
Neonatal DNA methylation profile in human twins is specified by a complex interplay between intrauterine environmental and genetic factors, subject to tissue-specific influence
- Lavinia Gordon1,
- Jihoon E. Joo2,3,
- Joseph E. Powell4,5,
- Miina Ollikainen6,
- Boris Novakovic2,3,
- Xin Li7,
- Roberta Andronikos3,7,
- Mark N. Cruickshank7,
- Karen N. Conneely8,
- Alicia K. Smith9,
- Reid S. Alisch10,
- Ruth Morley7,
- Peter M. Visscher4,5,11,
- Jeffrey M. Craig3,7,12,13 and
- Richard Saffery2,3,12
- 1Bioinformatics Unit, Murdoch Childrens Research Institute (MCRI), Parkville, Victoria 3052, Australia;
- 2Cancer and Developmental Epigenetics Group, MCRI, Parkville, Victoria 3052, Australia;
- 3Department of Paediatrics, University of Melbourne, Victoria 3052, Australia;
- 4University of Queensland Diamantina Institute, University of Queensland, Princess Alexandra Hospital, Brisbane, Queensland 4102, Australia;
- 5Queensland Institute of Medical Research, Brisbane, Queensland 4006, Australia;
- 6Hjelt Institute, Department of Public Health, FI-00014 University of Helsinki, Helsinki, Finland;
- 7Early Life Epigenetics Group, MCRI, Parkville, Victoria 3052, Australia;
- 8Department of Human Genetics, Emory University School of Medicine, Atlanta, Georgia 30322, USA;
- 9Department of Psychiatry & Behavioral Sciences, Emory University School of Medicine, Atlanta, Georgia 30322, USA;
- 10Department of Psychiatry, University of Wisconsin School of Medicine, Madison, Wisconsin 53719, USA;
- 11The Queensland Brain Institute, The University of Queensland, Brisbane, Queensland 4072, Australia
-
↵12 These authors contributed equally to this work.
Abstract
Comparison between groups of monozygotic (MZ) and dizygotic (DZ) twins enables an estimation of the relative contribution of genetic and shared and nonshared environmental factors to phenotypic variability. Using DNA methylation profiling of ∼20,000 CpG sites as a phenotype, we have examined discordance levels in three neonatal tissues from 22 MZ and 12 DZ twin pairs. MZ twins exhibit a wide range of within-pair differences at birth, but show discordance levels generally lower than DZ pairs. Within-pair methylation discordance was lowest in CpG islands in all twins and increased as a function of distance from islands. Variance component decomposition analysis of DNA methylation in MZ and DZ pairs revealed a low mean heritability across all tissues, although a wide range of heritabilities was detected for specific genomic CpG sites. The largest component of variation was attributed to the combined effects of nonshared intrauterine environment and stochastic factors. Regression analysis of methylation on birth weight revealed a general association between methylation of genes involved in metabolism and biosynthesis, providing further support for epigenetic change in the previously described link between low birth weight and increasing risk for cardiovascular, metabolic, and other complex diseases. Finally, comparison of our data with that of several older twins revealed little evidence for genome-wide epigenetic drift with increasing age. This is the first study to analyze DNA methylation on a genome scale in twins at birth, further highlighting the importance of the intrauterine environment on shaping the neonatal epigenome.
Epigenetics has been defined as “the structural adaptation of chromosomal regions so as to register, signal or perpetuate altered activity states” (Bird 2007). This is exemplified by the epigenetic mark of DNA methylation, which influences a gene's transcriptional potential and plays a role in differentiation (Reik 2007; Brunner et al. 2009; Huang and Fan 2010) and aging (Rakyan et al. 2010; Teschendorff et al. 2010). Disruption of epigenetic profile is a ubiquitous feature of cancers and is likely to play a role in the etiology of other complex diseases (van Vliet et al. 2007; Foley et al. 2009).
The DNA methylation profile is heritable through mitosis, but the fidelity of this transmission is imperfect (Bennett-Baker et al. 2003) and may contribute to differences in gene expression and phenotype observed between genetically identical individuals, whether isogenic strains of mice (Gartner and Baunack 1981; Pritchard et al. 2006) or human MZ twins (Fraga et al. 2005; Martin 2005; Kuratomi et al. 2008; Kaminsky et al. 2009; Javierre et al. 2010).
Animal studies have demonstrated that the environment can shape the epigenome, particularly during the intrauterine period, when it demonstrates the greatest plasticity (Gluckman et al. 2007, 2010; Ozanne and Constancia 2007). The importance of this period for human health is well documented, and mounting evidence implicates the intrauterine environment in the fetal “programming” of diseases of later life (Gluckman et al. 2007). Despite this, it remains unclear as to how influential the intrauterine period is in shaping the human epigenome, whether different genomic regions show varying sensitivities to this environment during this period, and the extent to which this interaction is sensitive to genetic influences.
Twin studies, which have traditionally enabled estimation of genetic and environmental components to phenotypic variance, have been used to estimate the effect of these factors on DNA methylation, at both a gene-specific level (Heijmans et al. 2007; Wong et al. 2010) or throughout the genome (Kuratomi et al. 2008; Kaminsky et al. 2009; Javierre et al. 2010; Rakyan et al. 2011a,b). Such studies are improving our understanding of the processes involved in the regulation of epigenetic variation and are disentangling the relative contributions of epigenetics, environment, and genetic variation, together with stochastic factors, in complex traits (Bell and Saffery 2012). This information is critical to understanding processes of development and evolution (Feinberg and Irizarry 2011) and for future potential epigenetic-based interventions in complex disease.
