III. This can be observed at the HOXA10 gene, where a CTCF site in the promoter region coincides with SUMO1/2 peaks (Supplemental Fig. S3E). Sumoylation of CTCF has been shown to repress its transcriptional activation function (MacPherson et al. 2009; Kitchen and Schoenherr 2010), in agreement with the idea that many SUMO peaks in cluster III correspond to sumoylated CTCF bound to promoters of inactive developmentally regulated genes. Moreover, a global analysis shows colocalization of a significant subset (27%) of all SUMO1 peaks with experimentally determined sites for both CTCF and its interacting partner, the cohesin subunit STAG1 (Supplemental Fig. S3F; Rubio et al. 2008).
In contrast, SUMO-occupied sites in H3K9me3-marked cluster IV are not enriched close to the TSS (Supplemental Fig. S3G), and these SUMO1/2 peaks coincide with a wider region of H3K9me3 (Supplemental Fig. S3H). Ontology analysis of the associated genes indicated mostly enrichment in zinc-finger transcription factors (Supplemental Fig. S3I). For example, at the ZNF134 and ZNF174 loci, peaks of SUMO1 and SUMO2 clearly colocalize with H3K9me3 at the 3′ end of the gene (Supplemental Fig. S3J,K). These sites also colocalize with those occupied by the TRIM28 (KAP1) corepressor and the SETDB1 histone methyltransferase that trimethylates H3K9. Accordingly, global clustering analysis identified a subset (12%) of SUMO1 sites that colocalize with TRIM28 (Supplemental Fig. S3F). These observations are in keeping with those showing that TRIM28 sumoylation is required for recruitment of SETDB1 on chromatin (Ivanov et al. 2007). However as previously reported (Blahnik et al. 2011), this mark does not lead to gene silencing as the ZNF134 and ZNF174 promoters display H3K4me3 and the presence of Pol II.
CTCF is therefore a major candidate substrate for SUMO1/2 in H3K27me3-repressed chromatin, whereas most SUMO1/2 sites associated with H3K9me3 likely represent sumoylated TRIM28 bound to the 3′ exon of ZNF genes.
SUMO is highly enriched at histone, ribosomal protein, and tRNA genes
We next asked whether specific gene families are associated with sites exhibiting highest levels of SUMO1 and SUMO2. The SUMO1 and SUMO2 TSS sites were ranked from the lowest to highest occupancy and the genes associated with the top quartile of sites (Fig. 3A,B, gray bars) were analyzed. For both SUMO1 and SUMO2, genes associated with the term nucleosome core are the most strongly enriched in the list (Fig. 3A,B; Supplemental Table S3). Visualization of genomic loci encoding clusters of histone genes on the UCSC browser confirms the high levels of both SUMO1 and SUMO2 at their promoters, as exemplified by the major histone cluster on chromosome 6 (Fig. 3C). In addition, the genes encoding proteins involved in protein biosynthesis are also highly represented in the top quartile of SUMO1/2-occupied sites (Fig. 3A,B; Supplemental Table S3). As the transcripts encoding ribosomal proteins are often highly expressed, their representation in the top quartile is in agreement with the positive correlation between SUMO and expression described above. In contrast and more interestingly, transcripts from the histone genes are not among the most highly expressed in asynchronized cells, and thus the high levels of SUMO at their TSS is likely to reflect a more specific function. It is also noteworthy that the positive correlation of SUMO1/2 presence at the TSS with the level of transcript expression was maintained even when the ribosomal protein and histone genes TSS were excluded from the analysis (Supplemental Fig. S4).
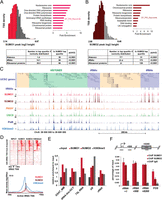
SUMO is highly enriched at histone, ribosomal protein, and tRNA genes. (A,B) Distribution of (A) SUMO1- and (B) SUMO2-marked TSS by peak height and TSS density showing the top quartile corresponding to the highest SUMO peaks in gray. Functional annotation of genes associated to the top quartile TSS using DAVID. The top overrepresented categories are shown. Table below summarizes the statistical enrichment of SUMO-bound TSS for histone, tRNA, and ribosomal protein annotations. (C) A Genome Browser view of ChIP-seq data across the histone and tRNA gene cluster in chromosome 6p. (D) Comparison of SUMO1, SUMO2, and H3K4me3 tag density at tRNA loci (upper) and their merged profiles restricted to the H3K4me3-positive subcluster with respect to distance from TSS (lower). (E) Relative enrichment of SUMO1, SUMO2, and H3K9me3 reads at the major satellite repeat (GSAT_MM), a tRNA, RN7SL1 (7SL), RNU6 (U6), and rRNA loci in the ChIP-seq compared to input. (F) ChIP-qPCR for SUMO1 and SUMO2 on three regions of the rRNA gene amplified with specific primers (Guetg et al. 2012). FOS promoter is used here as a positive control.
Analysis of the regions showing high levels of SUMO1/2 also revealed their strong enrichment at Pol III-transcribed tRNA genes, as clearly observed at the representative tRNA cluster on chromosome 6 (Fig. 3C), where prominent peaks of SUMO1 and SUMO2 are associated with a large fraction of the tRNA genes. Occupancy at these sites is comparable, or even higher for SUMO2, than at the neighboring histone genes. Global analysis of SUMO at the 625 annotated tRNA TSS indicated that SUMO is present only at active tRNAs as defined by the presence of H3K4me3 (Fig. 3D).
Analysis of different repetitive element sequences in the SUMO1 and SUMO2 ChIP-seq compared to input showed enrichment of SUMO1 and, to a lesser extent, of SUMO2 with the major satellite repeats, in agreement with binding of sumoylated CBX5 (HP1 alpha homolog [Drosophila]) to these elements (Maison et al. 2011), as well as to a tRNA family in agreement with our findings above (Fig. 3E). Enrichment of SUMO1 and SUMO2 was also observed at the Pol III-transcribed RN7SL1 and RNU6 genes as well as the Pol I-transcribed rRNA genes. The significant enrichment of SUMO1 and SUMO2 on the promoter and coding regions of rRNA genes was confirmed by ChIP-qPCR (Fig. 3F).