To investigate the components of epigenetic variation at birth, we have established a longitudinal cohort of 250 twin pairs with collection of extensive biospecimens and environmental data (Saffery et al. 2012) and have shown, in two tissues from 14 twin pairs at birth, that twins differ in levels of gene expression on a genome-wide scale (Gordon et al. 2011), most likely in response to epigenetic variability. Furthermore, we subsequently provided direct evidence that DNA methylation can vary considerably within a single locus in multiple tissues from MZ twin pairs collected at birth (Ollikainen et al. 2010). This supports the previously demonstrated genome-scale differences in methylation within MZ and DZ twin pairs in adults (Kaminsky et al. 2009). However, no study has yet focused on genome-scale methylation differences within twins at birth.
In this study, we used genome-scale DNA methylation profiling to measure the level of epigenetic variation present in three tissues from 22 MZ and 12 DZ twin pairs collected at birth. We then estimated the within-pair variation of methylation profile generally and in the context of specific genomic features, and estimated the contribution of genetic and common and unique environment to DNA methylation profile. We further defined gene pathways and networks subject to epigenetic change in association with birth weight with the aim of investigating a possible epigenetic link with risk for metabolic and cardiovascular disease.
Results
Genome-scale analysis of DNA methylation of three tissues from newborn twins
We used the Illumina Infinium HumanMethylation27 BeadChip array (HM27) platform to profile DNA methylation in cord blood mononuclear cells (CBMCs; 18 MZ, nine DZ pairs), human umbilical vascular endothelial cells (HUVECs, 14 MZ, 10 DZ pairs), and placenta (eight MZ and seven DZ twin pairs) (Table 1). The HM27 platform interrogates 27,578 CpG dinucleotides primarily associated with 14,475 transcription start sites and has a high technical reproducibility (Weisenberger et al. 2008; Bibikova et al. 2009; Rajendram et al. 2011). It contains separate probes to detect methylated and unmethylated sequences, and data from both probes are used to calculate a β-value between 0 and 1 (equivalent to 0%–100% methylation) (Bibikova et al. 2009). Initial quality control led to the removal of two twin pairs from HUVECs, one twin pair from CBMCs, and one twin pair from placenta (Table 1). We chose a highly conservative P-value probe cutoff of 0.001, lower than has previously been reported, to minimize the level of variability attributable to technical factors. After removing probes on the sex chromosomes and probes that had a detection P-value > 0.001, 19,350 probes for HUVECs, 19,204 for CBMCs, and 26,353 for placenta remained for subsequent analysis.
Twin pair characteristics
Twin pairs generally show a similar DNA methylation profile
In order to visualize the overall relationship between DNA methylation profiles of individuals, both within and between pairs, unsupervised clustering was applied to sample data using all HM27 probes that passed quality-control measures for each tissue separately (Supplemental Figs. S1–S3). Interestingly, twins within the same pair did not always cluster together, and the proportion of within-pair clustering varied considerably in a tissue-dependent manner. For example, only 29% of pairs clustered in HUVECS, whereas intrapair similarity predominated in CBMCS and placenta, with 58% and 71% of individual pairs clustering together respectively (Supplemental Table S1). In all three tissues, a greater proportion of MZ pairs clustered together relative to DZ pairs (Supplemental Table S1). In combination, these data provide prima facie evidence for both tissue-specific and genetic factors in determining neonatal epigenetic profile.
Identification of factors contributing to DNA methylation profile
To quantify within-pair relationships both for measures of discordance and similarity, we calculated Euclidean distance (ED) and Pearson's correlation coefficient (Fig. 1; Supplemental Table S2). Both ED and Pearson's correlation coefficient were plotted using logit-transformed β-values (i.e., M-values) with mean-subtracted transformed values used for the latter (Fig. 1). In all instances, MZ pairs showed a greater median within-pair similarity and lower median within-pair discordance than DZ pairs, confirming a role for underlying genetic factors in contributing to neonatal epigenetic profile. These data also show that a proportion of unrelated individuals have a higher degree of similarity in overall DNA methylation profile than some co-twins, highlighting the likely role of stochastic/nonshared environmental factors in determining the epigenetic profile. Surprisingly, when the effects of chorionicity were tested (within MZ pairs only), dichorionic (DC) pairs were found to be generally more similar (less discordant) epigenetically than monochorionic (MC) pairs in both CBMCs (eight MC, nine DC) and HUVECs (eight MC, five DC) (Fig. 1). Insufficient numbers precluded a similar examination in relation to the placental methylation profile. In addition, there were no sex-specific significant differences in discordance or similarity for all tissues (data not shown).
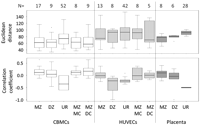
Relationship between within-pair methylation discordance/correlation, zygosity, and chorionicity. Box-and-whisker plots of within-pair methylation discordance (Euclidean distance) and Pearson's correlation coefficient of mean-corrected values in HUVECs, CBMCs, and placenta from MZ and DZ twins, same-sex unrelated (UR) individuals, and MC and DC MZ twins. Numbers of pairs within each category are shown above each graph.
Estimation of variance components of DNA methylation profile
Using methylation within our neonatal twin tissues as a variable phenotype, we estimated the narrow sense heritability (h2, the proportion of phenotypic variance due to additive genetic factors) and common (intrauterine) environmental variance
(c2) for all HM27 probe-associated CpGs within our data sets using the least squares estimation approach (Visscher 2004). The distribution of the h2 estimates obtained for each probe in the three tissues was overlaid against the expected null distribution with the variance
equal to the expected sampling variance (Visscher 2004) and a mean of zero (Fig. 2). Mean heritability across all probes was 0.12 (±0.0017, p = 0.0024) for CBMCs, 0.07 (±0.0014, p = 0.009) for HUVECs, and 0.05 (±0.0016, p = 0.017) for placenta with calculated P-values from empirical null distribution of 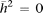 determined by a permutation analysis, suggesting a pattern of greater estimated heritability than that expected by chance.