Together, these observations show that the highest levels of SUMO are found not only with the Pol II-transcribed histone and ribosomal protein genes, but also the Pol III-transcribed tRNA, RN7SL1 and RNU6 genes, and the Pol I-dependent rRNA genes. Thus, genes ensuring sustained cell growth and proliferation are therefore major SUMO targets on chromatin.
Active sumoylation on chromatin
To next find out whether the SUMO chromatin profile represents mainly recruitment of presumoylated proteins or whether sumoylation of target proteins may occur on chromatin, we performed ChIP-seq profiling of the UBC9 conjugating enzyme and one of the SUMO E3 ligases, PIASY, which we previously showed to induce premature senescence (Bischof et al. 2006) (see below). The specificity of these antibodies was verified in the same way as for SUMO1/2 (Supplemental Fig. S1A).
Profiling of UBC9 revealed 7486 occupied loci (Fig. 4A) and a genomic distribution comparable to those of SUMO1 and SUMO2 with strong enrichment at the core promoters (Fig. 4B). However, the number of UBC9 sites is significantly less than the number of SUMO marks. Consequently, the vast majority of SUMO1/2 sites do not show a significant presence of UBC9 (Supplemental Fig. S5A). Conversely, UBC9 is also found at sites almost free of SUMO1/2 (Fig. 4A, part of cluster I and clusters II and IV), suggesting that, at these loci, it may be in an inactive state or that conjugation by SUMO is highly transient. UBC9 is present at active core promoters marked by high levels of Pol II and H3K4me3 (Fig. 4A, cluster I), most of which are also occupied by SUMO1/2-conjugated proteins. In contrast, the vast majority of SUMO1/2-occupied promoters is not bound by UBC9 (Fig. 4C).
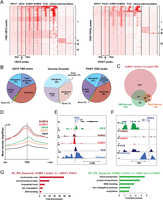
Chromatin profiles of UBC9 and PIASY. (A) Association of UBC9 (left) and PIASY (right) with SUMO1, SUMO2, Pol II, H3K4me3, H3K27me3, and H3K9me3 in WI38 proliferating cells. Comparison of the tag density in the ±5-kb region around UBC9- or PIASY-bound loci. Clustering identifies four classes as indicated. (B) Pie chart representation of UBC9 and PIASY binding site distribution in five different genomic regions as described in Fig. 1B. (C) Venn diagram representing overlap between SUMO1- and/or SUMO2-, UBC9- and PIASY-marked TSS. (D) Merged profiles of the SUMO machinery read density with respect to distance from TSS. (E,F) A Genome Browser view of the indicated ChIP-seq data at the FOS (E) and HRAS loci (F). (G) DAVID functional annotation of TSS marked by SUMO, UBC9, and PIASY (left) or SUMO, UBC9, but no PIASY (right). The top overrepresented categories are shown.
UBC9 is also present at repressed regions marked by H3K27me3 and H3K9me3 (Fig. 4A, clusters II and III). In cluster II, UBC9 is not associated with a high SUMO1/2 signal, whereas in cluster III, UBC9 colocalizes with the SUMO1/2 that marks TRIM28 on the genes encoding zinc finger proteins, suggesting that active sumoylation may occur at these sites. Yet, the absence of UBC9 at the majority of SUMO1/2-occupied sites supports the idea that proteins are predominantly recruited in a presumoylated state and that active sumoylation on chromatin occurs only at a specific set of sites in normally growing fibroblasts.
This idea is further supported by profiling of PIASY, in which only 3520 bound sites were detected distributed throughout the genome (Fig. 4A). PIASY-occupied sites are associated with active core promoters (Fig. 4B) as well as with regions marked by H3K27me3 and H3K9me3 (Fig. 4A, clusters II and III), the latter coinciding with SUMO1/2-conjugated proteins, similar to that observed for UBC9. Moreover, most SUMO sites are devoid of PIASY (Fig. 4C; Supplemental Fig. S5A), a finding in keeping with the existence of multiple SUMO E3 ligases.
Interestingly, while both UBC9 and PIASY are present at a subset of core promoters, their precise binding sites do not coincide with those of SUMO1/2 at these promoters. The major peaks of UBC9 and PIASY lie downstream from the TSS, whereas those of SUMO1/2 are immediately upstream of the TSS (Fig. 4D). A representative example is shown at the FOS gene promoter (Fig. 4E). The UBC9 molecules present at such promoters may therefore not necessarily be involved in sumoylation of the proteins detected upstream of the TSS. Alternatively, UBC9 and the substrate may be brought into proximity by the chromatin architecture. In some cases, UBC9 is also found highly enriched within genes, as exemplified by the HRAS locus (Fig. 4F), suggesting that it may function in transcriptional elongation and/or splicing at these loci.
Comparison of TSS occupancy by SUMO1 and/or SUMO2, UBC9, and PIASY showed that only a small number of sites are co-occupied by all of these components (Fig. 4C). Strikingly, ontology analysis of the genes associated with these TSS indicated a remarkable enrichment for the histone genes (Fig. 4G; Supplemental Table S4). Browser views documenting the high levels of UBC9 and PIASY colocalizing with SUMO1/2 at the two histone gene clusters on chromosomes 1 and 6 are shown in Supplemental Figure S5B,C. Interestingly, histone loci, such as those on chromosome 6, represent by far the major site of enrichment for PIASY on the genome (Supplemental Fig. S5D), suggesting an important role for PIASY at these TSS. In addition, we observed a clear colocalization of UBC9 with sumoylated proteins at the tRNA genes, whereas PIASY is barely visible, suggesting that it may be replaced by another E3 ligase in this region (Fig. 3C; Supplemental Fig. S5E,F). A similar situation is seen at the ribosomal protein genes (see for example RPL26 in Supplemental Fig. S3G) or the gene encoding the noncoding RNA RMRP (Supplemental Fig. S5H). Accordingly, genes involved in protein biosynthesis were the most highly enriched among those co-occupied by SUMO1/2 and UBC9 (Fig. 4G).
The general lack of UBC9 at SUMO-marked sites suggests that most proteins are recruited in a presumoylated fashion, but their colocalization on the histone, tRNA, and ribosomal protein genes indicates that these loci constitute major sites for active sumoylation on chromatin.
Sumoylation negatively regulates expression of histone and growth control genes
To address the role sumoylation may have on the expression of the above genes that display high levels of SUMO and its associated machinery, we used si/shRNAs to silence expression of UBC9, SUMO, and PIASY. Lentiviral-mediated expression of an shRNA against UBC9 strongly down-regulated its expression, leading to a dramatic loss of sumoylated proteins such as SP100 compared to infection with a control nontargeting shRNA (Fig. 5A,B). Loss of active sumoylation up-regulated histone gene expression at both the mRNA and protein levels, whereas that of control genes such as GAPDH was unaffected (Fig. 5A,B). SiRNA-mediated SUMO (SUMO1 + SUMO2/3) knockdown also increased histone gene expression, although to a lesser extent, that may reflect the lower knockdown efficiency (Supplemental Fig. S6A). Accordingly, increased Pol II levels at histone gene promoters was also observed in genetically modified murine embryo fibroblasts treated to knockout the Ubc9/Ube2I gene (Ubc9 KO MEFs) (Supplemental Fig. S6B; Demarque et al. 2011). Similarly, PIASY silencing also increased histone gene expression (Fig. 5C), whereas upon stable PIASY overexpression, the histone genes are strongly overrepresented among the repressed genes, along with those involved in cell division (Fig. 5D, Supplemental Fig. S6C; Supplemental Table S5). Thus, although loss of the sumoylation machinery up-regulates histone gene expression, PIASY overexpression represses their expression. These observations together with the ChIP-seq data unravel an important role for active sumoylation at the TSS of the histone genes that acts to down-regulate their expression.
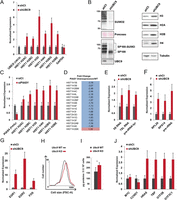
Sumoylation controls expression of histone and growth control genes. (A) WI38 cells were infected with lentiviruses expressing control shCt or shUBC9 shRNAs. Five days post-selection, the expression of the indicated genes was analyzed by RT-qPCR. (B) Western blot analysis of WI38 cells expressing shCt or shUBC9 showing expression of histones H3, H2A, H2B, and H4. Tubulin and Ponceau were used as loading controls. Depletion of UBC9 and concomitant loss of global sumoylation and sumoylated SP100 are shown as controls for knockdown efficiency. (C) WI38 cells were transfected with a control siCt or siPIASY siRNAs, and the expression of the indicated genes was analyzed by RT-qPCR. (D) Affymetrix analysis of histone mRNA differential expression in retrovirally infected WI38 cells overexpressing PIASY or a control vector (WT). (E–G) As in A. (H) Forward scatter analysis (FSC) of Ubc9+/+ (WT) and Ubc9fl/-;T2 (KO) MEFs treated for 7 d by tamoxifen (Demarque et al. 2011) (four embryos/genotype); mean size (FSC units): 628 ± 17.5 (Ubc9−/−) versus 594.3 ± 7.1 (Ubc9+/+); P-value = 0.006; one representative example is shown. (I) Total protein levels as measured by OD normalized to cell number of Ubc9+/+ (WT) and Ubc9fl/-;T2 (KO) MEFs treated for 7 d by tamoxifen (four embryos/genotype); mean amount (μg/mL): 219.3 ± 32.9 (Ubc9−/−) versus 193.5 ± 63.2 (Ubc9+/+); P-value = 0.019. (J) As in A. For all RT-qPCR, experiments were carried out in triplicate and data are represented as mean ±SEM (n = 3).
We next investigated the effect of the different knockdowns on the expression of other classes of genes. Expression of the Pol III-transcribed RNA5S and RN7SL1 RNAs, as well as tRNA tyr, are all up-regulated upon UBC9 knockdown (Fig. 5E), along with the Pol I-transcribed rRNA and the Pol II-transcribed ribosomal protein genes RPL26 and RPS14 (Fig. 5F). However, the strongest increase is seen with critical immediate early growth-promoting genes such as EGR1/2 and FOS that are strongly induced (Fig. 5G). The expression of all the above genes was also up-regulated upon SUMO knockdown (Supplemental Fig. S6D–F). In line with these findings, we observed a small but consistent increase in both cell size and global protein levels in Ubc9 KO MEFs when compared to control cells (Fig. 5H,I). Interestingly, UBC9 knockdown also increased expression of HRAS and of transcription factors such as TBP, GTF2B (also known as TFIIB), and the TFIIIC subunit GTF3C1 (Fig. 5J). Nevertheless, not all growth and proliferation regulating genes are regulated in this way as expression of MYC is unchanged and CCND1 is down-regulated upon UBC9 knockdown (Fig. 5J). Together, these data show that sumoylation negatively regulates the expression of a large set of genes transcribed by all three nuclear RNA polymerases involved in control of growth and proliferation.
UBC9 silencing induces important changes in gene expression and an atypical senescent state
We next investigated more precisely the effects of UBC9 depletion on global gene expression and proliferation in WI38 fibroblasts. Comparative microarray-based gene expression profiling of vector control-infected cells and cells expressing shUBC9 detected 1945 differentially expressed genes, 1021 of which are up-regulated and 924 of which are down-regulated (Supplemental Table S6). Therefore, loss of UBC9 dramatically alters the gene expression program with sumoylation functioning equally as both activator and repressor. Among the 1945 genes exhibiting changes in expression, ∼30% were marked by SUMO1 and/or SUMO2 in their promoters, suggesting that both direct and indirect chromatin functions of sumoylation account for its transcriptional effects. Ontology analysis of the 321 SUMO-marked genes showing up-regulation identified histone, cell cycle, and ribosomal protein genes among the most significantly enriched, confirming the data above, whereas the 285 down-regulated genes labeled by SUMO were overrepresented for cytoskeleton, proteasome, and cell adhesion genes (Fig. 6A,B).