Because the distribution of c2 did not differ from a random distribution (median = 0), we conclude that shared environment does not contribute significantly
to methylation variation within our twins in utero and that the remaining variance in methylation is due to the sum of unique
intrauterine environment and stochastic factors encountered by the tissues in each twin. An investigation of probes with the
highest (top 5%) heritability estimates (Supplemental Table S3) revealed 961 probes in CBMCs with h2 values ranging from 0.48 to 0.94; 968 probes in HUVECs with h2 = 0.37–0.95, and 1304 probes in placenta with h2 = 0.49–0.97. In addition, only 3%–10% of highly heritable probes were shared between two tissues (28 between placenta and
HUVECs, 109 between HUVECs, and CBMCs and 58 between CBMCs and placenta). Only three probes were highly heritable in all three
tissues (cg15052901/SLC24A4, h2 = 0.49–0.62; cg14217157/WHSC2, h2 = 0.52–0.63; cg01593886/COL1A1, h2 = 0.51–0.72). Ontology analysis of genes linked to highly heritable probes revealed enrichment of genes involved in differentiation
and development for CBMCs and HUVECs, metabolism, and biosynthesis in HUVECs and signaling in placenta (Supplemental Table
S4).
determined by a permutation analysis, suggesting a pattern of greater estimated heritability than that expected by chance.
Because the distribution of c2 did not differ from a random distribution (median = 0), we conclude that shared environment does not contribute significantly
to methylation variation within our twins in utero and that the remaining variance in methylation is due to the sum of unique
intrauterine environment and stochastic factors encountered by the tissues in each twin. An investigation of probes with the
highest (top 5%) heritability estimates (Supplemental Table S3) revealed 961 probes in CBMCs with h2 values ranging from 0.48 to 0.94; 968 probes in HUVECs with h2 = 0.37–0.95, and 1304 probes in placenta with h2 = 0.49–0.97. In addition, only 3%–10% of highly heritable probes were shared between two tissues (28 between placenta and
HUVECs, 109 between HUVECs, and CBMCs and 58 between CBMCs and placenta). Only three probes were highly heritable in all three
tissues (cg15052901/SLC24A4, h2 = 0.49–0.62; cg14217157/WHSC2, h2 = 0.52–0.63; cg01593886/COL1A1, h2 = 0.51–0.72). Ontology analysis of genes linked to highly heritable probes revealed enrichment of genes involved in differentiation
and development for CBMCs and HUVECs, metabolism, and biosynthesis in HUVECs and signaling in placenta (Supplemental Table
S4).
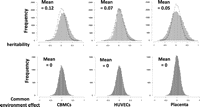
Analysis of variance components of DNA methylation in CBMCs, HUVECs, and placenta. Data from all probes were used to plot histograms of heritability (h2) and common (intrauterine) environmental variance (c2). Distributions of h2 were also compared with the random distribution (dotted lines).
Within-pair discordance varies with genomic location
Because the annotation of the HM27 array focuses on CpG islands and gene promoters (Bibikova et al. 2009), we examined how within-pair methylation discordance, measured using median within-pair standard deviation, varies in relation to these landmarks. Previous studies in unrelated individuals (Bock et al. 2008) and twins (Kaminsky et al. 2009; Sandoval et al. 2011) have suggested that such regions are more refractory to methylation variation than non-island CpGs. To test this in our data, we compared median within-pair methylation discordance for CpG islands, CpG island “shores” (sequences up to 2 kb from CpG islands), CpG island “shelves” (sequences within 2 kb and 4 kb of CpG islands), and CpGs >4 kb from CpG islands (Fig. 3; Sandoval et al. 2011). In agreement with previous studies (Doi et al. 2009; Irizarry et al. 2009), we found that mean absolute methylation levels in our samples increased as a function of distance from CpG islands (data not shown). We also found that median within-pair methylation discordance increased with increasing distance from CpG islands (up to 4 kb) in all tissues for both MZ and DZ pairs, with no evidence for a further increase at distances >4 kb.
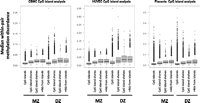
Relationship between methylation discordance to location within CpG islands, shores, and shelves. Median within-pair methylation discordance is plotted as box-and-whisker plots against probes depending on location in relation to CpG islands. Data are plotted for CpG islands, CpG island shores (0–2 kb from islands), CpG island shelves (2–4 kb from islands), and “open sea” probes (>4 kb from CpG islands).
Relationship between within-pair methylation discordance and age: no evidence for genome-scale epigenetic drift
We have presented evidence that the nonshared intrauterine environment contributes to methylation discordance at birth for both MZ and DZ twins. However, little is known about how nonshared environment can influence epigenetic discordance postnatally. We and others have shown using array analysis that within-twin pair discordance in the gene expression profile increases as a function of age (Fraga et al. 2005; Gordon et al. 2011). Additionally, limited evidence from low-resolution analysis also suggests that within-pair DNA methylation discordance increases with age (a phenomenon termed “epigenetic drift”) (Fraga et al. 2005). However, a recent Infinium HM27 array analysis of saliva from adult twins did not find evidence for such drift (Bocklandt et al. 2011). We plotted within-pair methylation discordance using Euclidean distance for blood-derived data from twins from birth to 73 yr of age from our data and 94 MZ pairs and 17 DZ pairs from Infinium HM27 data sets from a previously published data set (Rakyan et al. 2010), with additional sets of unpublished data (Fig. 4; Table 2). We found no evidence for epigenetic drift throughout the life course in either MZ or DZ pairs.
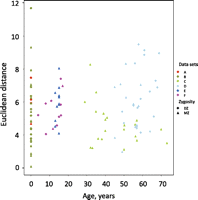
Relationship between within-pair methylation discordance with age in blood-derived tissues. Euclidean distance is plotted, as in Methods, for twins from birth to 73 yr of age from our data and 94 MZ pairs and 17 DZ pairs from previously published and unpublished Infinium HM27 data sets (see text and Table 2).
Details of the Infinium HumanMethylation27 data sets used for Figure 4
Identification of highly discordant gene classes
Despite the overall low median discordance in methylation apparent in all twin pairs (Supplemental Fig. S4), almost all pairs had several HM27 probes with a high level of methylation discordance in excess of ∼20% (Δβ > 0.2). To investigate the potential biological relevance of such probes, we ranked each gene-associated CpG by median within-pair standard deviation, a measure that summarizes the typical discordance between co-twins for that gene, separately for MZ and DZ pairs, for all three tissues (Supplemental Table S5). Using a combination of Gene Ontology (Supplemental Table S6) and pathway analysis (Supplemental Table S7), we found that genes associated with development and morphogenesis were over-represented in MZ and DZ pairs from all three tissues, closely followed by genes involved in response to environment and the cell cycle/cell division. In contrast to our previous analysis of gene expression (Gordon et al. 2011), we found no evidence that imprinted differentially methylated regions (DMRs) (Choufani et al. 2011) and housekeeping genes are significantly variably methylated within MZ twin pairs (p > 0.14 and p > 0.37 for all tissues, respectively).
Identification of discordantly methylated genes associated with birth weight
We are particularly interested in identifying epigenetic variation in genes potentially associated with birth weight because of the numerous previous studies linking low birth weight with later development of cardiovascular, metabolic, and other complex diseases (Barker 1990; Morley and Dwyer 2005; Burdge et al. 2007; Gluckman et al. 2007). We have previously identified genes whose expression levels correlate with birth weight in a subset of tissues from newborn twins and shown that these genes are enriched for functions and pathways associated with metabolism, growth, and cardiovascular disease (Gordon et al. 2011). In the present study, we performed a similar regression analysis of methylation M-values on birth weight, using a statistical model that estimates association based on within-pair variation of both DNA methylation and birth weight. Methylation of a small number of genes (seven in DZ CBMCs, one in MZ HUVECs) was significantly associated with birth weight after adjustment for multiple testing (adjusted P-value < 0.1) (Supplemental Table S8) and included genes with links to metabolism, growth, and cardiovascular disease. For example, APOLD1 (apolipoprotein L domain containing 1), identified in HUVECs and EDG1 (sphingosine-1-phosphate receptor 1), identified in CBMCs, both regulate vascular function in mice and humans (Liu et al. 2000; Regard et al. 2004; Simonsen et al. 2010; Gordon et al. 2011). Even though no genes in placenta reached significance after multiple testing, polymorphisms in the genes ranked 1 and 2, HLA-B (major histocompatibility complex class 1B) and SCD (stearoyl-CoA desaturase), have been associated with low birth weight and metabolic disease, respectively, in humans (Warensjo et al. 2007; Sampath and Ntambi 2008; Capittini et al. 2009; Shin et al. 2010).
To investigate pathways and processes that may be subject to epigenetic variation in association with birth weight, genes from the above regression analysis were ranked by adjusted P-values for birth weight and analyzed using Gene Ontology (Supplemental Table S9) and pathway analysis (Supplemental Table S10). An over-representation of genes associated with metabolism and biosynthesis was found for all three tissues for both MZ and DZ pairs, genes associated with cardiovascular function/disease were over-represented in all three tissues from MZ pairs and HUVECs from DZ pairs, and genes associated with growth and proliferation were over-represented in only CBMCs for both MZ and DZ pairs. To test for an association of specific classes of genes previously linked to birth weight and metabolic function, we performed gene set test analysis on a list of 167 imprinted DMRs (Choufani et al. 2011), 247 cardiovascular-associated genes (http://www.ucl.ac.uk/silva/cardiovasculargeneontology) (Lovering et al. 2008), and 41 genes previously linked with obesity by genome-wide association studies (Thorleifsson et al. 2009; Willer et al. 2009; Heid et al. 2010; Speliotes et al. 2010). No evidence of association of imprinted or cardiovascular-associated genes with birth weight was found in any tissue from MZ or DZ twins (p > 0.15 and p > 0.2, respectively), whereas some evidence for a moderate relationship between obesity-associated genes and birth weight was found in HUVECs from DZ (p = 0.01) and MZ pairs (p = 0.11), but not in other tissues (p > 0.56).
To further validate the observed associations, we performed locus-specific DNA methylation analysis on three genes using the Sequenom EpiTYPER platform, which we have previously found to be accurate and reproducible (Novakovic et al. 2010, 2011; Ollikainen et al. 2010). Methylation assays were designed to interrogate specific CpG sites (plus adjacent sites) measured by the HM27 platform, with measurements performed using the same sample set. Absolute DNA methylation was highly correlated across the two platforms (Spearman's correlation coefficient 0.485–0.947) (Supplemental Fig. S5) and validated the relationship between within-pair methylation discordance and birth weight discordance (Supplemental Figs. S6–S8). This analysis also highlighted that the observed association of birth weight with methylation level is regional rather than localized to a specific CpG site.