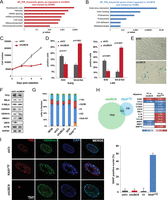
Depletion of UBC9 induces altered gene expression program together with a senescence-related phenotype. (A,B) Top selected categories identified by DAVID ontology analysis of up-regulated (A) and down-regulated (B) genes marked by SUMO1 and/or SUMO2 in their promoters in shUBC9 WI38 cells. (C) Growth curve of WI38 cells following infection with lentiviruses expressing shCt or shUBC9 shRNAs. (D) Percentage of EdU and SA-β-Gal positive WI38 cells at 4 d (early) and 8 d (late) post-infection. (E) Representative micrographs showing SA-β-Gal staining. (F) Western blot analysis of shCt or shUBC9 WI38 extracts for the indicated senescence markers. Actin and GAPDH were used as loading controls. (G) WI38 cells infected with lentiviruses expressing shCt, shUBC9, GFP, or HRASG12V were stained with propidium iodide and subjected to cell cycle analysis by flow cytometry. (H) Venn diagram showing overlap between Affymetrix gene expression profiles of HRASG12V-induced senescent and shUBC9 WI38 cells. Heat map below represents fold changes (FC) of selected genes in the two data sets. (I) Immunostaining of WI38 infected cells with control shCt, shUBC9, or HRASG12V as indicated and stained for PML (red), H3K9me3 (green), and DAPI for SAHF visualization. The unique PML aggregate in the shUBC9 cells is used as a positive control for knockdown efficiency (Zhong et al. 2000). (Right) Graph presenting the associated percentages of SAHF positive nuclei.
Compared to control-infected cells, shUBC9-expressing cells showed reduced proliferation as measured by cell counting (Fig. 6C) or by EdU incorporation at early and late times after infection (Fig. 6D). In addition, UBC9 knockdown strongly increased the number of cell staining with beta-galactosidase, and cell morphology was dramatically altered (Fig. 6D,E). A similar phenotype can be induced by siRNA silencing of SUMO or UBC9 (Supplemental Fig. S7A–D). All of these features constitute hallmarks of the senescent state. Nevertheless, although Western blot analysis showed a clear loss of UBC9 expression in shUBC9-expressing cells, no increase in the expression of senescence marker genes TP53, CDKN2A (also known as P16), or CDKN1A (also known as P21) was observed (Fig. 6F). Similarly, no increase in phosphorylation of RELA, the subunit of NFKB normally associated with activation of the secretome genes, was observed, whereas HMGA1 expression was increased as reported previously (Fig. 6F; Narita et al. 2006). Finally, FACS analysis failed to reveal any arrest in a particular phase of the cell cycle, unlike HRASG12V-transduced cells that showed a typical increase in S-phase (Fig. 6G).
These results indicate that UBC9 silencing induces an atypical senescent state. This is further confirmed by the limited overlap in gene expression changes when comparing the transcriptome microarrays of UBC9 knockdown and HRASG12V-transduced cells with respect to proliferating cells (Fig. 6H). Notably, in the HRASG12V-induced senescent cells, cell cycle gene expression is strongly reduced and secretome genes are up-regulated, whereas almost none of these changes are seen in the UBC9-silenced cells (Fig. 6H). Moreover, although the formation of senescence-associated heterochromatin foci (SAHFs) is strongly stimulated in senescent cells, no increase in SAHFs was observed upon UBC9 depletion (Fig. 6I). Together, these data indicate that loss of sumoylation triggers massive alterations in the gene expression program associated with a growth arrest that is related, but not identical, to senescence.
A general reorganization of the SUMO chromatin landscape in senescent cells
To next assess the potential dynamic nature of the SUMO chromatin landscape, we determined the genomic profiles of the SUMO machinery together with Pol II and histone marks in cells undergoing senescence, a cellular state characterized by marked changes in higher order chromatin organization (Narita et al. 2003). Senescence was triggered in WI38 fibroblasts by retroviral-mediated expression of HRASG12V, and ChIP-seq data were compared to those of proliferative cells. Strikingly, comparison of the SUMO1 and SUMO2 profiles showed a marked loss across most of the genome (Fig. 7A; Supplemental Fig. S8A). Of the more than 25,000 SUMO1 peaks observed in the proliferating cells, only 4691 remain in the senescent cells (Supplemental Fig. S1B). Loss of SUMO is seen at the majority of TSS, whereas overall Pol II and H3K4me3 profiles do not change upon entry into senescence (Fig. 7A). A similar situation was seen for SUMO2, although the overall reduction is less marked than for SUMO1 (Supplemental Figs. S1B, S8A). The release in SUMO is exemplified by the FOS and RPL26 promoters (Supplemental Fig. S8B). The decrease in SUMO1/2 at several TSS was confirmed by ChIP-qPCR using independent chromatin preparations from proliferative and both HRASG12V-induced and replicative senescent cells (Fig. 7B), suggesting that SUMO loss is a general feature of senescent cells.
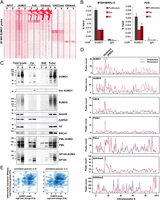
The SUMO machinery is released from chromatin in senescent cells. (A) Comparison of SUMO1 association with Pol II, H3K4me3, H3K27me3, and H3K9me3 in proliferating (P) and HRASG12V-induced senescent (S) WI38 cells. (B) ChIP-qPCR for SUMO1 in proliferating (P), HRASG12V-induced senescent (RAS), and replicative senescent (RS) WI38 cells on MTBP/MRPL13 and FOS promoters. (C) Proliferating and senescent cells were fractionated, and the presence of the indicated proteins in each fraction was quantitated by Western blot. Equal amounts of proteins were loaded for total lysate, Cyt (cytoplasmic fraction), SNE (soluble nuclear extract), and pellet (insoluble fraction). (D) Plots showing peak density in proliferating (red) and senescent (blue) cells over chromosome 6. (E) Scatter plot comparison of differential gene expression as measured by RNA-seq and differential occupancy of SUMO1 (left) or SUMO2 (right) (peak height fold change) in proliferating versus senescent cells. Differential expression is shown as log2 of reads per RPKM.
Despite the above observations, Western blot analysis revealed an overall increase in levels of SUMO1- and SUMO2-conjugated proteins in senescent versus proliferating cells (Fig. 7C). Protein fractionation into cytoplasmic, soluble nuclear, and nuclear pellet fractions indicated an increased SUMO signal in all of the fractions, including the nuclear pellet where most of the chromatin-associated proteins are found (Fig. 7C). Importantly, the matrix-associated PML and SP100 proteins, that constitute major SUMO substrates of the PML nuclear bodies (NBs) (Seeler and Dejean 2003), are present in the nuclear pellet fraction, where increases in their sumoylated forms are observed (Fig. 7C). Increased PML sumoylation is corroborated by immunofluorescence staining, indicating a striking enlargement of PML NBs in the senescent cells (Supplemental Fig. S8C). Diminished sumoylation of chromatin-associated substrates in this fraction is therefore most likely to be masked by the presence of the PML NBs and other insoluble SUMO susbtrates. However, in agreement with previous reports (Tu et al. 2011), a reduction in BRCA1 is seen in the nuclear pellet fraction of the senescent cells (Fig. 7C). Thus, although global sumoylation increases in senescent cells, the ChIP-seq experiments reveal a general loss of sumoylated chromatin-associated proteins in the senescent state.