Discussion
Despite the growing awareness of the importance of interindividual epigenetic variation in modulating the risk associated with human disease, it remains largely unclear as to when and how such variation arises in humans. In this study, we capitalized on the strengths of a twin study design to present the first evidence for widespread, genome-scale epigenetic discordance between genetically identical humans at birth. These findings highlight the importance of the intrauterine environment in determining the neonatal DNA methylation profile and reveal the presence of tissue-specific variability in response to such factors.
Nonshared environmental/stochastic factors predominate in determining neonatal DNA methylation
Unsupervised clustering and pair-specific analysis of discordance and correlation revealed compelling evidence for a limited genetic contribution to the neonatal methylation variability, supported by analysis of variance components of DNA methylation, which produced mean levels of heritability of 0.05–0.12 depending on tissue (Fig. 2). However, the HM27 platform we used for this analysis contains probes associated with gene promoters and CpG islands, and previous studies have demonstrated that genomic regions showing a high heritability of DNA methylation are under-represented in CpG islands (Gertz et al. 2011). As such, our heritability estimates are potentially conservative and may not be representative of the genome as a whole. Additionally, it is important to remember that epigenetic heritability estimates will not only be population-specific, but also cell-, tissue-, time-, and locus-specific. These will also be largely dependent on the sensitivity, resolution, and coverage of the specific epigenetic assay used for measurement.
The suggestion that the DNA methylation profile is only minimally influenced by genetic variation agrees with a previous array-based, genome-scale study of DNA methylation in buccal cell DNA from teenage twins (Kaminsky et al. 2009) and with an analysis of DNA methylation at 1760 CpG sites in CD4+ lymphocytes from adult twins (Gervin et al. 2011). One study that measured DNA methylation at ∼1500 CpG sites in whole blood from 43 MZ and 43 DZ twin pairs found that 23% of all CpG sites displayed significant heritability of methylation level (Boks et al. 2009). Although low power makes comparison difficult, the range of heritability within the top 5% of probes in the Boks and colleagues study (0.62–0.94) is similar to that found for the top 5% of probes for CBMCs in our study (0.48–0.94). Studies of allele-specific methylation (ASM) have also found evidence for a high heritability of DNA methylation at a subset of genomic loci, with proportions varying with the method of analysis and the tissue examined (Kerkel et al. 2008; Boks et al. 2009; Zhang et al. 2009, 2010; Meaburn et al. 2010; Schalkwyk et al. 2010; Shoemaker et al. 2010; Gertz et al. 2011).
Surprisingly, we found little evidence for an effect of common environment on the overall DNA methylation profile at birth using variance component analysis. This does not rule out the possibility that a minority of genes are influenced by a common environment. Indeed, previous genome-scale studies of DNA methylation have found a range of probes significantly associated with maternal environmental, from 0.6% (Breton et al. 2009) and 1.1% (Fryer et al. 2011) to 23% (Katari et al. 2009). Because the accuracy of the estimation of effect size depends on study power, further investigation in larger numbers of twins is needed (Visscher 2004).
Given the small genetic effect and lack of evidence for widespread common environmental effects observed in our study, the largest residual variance component contributing to overall DNA methylation profile represents cumulative nonshared (individual intrauterine) environment and stochastic factors. There is prior evidence that nonshared environmental factors can influence phenotype (Bergvall and Cnattingius 2008; Plomin 2011; Torche and Echevarria 2011). Such factors in twin pregnancies include discordant placental weight, discordant placental umbilical cord insertion (both resulting in differential fetal blood supply), or differential exposure to infection (Stromswold 2006; Antoniou et al. 2011). Further evidence that factors specific to each twin can influence methylation comes from our finding that genes involved in response to environment are discordantly methylated (this study) or expressed (Gordon et al. 2011) within twin pairs.
Our finding that MZ twins sharing a single placenta (MC) were more discordant for methylation profile than MZ twins with their own placenta (DC) is counterintuitive but similar to that reported previously (Kaminsky et al. 2009). It was suggested that the earlier embryo splitting associated with DC pairs potentially reflects epigenetically divergent cell populations in the pre-splitting blastomere (Kaminsky et al. 2009), although there is no direct evidence supporting this hypothesis. An alternative explanation could be that MC twins are more likely to experience competition for resource allocation, with or without associated vascular connections that in extreme cases can lead to a large flow of blood in one direction known as twin-to-twin transfusion syndrome (TTTS). Although each of these scenarios is possible, it is worth noting that we have shown previously that the effect of chorionicity on DNA methylation can be tissue-dependent (Ollikainen et al. 2010; Novakovic et al. 2011). Clearly, larger studies are needed to resolve this issue.
Extensive evidence exists for a role of stochastic influence in determining DNA methylation and other epigenetic marks during early development (for review, see Whitelaw et al. 2010). An elegant example of this was reported by Feinberg and colleagues, who demonstrated that the genome-wide DNA methylation profile in the livers of newborn inbred mice raised in identical environments was hypervariable at certain loci, termed variably methylated regions (VMRs) (Feinberg and Irizarry 2011). VMRs were enriched in genes involved in development and morphogenesis both in mice and unrelated humans (Feinberg et al. 2010), and such genes are also discordantly methylated within our twin pairs (Supplemental Tables S9, S10). Because early development is associated with rapid cell division, this could explain our finding that genes involved in the cell cycle are discordantly methylated within twins, which was also found in adult twins (Kaminsky et al. 2009). We also found that genes associated with response to environment are hypervariable within MZ and DZ twin pairs in multiple tissues (Supplemental Tables S9, S10), which agrees with previous studies of expression discordance within MZ twin pairs (Sharma et al. 2005; Choi and Kim 2007).