Closer examination of the ChIP-seq data reveals that SUMO is not lost at all genomic locations. Rather, a new small set of senescence-specific SUMO1 sites is observed (Fig. 7A). A similar situation is observed for SUMO2, where a number of sites are mostly unchanged in addition to the appearance of new regions specific to the senescent state (Supplemental Fig. S8A). Senescence is also marked by a global redistribution of UBC9 and PIASY that are also lost from the majority of the regions marked in proliferating cells, whereas a new set of sites become occupied in senescent cells (Supplemental Fig. S8A,B).
Strikingly, analysis of SUMO machinery distribution along chromosome 6 revealed that both SUMO2 and UBC9 are retained at the canonical histone and tRNA gene clusters in senescent cells, indicating that active sumoylation is selectively maintained at these loci (Fig. 7D, brackets; Supplemental Fig. S8D). Maintenance of SUMO2 and UBC9 is also seen at canonical histone and tRNA gene clusters on other chromosomes but not on noncanonical histone genes, such as H3F3B (also known as H3.3B) or H2AFZ (also known as H2A.Z), whose expression is replication-independent (data not shown). SUMO1 is lost at almost every site on chromosome 6 and strongly reduced at the histone and tRNA clusters, whereas PIASY was entirely removed from the histone locus (Fig. 7D; Supplemental Figs. S5D, S8D).
In contrast to the general depletion of the SUMO machinery from chromatin in senescent cells, H3K9me3 is only marginally altered with a rather slight increase as exemplified by chromosome 6 (Fig. 7D), a finding in agreement with recent reports (Chandra et al. 2012; Chicas et al. 2012). Similarly, H3K4me3 and Pol II landscapes look very similar in proliferative and senescent cells, with selective increase or loss of signal at specific loci, but no overall reduction comparable to that seen with the sumoylation machinery (Fig. 7D). This finding contrasts with a previous study reporting a global decrease in H3K4me3 in senescent cells (Chicas et al. 2012). Whether this reflects a cell type-specific difference remains to be determined. Detailed analysis of H3K4me3 signals in the proliferative and senescent states identified sites showing significant loss or gain in signal (Supplemental Fig. S8E). Genes showing decreased H3K4me3 are generally down-regulated in senescent cells and associated with ontology terms such as cell cycle and mitosis and the upstream master regulators E2F, RB1, and TP53. In contrast, increased H3K4me3 is associated with ontology terms that reflect the up-regulation of genes involved in cell morphology and secretome activation, notably genes enriched in the NFKB upstream regulator, a major activator of the secretory program (Supplemental Fig. S8E; Chien et al. 2011).
Gene expression changes in senescent cells were next profiled by RNA-seq, identifying 1130 up-regulated and 1281 down-regulated transcripts (Supplemental Table S7). A global comparison of the RNA-seq and ChIP-seq data did not reveal a significant correlation between changes in gene expression and gain or loss in SUMO1/2 (Fig. 7E). This, taken together with the chromatin profiles showing strong reduction of SUMO1/2 at promoters where no change in Pol II is observed, indicates that decreased SUMO occupancy in senescence is therefore not directly related to either changes or maintenance of gene expression, but may rather reflect other functions on chromatin in this specific state (see Discussion).
Together, these data reveal a dramatic redistribution of the SUMO chromatin landscape in senescent cells. The SUMO machinery is massively released from chromatin, whereas SUMO2 and UBC9 are specifically retained at the histone and tRNA genes. These data suggest that the reconfiguration of the SUMO chromatin profile, notably its selective retention at the histone and tRNA genes, is conducive for the stability of the senescent state by maintaining a negative control on these loci.
Discussion
Although transcription factors are one of the most abundant classes of SUMO substrates, whether and where sumoylated transcription factors associate with chromatin and their global distribution remain largely unexplored. Here, we show that SUMO components are widely distributed over the genome at discrete sites, likely corresponding to binding of multiple transcription factors and at the TSS of active promoters for all three nuclear RNA polymerases. Highest enrichment is seen at promoters for histone genes and genes involved in protein biogenesis as well as tRNAs and rRNAs, where in all cases sumoylation acts as a repressor. However, the SUMO landscape is highly dynamic as a widespread loss of chromatin-associated SUMO, that excludes the histone and tRNA loci, is observed during senescence and is likely to contribute to its stability.
An important finding is that of a major peak of SUMO1/2 immediately upstream of the TSS that correlates positively with Pol II occupancy and transcript levels. Although it has been reported that human histone H4 is a substrate for sumoylation (Shiio and Eisenman 2003), as are the four core histones in yeast (Nathan et al. 2006), the TSS peak lies in the nucleosome-depleted region, suggesting that it is not a histone modification but concerns one or more components of the Pol II PIC. Many PIC components can be sumoylated, such as TAF5 and TAF12 of the TFIID complex, GTF2A1 (also known as TFIIA), GTF2E1 and GTF2E2 (also known as TFIIE), and subunits of the mediator complex, as well as other factors reported to be present at active promoters, such as topoisomerase (DNA) I, PARP1 and chromatin remodeling complexes (Panse et al. 2004; Zhao et al. 2004; Rosas-Acosta et al. 2005; Hornbeck et al. 2012). Our results suggest that one or several of the PIC components are major SUMO substrates at active promoters.