Evidence for a tissue-specific effect on heritability of methylation profile
We found no compelling evidence for a common set of highly heritable DNA methylation variants across different tissues, supporting similar findings in a study of ASM in multiple human cell lines (Shoemaker et al. 2010) and recent data using gene expression as a phenotypic outcome (Powell et al. 2012). This contrasts with a subset of recently described ASM events described in a single individual that appear to be shared across kidney and skeletal muscle (Gertz et al. 2011). Such tissue-specific differences in the heritability of DNA methylation (and expression) may arise due to the possibility that different CpGs or combinations of CpGs may be involved in control of the expression of the same gene in different tissues, in association with the biological function of specific cells (Altschuler and Wu 2010).
Genomic regions with high within-pair methylation discordance; constraint at CpG islands?
We found that in all tissues and in all twins, within-pair differences in DNA methylation increased with increasing distance from CpG islands (Fig. 3). This agrees with a similar finding for highly variably methylated regions (VMRs) between unrelated individuals (Zhang et al. 2010) and with data showing that interindividual differences in methylation are lowest in unmethylated CpG islands and highest in methylated regions of the genome (Bock et al. 2008). Furthermore, our data have shown that such regions are hypervariable irrespective of zygosity, in support of our findings of only a minor genetic effect in determining the overall DNA methylation profile. In combination, these data suggest that methylation levels are more constrained in CpG-dense genomic regions, possibly because of a stabilizing influence from neighboring CpGs (Bock et al. 2008).
What function does variable methylation serve?
Because MZ twins share the same mother and genotype, factors present with the residual variance component “nonshared environment” are most likely driving variation here (see above). Although the relative proportions of nonshared (intrauterine) environment versus stochastic influences on the neonatal epigenome remain to be demonstrated, Feinberg and colleagues have proposed a model in which a genetically inherited propensity to stochastic variability in DNA methylation has evolved to increase fitness in a varying environment (Feinberg and Irizarry 2011). Furthermore, a combination of influences have been proposed to explain the phenomenon of metastable epialleles, which are variably expressed due to epigenetic modifications that are established in a stochastic manner during early development, and that are also environmentally labile (for review, see Bernal and Jirtle 2010).
DNA methylation in relation to low birth weight: A mechanistic link with complex disease in later life
We found that the genes whose methylation was tightly associated with birth weight were enriched for functions and pathways associated with growth, metabolism, and cardiovascular disease (Supplemental Tables S9, S10). A subset of these was confirmed by locus-specific DNA methylation analysis that revealed up to 60% methylation discordance between heaviest versus lightest twins (Supplemental Figs. S6–S8). Taking into account our previous data from expression arrays, we speculate that DNA methylation and expression levels of key genes associated with cardiovascular and metabolic function can be set in utero to confer elevated risk for disease in later life and that this setting is linked with low birth weight.
Lack of evidence for genome-wide epigenetic drift throughout postnatal life
Our data support the idea that a combination of stochastic and nonshared intrauterine environment can generate a net within-pair difference in DNA genome-wide epigenomic profiles at birth. But do such factors influence the epigenome after birth in a similar way? The cumulative effects of environmental and stochastic variation on changing epigenetic profile (known as “epigenetic drift”) were first described by a study that examined both genome-wide and locus-specific DNA methylation variation in a small number of young and middle-aged MZ twins, which found a greater within-pair discordance in the latter (Fraga et al. 2005). Our cross-sectional analysis of genome-scale data from HM27 arrays from blood-derived DNA from twins from birth to >70 yr of age does not support a generalized age-related epigenetic drift at the genome-scale level (Fig. 4) and agree with other recent data from saliva using the same platform (Bocklandt et al. 2011). These discrepancies are most likely due to differences in methodology, sample size, and genomic regions the genome studied. Despite our lack of compelling evidence for epigenetic drift over the life course, others have shown that DNA methylation can vary over time, using cross-sectional (Boks et al. 2009; Christensen et al. 2009; Rakyan et al. 2010) or longitudinal (Bjornsson et al. 2008; Feinberg et al. 2010) approaches. Further studies are needed that focus on both epigenetic change and epigenetic discordance within twin pairs over time, ideally addressing the suggestion that epigenetic drift could be driven by differences in environment (Fraga et al. 2005).
Strengths and weaknesses of this study
In focusing on newborn twins, we have isolated effects accumulated solely during intrauterine life. This is a unique time during the life span in which individuals share the same maternal environment, thus minimizing differences in shared environment. This has also enabled us to use traditional modeling of variance components to estimate the relative influence of genetic, common, and nonshared environment throughout the genome. Because only one other study has measured chorionicity in twins (Kaminsky et al. 2009), this has enabled us to investigate the effects of sharing or having separate placentas on the methylome. Studying twins at birth has also enabled us to identify a possible epigenetic mechanism linking low birth weight and risk for complex disease in later life. This has implications for future minimization of the poor health outcomes associated with low birth weight, through reversing the associated epigenetic changes. Technologically, we have used a reproducible and accurate method of genome-scale measurement of DNA methylation, which compares well to and is more quantifiable than immunoprecipitation or enzyme-based methods (Bock et al. 2010). We have also looked at multiple tissues, which has enabled us to determine whether methylation variation occurs in a tissue-specific manner. Twin studies sometimes attract the criticism that they may not be applicable to the rest of the population. However, apart from sharing a placenta, and a relatively smaller birth size and lower gestational age, twins have similar health outcomes to singletons (Morley et al. 2003; Morley and Dwyer 2005). Greater than 95% of twins and all singletons have their own amniotic sac and therefore would have similar issues of nonshared environment, albeit with different magnitudes of variation in placenta weight, cord placement, and cord thickness (Antoniou et al. 2011).