The promoters showing simultaneous presence of SUMO1, SUMO2, UBC9, and PIASY are remarkably enriched in histone genes that are among the most highly marked sites for each of the four components. Moreover, histone gene promoters show the highest enrichment for PIASY on the whole genome, suggesting some specific function for PIASY at these promoters. The SUMO substrate(s) among the several specific and more general transcription factors that regulate coordinated expression of canonical histones remain(s) to be identified (for review, see Marzluff et al. 2008). Of note, PIASY was reported to act as a SUMO E3 ligase for CASP8AP2 (also known as FLASH), a processing factor of histone mRNAs, and IRF2, an activator of histone gene promoters, with sumoylation being repressive in both cases (Han et al. 2008; Alm-Kristiansen et al. 2009). Active sumoylation also appears to occur at Pol III-transcribed tRNA genes that are strongly enriched for both SUMO and UBC9. Several subunits of Pol III have been shown to be sumoylated in both mammalian cells and yeast (Panse et al. 2004; Zhao et al. 2004). A recent report also mentions that sumoylation of the transcriptional repressor MAF1 controls its recruitment to tRNA gene promoters and their subsequent repression in an RNA pol III-dependent manner (Rohira et al. 2013). Therefore, Pol III subunits, MAF1, or other Pol III-associated transcription factors may be the substrates detected in our analysis. Interestingly, the PML NBs, which constitute major sites for the SUMO machinery, have been shown to associate specifically with the MHC gene cluster, a region neighboring the histone and tRNA loci on chromosome 6 (Shiels et al. 2001). Whether the PML NBs may contact, even transiently, the histone/tRNA loci remains to be determined.
Silencing of UBC9, SUMO1/2, or PIASY all up-regulate histone gene expression, whereas PIASY overexpression represses it. Transcription of canonical histone genes is tightly regulated during the cell cycle; and coupling both the timing and level of their expression with DNA synthesis is critical for cell viability, and their misregulation can lead to cell cycle defects (Gunjan et al. 2005). Our data therefore suggest that active sumoylation at their TSS fine tunes either the overall level and/or timing of histone expression to ensure a normal cell cycle. Histone gene transcription is rapidly turned down at the end of S phase, and the mechanisms underlying this repression remain poorly understood. Recently, the histone chaperone HIRA was shown to be recruited in a H2BTyr37 phosphorylation-dependent manner upstream of the major histone gene cluster, thereby suppressing histone expression (Mahajan et al. 2012). However, our finding of specific peaks for SUMO and its machinery at each individual histone TSS, rather than a unique enrichment site at the upstream HIRA-bound locus, makes it unlikely that SUMO controls the whole histone cluster at a distance; yet whether HIRA governs the sumoylation status of the entire locus remains an intriguing possibility. Depletion of the sumoylation machinery also up-regulated tRNA expression along with that of other Pol III-transcribed genes and the Pol I-transcribed rRNA. A similar effect was seen for many proteins involved in translation and some immediate early growth control genes. Modulating levels of sumoylation at the TSS of these genes hence provides a key mechanism to coordinate regulation of a network of histone and protein biogenesis genes transcribed by all Pol I, II, and III polymerases, thereby controlling cell growth and proliferation (White 2005).
In a recent report, Liu et al. (2012) used ChIP-seq to profile SUMO1 in HeLa cells showing, in agreement with our data, its presence at active TSS. Nevertheless, the authors proposed that SUMO1 acts to positively regulate transcription of Pol II-transcribed ribosomal protein genes, whereas we show that sumoylation represses expression of a large set of protein biosynthesis genes transcribed by all three nuclear polymerases. A likely explanation of these conflicting results is that knocking down a single SUMO paralog does not recapitulate a global loss of sumoylation as achieved by UBC9 knockdown. Indeed, Liu et al. (2012) observed opposite results upon depletion of UBC9 versus SUMO1 for a number of analyzed genes.
Another major finding of our study is the widespread but not universal loss of SUMO1/2, UBC9, and PIASY from the chromatin in senescent cells. An important exception is the promoters of the histone and tRNA genes where, although SUMO1 and PIASY are depleted, SUMO2 and UBC9 levels remain high. The presence of UBC9 indicates that active sumoylation is maintained at these loci. In contrast, PIASY is removed from these TSS in senescent cells, but the persistence of SUMO2 indicates that it may be replaced by another SUMO E3 ligase. As sumoylation represses the expression of these genes, the maintenance of SUMO2 at these sites may be critical to restrain their expression either because of the diminished requirement for histones and tRNA in the post-mitotic senescent state or, more importantly, because their strong expression in the absence of DNA replication would be detrimental for cell viability. Hence, a general loss of chromatin-associated sumoylation but its active maintenance to repress histone and tRNA genes may be necessary for establishment and/or maintenance of the senescent state. It remains to be determined whether the same or distinct substrates are sumoylated at these loci in proliferating and senescent cells.
The general depletion of SUMO in senescent cells may be explained by dissociation of the SUMO substrate proteins from chromatin. Yet transcription in senescent cells presumably requires many of the activators, repressors, and PIC components that are substrates for SUMO in proliferating cells. Thus, an alternative to global loss of SUMO substrates is that they remain present on the chromatin, but are no longer sumoylated. Sumoylation may be required to promote the orderly de novo reassembly of transcription factor complexes and PIC formation on post-mitotic chromatin. In this scenario, nonmitotic senescent cells would have a reduced requirement for sumoylated proteins which are progressively replaced by their nonsumoylated counterparts. In a similar manner, it has been proposed that CBX5 sumoylation is essential for initial seeding of pericentric heterochromatin with CBX5, but not for the maintenance of these CBX5 domains once formed (Maison et al. 2011).
Finally, we show that UBC9 depletion induces a senescent-like state that may result from nonchromatin-associated functions of SUMO, but also from loss of transcription factor sumoylation on chromatin, thus promoting cell cycle exit. Nevertheless, UBC9 depletion does not fully recapitulate the situation seen in senescent cells as sumoylation is normally maintained at the histone and tRNA genes in this state. Here, however, unscheduled up-regulation of histone and tRNA genes could be an additional cause for cell cycle arrest.
Our study provides the first comprehensive view of the SUMO machinery on chromatin establishing SUMO as a bona fide chromatin-associated modification and unveils a previously unappreciated role for this process, that, in marking chromatin by a unique modifier, acts as a major negative regulator of genes essential for cell growth and proliferation. This work will hopefully pave the way toward characterization of the large number of possible SUMO substrates involved. These will most likely not only differ depending on the gene promoter, but also there are likely multiple substrates at a given promoter. In this context, it is noteworthy that a recent study (Psakhye and Jentsch 2012) underscored the importance of multiple sumoylation events occurring on a protein group rather than on individual proteins.
Methods
Vectors, viruses, transfections, and cell culture
Culturing of human diploid WI38 fibroblasts (ATCC) and infection by retroviral-mediated gene transfer with either pBABE-puro-HRASG12V, pBABE-puro-HA-PIASY, or empty vector as a control were performed at a physiological oxygen concentration of 3% as previously described (Bischof et al. 2006) or at 21% for growth analysis.
For stable UBC9 knockdown (shRNA), synthetic hairpin oligos were cloned into the AgeI-EcoRI sites of pLKO1 vector (Sigma). Lentiviral infections were carried out by standard methods, and infected cells were selected for 24–48 h with puromycin (4 ug/mL). pTRIP-GFP and pTRIP-Rasv12 were a kind gift from Pierre Charneau. shRNA target sequences are available on request. siRNA transfection was performed according to siIMPORTER standard protocol (Millipore) with siRNAs targeting SUMO-1, -2, and -3 as previously described (Lallemand-Breitenbach et al. 2008) or with oligo2 from Dharmacon for UBC9 and PIASY targeting.
Senescence analysis
The senescent phenotype was assessed by EdU incorporation (Click-iT, Invitrogen), senescence-associated beta-galactosidase (SA-β-Gal) activity, and SAHF formation as previously described (Bischof et al. 2006).
MEFs and cell size
Primary mouse embryonic fibroblasts (E12.5) isolated from Ubc9+/+ or Ubc9fl/-;T2 C57BL/6J mice (Demarque et al. 2011), were cultured in complete medium (DMEM Glutamax, 10% SVF) containing 100 nM of tamoxifen during 7 d. Cell size was analyzed by flow cytometric examination of a forward scatter (FSC) dot plot. Analyses were performed on a FACSCalibur flow cytometry system (BD), acquiring a minimum of 10,000 total events and using CellQuest Pro software, after gating viable cells.
Cell cycle analysis
Cells were harvested, fixed with 70% ethanol, and stained with 30 μg/mL of propidium iodide overnight before FACS analysis (Becton Dickinson FACSCalibur, 10,000 events/sample).
ChIP experiments
ChIP was performed on proliferating cells infected with empty vector or HRASG12V-induced WI38 senescent cells 5 d post-selection. Cells were crosslinked directly in the growing medium with formaldehyde 1% for 15 min at room temperature. The reaction was stopped by adding glycine to a final concentration of 0.125 M for 10 min at room temperature. Fixed cells were rinsed twice with PBS-N-Ethylmaleimide (10mM) and resuspended in lysis buffer (10 mM EDTA, pH 8, 50 mM Tris-HCl, pH 8, SDS 1%). Lysate was sonicated for 30 min (30 sec on/30 sec off) in Diagenode water bath-sonicator and centrifuged at 14,000 rpm for 10 min. The cleared supernatant was used immediately in ChIP experiments or stored at -80°C. Sonicated chromatin of 2–4 million cells was diluted 10 times in ChIP Dilution Buffer (SDS 0.01%, Triton X-100 1.1%, 1.2 mM EDTA, pH 8, 16.7 mM Tris-HCl, pH 8, and 167 mM NaCl) and precleared for 1 h rotating at 4°C, with 50 μL of equilibrated Protein A/G UltraLink Resin (Thermo Scientific) before an overnight incubation with 5 μg of specific antibody. The bound material was recovered after a 2-h incubation, rotating at 4°C, with 30 μL of Protein A/G UltraLink Resin. For histone marked ChIP; the beads were washed for 5 min, once in Low Salt Buffer (SDS 0.1%, Triton X-100 1%, 2 mM EDTA, pH 8, 20 mM Tris-HCl, pH 8, and 150 mM NaCl), twice in High Salt Buffer (SDS 0.1%, Triton X-100 1%, 2 mM EDTA, pH 8, 20 mM Tris-HCl, pH 8, and 500 mM NaCl), twice in LiCl Buffer (0.25 M LiCl, NP-40 1%, Na Deoxycholate 1%, 1 mM EDTA, pH 8, and 10 mM Tris-HCl, pH 8), and twice in TE or for other ChIPs, 4 × 5 mins with ChIP dilution buffer. ChIPed material was eluted by two 15-min incubations at room temperature with 250 μL Elution Buffer (SDS 1%, 0.1 M NaHCO3). Chromatin was reverse-crosslinked by adding 20 μL of NaCl 5M and incubated for 4 h at 65°C minimum, DNA was submitted to RNase and proteinase K digestion, and extracted with phenol-chloroform. DNA was ethanol precipitated in the presence of 20 μg of glycogen and resuspended in 50 μL of H20 in a siliconized tube.
For ChIP-qPCR experiments, fold enrichment of targets in ChIPed DNA relative to input was calculated from an average of three replicate qPCR reactions.
ChIP-seq analysis
ChIP-seq was performed using an Illumina GAII sequencer, and raw data were analyzed by the Illumina ELAND pipeline v1.6. Peak detection was performed using the MACS software (http://liulab.dfci.harvard.edu/MACS/) (Zhang et al. 2008), and the peaks annotated using GPAT (http://bips.u-strasbg.fr/GPAT/Gpat_home.html) (Krebs et al. 2008) using a window of +/− 20 kb with respect to the coordinates of the beginning and end of Ensembl transcripts (Flicek et al. 2013). Global clustering of the ChIP-seq data and quantitative comparisons were performed using the seqMINER program (http://bips.u-strasbg.fr/seqminer/) (Ye et al. 2011). As reference coordinates, we used annotated transcription start sites of human genes or the MACS determined peaks for the factors as indicated in each figure. Tag densities from each ChIP-seq data set were collected in a window of 10 kb around the reference peak. The collected values were subjected to k-means clustering coupled to linear-based normalization. The normalization procedure reduces bias in the clustering due to inherent differences between ChIP-seq experiments.
Sequences under SUMO1 and SUMO2 peaks were analyzed using the MEME ChIP program and AME output (http://meme.nbcr.net/meme/cgi-bin/meme-chip.cgi).
In order to estimate the number of SUMO sites mapping to different gene features, it was necessary to assign one site to one and only one gene feature. We used the following hierarchy to assign sites to features: core>upstream>intron>exon>intergenic. Core was defined as 500 bp upstream of and downstream from the TSS, upstream was defined as 500 bp upstream to a maximum of 20,000 bp upstream of the TSS. Binding sites that could not be mapped to within 20,000 bp upstream of any TSS and were not assigned to any intron or exon were termed intergenic.