The main weakness of this and similar studies (e.g., Boks et al. 2009) is the relatively small sample size. However, our primary focus is not on specific genes, but on groups of genes with shared ontology, and on compound measurements of similarities and differences between the methylomes of twins. We have also focused on gene promoters and CpG islands, which constitute a relatively small proportion of the genome, and acknowledge that some of our findings may not be reflected in the rest of the genome.
In our calculations of within-pair discordance and heritability, it is possible that genetic differences within DZ pairs result in differing probe-hybridization efficiencies within such pairs. However, our unpublished studies have shown that such sequence variants do not affect the estimation of DNA methylation at the CpG associated with each probe, essentially because any changes to hybridization kinetics will affect the methylation-specific probes and the non-methylation-specific probes in the same manner. But what of the effect of SNPs at CpG sites assayed by Infinium HM27 probes? Of a small number of probes for which this is the case (Chen et al. 2011), only one appeared in our list of ∼1000 genes with a high heritability (data not shown).
In summary, our study uses biological samples from twins at birth and contributes to the understanding of prenatal human development and the factors by which it is influenced. It is essential to understand these factors in healthy individuals in order to compare with states of disease and disease predisposition.
Methods
Subjects and tissues
Sample collection from twins at the time of delivery was performed with appropriate human ethics clearances from the Royal Women's Hospital (06/21), Mercy Hospital for Women (R06/30), and Monash Medical Center (06117C), Melbourne. The twin pairs chosen for methylation array analysis are shown in Table 1. They shared a similar sex ratio, gestational age, and birth weight to the full group of 250 pairs. For CBMCs, we studied 18 MZ and nine DZ pairs; from HUVECs, 14 MZ and 10 DZ pairs; and from placenta, eight MZ and seven DZ pairs. HUVECs and CBMCs were examined in combination for 19 pairs, and all three tissues were profiled in seven pairs (Table 1).
Sample preparation and DNA extraction
CBMCs were purified using density gradient centrifugation, CD31-positive HUVECS were isolated using collagenase and magnetic sorting, and full-thickness placental samples were isolated as described previously (Novakovic et al. 2010; Gordon et al. 2011). DNA was extracted using phenol:chloroform as described previously (Novakovic et al. 2010).
Infinium methylation analysis
DNA samples were processed using the MethylEasy Xceed bisulphite conversion kit (Human Genetic Signatures), according to the manufacturer's instructions. Genome-wide DNA methylation analysis was performed by the Australian Genome Research Facility (Melbourne, Australia) or ServiceXS (Leiden, The Netherlands). Infinium HM27 BeadChip arrays (Illumina) were hybridized and scanned as per the manufacturer's instructions. Raw data were exported from BeadStudio (Illumina). All statistical analysis was performed in R (version 2.12) (R_Development_Core_Team 2009) using packages from the Bioconductor project (Gentleman et al. 2004) and in-house scripts. Data quality was confirmed using arrayQualityMetrics (Kauffmann et al. 2009). Probes on the X and Y chromosomes were removed from further analysis to eliminate sex-specific differences in methylation. The lumi package, which is specifically designed for Illumina data, was used to calculate the log2 ratio for methylated probe intensity to unmethylated probe intensity, the M-value (Du et al. 2008, 2011). These values underwent background correction and normalization using lumi. Possible batch effects from samples processed at different times were compensated for with “color adjustment” from lumi. Any probe within a sample with a highly conservative detection P-value of 0.001 or greater was excluded from further analysis. For correlation coefficients, M-values with batch effects removed were used to calculate a mean M-value for each probe. This was then subtracted across all of the samples to create a mean-corrected set of M-values. The correlation coefficients for each pair for MZ, DZ, MZMC, and MZDC were then calculated from these values. Correlation coefficients were also calculated only within each array, for every unrelated individual (UR) comparison, i.e., pairwise comparison of unrelated individuals. Euclidean distances were also calculated using M-values from all probes.
Differential methylation analyses were performed using the limma package (Smyth 2005), which is designed for the analysis of microarray data. Array quality weights were estimated to allow for the possibility of variable DNA quality between the samples (Ritchie et al. 2006), and these weights were incorporated in all differential methylation analyses. To study discordance between co-twins, a linear model was fitted to the M-values for each gene with twin-pair as a predictive factor, equivalent to a one-way ANOVA analysis in which variability is broken up into between-twin and within-twin components. This analysis yielded a within-twin standard deviation for each CpG across all twin pairs, which was taken to summarize the level of discordance between co-twins for that gene. To examine the relationship between birth weight and DNA methylation, genewise linear models were fitted with twin-pair as a factor and log-weight as a covariate. The linear models also included a correction for possible batch effects. The association of birth weight on each gene was assessed using moderated t-statistics (Smyth 2004). Genes were judged to be differentially methylated if the false discovery rate was under 0.1 after adjustment for multiple testing using the Benjamini and Hochberg algorithm (Hochberg and Benjamini 1990).
Limma was also used to conduct several gene set tests. These tests use a Wilcoxon rank-sum test to assess whether a particular set of genes tends to be more highly ranked according to some given criterion than the remaining genes (Michaud et al. 2008). To test whether certain functional categories of genes tend to be more discordant than others, genes were ranked by within-pair standard deviation. This approach was applied to 45 imprinted genes and to 568 housekeeping genes (Eisenberg and Levanon 2003). To test whether certain functional groups of genes are associated with birth weight, genes were ranked by moderated t-statistic when testing for birth weight as a covariate. This approach was applied to a list of 247 cardiovascular-associated genes from the Cardiovascular Gene Ontology Annotation Initiative (Lovering et al. 2008).