Functional annotations were carried out using DAVID, IPA Ingenuity software, and GREAT (McLean et al. 2010).
RNA preparation and RT-qPCR
Total RNA was prepared using RNeasy kit (Qiagen) 5 d post-infection for HRASG12V-expressing WI38 cells, 24 h post-selection for PIASY-overexpressing WI38 cells, 5 d post-selection for shUBC9-infected WI38 cells, and 4 d post-transfection for siRNA-treated cells. cDNAs were synthesized using a random primer kit according to manufacturer's instructions (Applied Biosystems). Quantitative-PCR (qPCR) and RT-qPCR were performed using the SYBR Green PCR kit from Applied Biosystems on the CFX96 cycler (Bio-Rad). Sequences for primers are available in Supplemental Table S8. Relative quantities of gene expression levels were normalized to PPIA and HPRT1.
mRNA-seq
The mRNA-seq libraries were prepared following the Illumina protocol with some modifications. Briefly, mRNA was purified from total RNA using oligo-dT magnetic beads and fragmented using divalent cations for 5 min at 95°C. The cleaved mRNA fragments were reverse transcribed to cDNA using random primers. This was followed by second strand cDNA synthesis using DNA Polymerase I and RNase H. The double strand cDNA fragments were blunted, phosphorylated, and ligated to single-end adapter dimers follwed by PCR amplification (30 sec at 98°C; [10 sec at 98°C, 30 sec at 65°C, 30 sec at 72°C] × 13 cycles; 5 min at 72°C). After PCR amplification, surplus PCR primers and dimer adapters were removed by purification using AMPure beads (Agencourt Biosciences Corporation). Size selection was performed by electrophoresis on a 2% agarose gel and DNA fragments in the range of ∼250–350 bp were excised and purified using QIAquick Gel Extraction Kit (Qiagen). DNA libraries were checked for quality and quantified using 2100 Bioanalyzer (Agilent). The libraries were loaded in the flowcell at 6 pM concentration and clusters generated and sequenced on the Illumina Genome Analyzer II* as single-end 72-base reads. Image analysis and base calling were performed using the Illumina Pipeline version 1.6 and sequence reads mapped to reference genome mm9/NCBI37 using TopHat (Trapnell et al. 2009). Quantification of gene expression was performed using the DESeq Bioconductor package as previously described (Anders and Huber 2010; Benhaddou et al. 2012).
Microarray
Microarray analysis was performed on the Affymetrix GeneChip Human Gene 1.0 ST Array containing probe sets for 28,860 transcripts. Three biological replicates were run for each condition. For each array, the cell intensity files (CEL) were generated with an Affymetrix GeneChip Scanner 3000. Gene-level expression values were derived from the CEL file probe-level hybridization intensities using the model-based Robust Multichip Average algorithm (RMA). Statistical analysis to compare replicates' arrays was done with the local poor error test. The P-values were adjusted using the Bonferroni algorithm and a threshold of P < 0.05 is used as the criterion for expression. Functional annotation was performed by DAVID software.
Antibodies, immunofluorescence, immunoblotting, and subcellular fractionation
ChIP was performed with the following antibodies: in-house rabbit polyclonal anti-SUMO1 and anti-PIASY (see Supplemental Fig. S1A), rabbit polyclonal anti-SUMO2 (Azuma et al. 2003), mouse monoclonal anti-UBC9 (BD Transduction Laboratories) (see Supplemental Fig. S1A), rabbit polyclonal anti-PolII (sc-9001), H3K4me3 (04-745, Upstate), H3K27me3 (07-449, Upstate), and H3K9me3 (07-442, Upstate).
For immunoblotting and immunofluorescence experiments, we used rabbit polyclonal anti-H3 (ab8898), rabbit polyclonal anti-H2A (ab18255), rabbit polyclonal H2B (ab9110), rabbit polyclonal anti-H4 (ab7311), rabbit polyclonal anti-LaminB1 (ab16048), mouse monoclonal anti-GAPDH (ab8245), mouse monoclonal anti-BRCA1 (sc-6954), rabbit monoclonal anti-Tubulin (ab52866), all from Abcam; in-house rabbit polyclonal anti-SP100 and anti-PML, mouse monoclonal anti-TP53 (DO-1, Santa Cruz); rabbit monoclonal anti-RELA (3987S) and anti-P-RELA (3033P) from Cell Signaling Technology, rabbit polyclonal anti-CDKN2A (sc-759, Santa Cruz), anti-CDKN1A (sc-469, Santa Cruz), rabbit polyclonal anti-HMGA1 (kind gift from M. Narita), rabbit polyclonal anti-ACTB (A2066, Sigma), and rabbit monoclonal anti-Ki67 (ab16667, Abcam).
Immunoblot and immunofluorescence were performed according to standard procedures. All fluorochrome-tagged secondary antibodies were from Invitrogen. Subcellular fractionation was prepared according to published methods (Méndez and Stillman 2000; Narita et al. 2006). Soluble proteins in cytoplasm, soluble nuclear fraction, and chromatin-bound proteins in the pellet fraction were detected by Western blot.
Data access
ChIP-seq and RNA-seq data were deposited in the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE42213. Affymetrix data were deposited on ArrayExpress (http://www.ebi.ac.uk/arrayexpress/) under accession numbers E-MEXP-3778, E-MEXP-3777, and E-MEXP-3776.
Acknowledgments
We thank R. Santoro and M. Werner for rRNA/tRNA reagents and advice. We acknowledge A. Andrieux for technical assistance. This work was supported by grants from LNCC, Odyssey-RE, INCa, Université de Strasbourg, ANR, ERC, and EEC (EuTRACC). The IGBMC high-throughput sequencing facility is a member of the “France Génomique” consortium (ANR10-INBS-09-08). H.N.-K. was supported by the MRT and ARC.
Footnotes
-
↵7 Corresponding authors
E-mail irwin{at}igbmc.fr
E-mail anne.dejean{at}pasteur.fr
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.154872.113.
- Received January 15, 2013.
- Accepted July 26, 2013.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported), as described at http://creativecommons.org/licenses/by-nc/3.0/.