Estimating variance components of DNA methylation
Using genetic (twin) models, we are able decompose the observed variance in methylation levels into its additive genetic and common environmental components (Neale and Cardon 1992). Additive genetic variance (A) denotes the variance resulting from the sum of allelic effects throughout the genome, whereas common environmental variance (C) relates to the environmental influences shared within twin pairs. The remaining variance (E) is nonshared (individual) environmental effects and includes error terms. We can estimate A, C, and E for each methylation probe based on the resemblance between MZ twins and DZ twins, who share, on average, 50% of segregating loci. For each methylation probe, we calculate the intraclass correlation between MZ (rMZ) and DZ (rDZ) twin pairs using a least-squares estimator (Visscher 2004). Variance components A, C, and E are calculated as follows: A = 2(rMZ − rDZ); C = rMZ − A; E = 1 − rMZ. Heritability (h2) is calculated as h2 = A/P, where P is the sum of A, C, and E (observed phenotypic variance).
Locus-specific methylation analysis
Sequenom MassARRAY EpiTYPING was performed as previously described (Ollikainen et al. 2010; Novakovic et al. 2011). The primers are listed in Supplemental Table S11. In brief, amplification was performed after bisulfite conversion of genomic DNA with the MethylEasy Xceed bisulphite conversion kit (Human Genetic Signatures). All PCR amplifications and downstream processing were performed at least in duplicate, and the mean methylation level at specific CpG sites was determined. Raw data obtained from MassArray EpiTYPING were cleaned systematically using an R-script to remove samples that failed to generate data for >70% of CpG sites tested. Also, technical replicates showing ≥5% absolute difference from the median value of the technical replicates were removed, and only samples with at least two successful technical replicates were analyzed. Samples were compared across each analyzable CpG site in the amplicon, as well as the mean across the whole amplicon.
Gene Ontology and pathway analysis
Gene Ontology (GO) analyses were conducted using GOrilla software (Eden et al. 2009), with the default options and searching all ontologies. This program identifies enriched GO terms from ranked gene lists using all ranked genes, an approach that is analogous to the limma Wilcoxon gene set tests. Genes were ranked for GOrilla analyses using the same ranking statistics as described above for the gene sets tests. Pathway analysis was performed using Ingenuity Pathways Analysis (IPA) software (Ingenuity Systems). The functional analysis identified the biological functions and/or diseases that were most significant for each of the data sets. IPA was used to identify enriched canonical pathways, gene networks, and functional classes. Genes corresponding to all Infinium probes that passed QC were used as a reference set.
Public microarray data analysis
Illumina HumanMethylation27 BeadChip twin data were obtained from various sources (see Table 2). The data sets used were restricted to blood and, wherever possible, phenotypically normal twins. X- and Y-chromosome probes were removed from the analysis, and correlation coefficients and Euclidean distances were calculated from all probes as described above (Fig. 4).
Data access
The data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE36642.
Acknowledgments
We thank the following people for kindly sharing their unpublished data: Robert Lyle and Kristina Gervin, Department of Medical Genetics, Oslo University Hospital and University of Oslo, Norway; Jennifer Harris, Division of Epidemiology, Norwegian Institute of Public Health, Oslo, Norway; Jordana Bell and Pei-Chien Tsai, Department of Twin Research and Genetic Epidemiology, King's College and St. Thomas' Hospital, London, England; and Jonathan Mill and Chloe Wong, King's College, London, England. We also thank John Carlin for his contributions to establishing the PETS cohort and for biostatistical support; obstetricians Mark Umstad, Royal Women's Hospital, Melbourne; Euan Wallace, Monash Medical Centre, Melbourne; and Mark Permezel, Mercy Hospital for Women, Melbourne for their contributions to establishing the PETS cohort and access to study participants; Sarah Healy, Tina Vaiano, Nicole Brooks, Jennifer Foord, Sheila Holland, Anne Krastev, Siva Illancheran, and Joanne Mockler for recruitment and sample collection; and Technical Officer Anna Czajko, Study Coordinator Geraldine McIlroy, and all mothers and twins that participated in this study. Finally, we are grateful to all of the families at the participating SFARI Simplex Collection (SSC) sites, as well as the principal investigators (A. Beaudet, R. Bernier, J. Constantino, E. Cook, E. Fombonne, D. Geschwind, D. Grice, A. Klin, D. Ledbetter, C. Lord, C. Martin, D. Martin, R. Maxim, J. Miles, O. Ousley, B. Peterson, J. Piggot, C. Saulnier, M. State, W. Stone, J. Sutcliffe, C. Walsh, E. Wijsman). We also appreciate the access to phenotypic data on SFARI Base. Approved researchers can obtain the SSC population data set described in this study by applying at https://base.sfari.org. This work was supported by grants from the Australian National Health and Medical Research Council (grant numbers 437015 and 607358 to J.C. and R.S.), the Bonnie Babes Foundation (grant number BBF20704 to E.J.), the Sigrid Juselius Foundation (to M.O.), the Academy of Finland (to M.O.), the Finnish Cultural Foundation (to M.O.), the Financial Markets Foundation for Children (grant no. 032-2007), and by the Victorian Government's Operational Infrastructure Support Program.
Footnotes
-
↵13 Corresponding author
E-mail jeff.craig{at}mcri.edu.au
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.136598.111.
- Received December 16, 2011.
- Accepted April 26, 2012.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








