
Whole genome sequencing of matched primary and metastatic acral melanomas
- Samra Turajlic1,8,
- Simon J. Furney1,8,
- Maryou B. Lambros2,
- Costas Mitsopoulos3,
- Iwanka Kozarewa4,
- Felipe C. Geyer2,
- Alan MacKay2,
- Jarle Hakas3,
- Marketa Zvelebil3,
- Christopher J. Lord4,
- Alan Ashworth4,
- Meirion Thomas5,
- Gordon Stamp6,
- James Larkin7,
- Jorge S. Reis-Filho2 and
- Richard Marais1,9
- 1Signal Transduction Team, Division of Cancer Biology, Institute of Cancer Research, London SW3 6JB, United Kingdom;
- 2Molecular Pathology Team, The Breakthrough Breast Cancer Research Centre, Institute of Cancer Research, London SW3 6JB, United Kingdom;
- 3Cancer Informatics, The Breakthrough Breast Cancer Research Centre, Institute of Cancer Research, London SW3 6JB, United Kingdom;
- 4Division of Breast Cancer Research, The Breakthrough Breast Cancer Research Centre, Institute of Cancer Research, London SW3 6JB, United Kingdom;
- 5Department of Surgery, Royal Marsden Hospital, London SW3 6JJ, United Kingdom;
- 6Department of Histopathology, Royal Marsden Hospital, London SW3 6JJ, United Kingdom;
- 7Melanoma Unit, Royal Marsden Hospital, London SW3 6JJ, United Kingdom
-
↵8 These authors contributed equally to this work.
Abstract
Next generation sequencing has enabled systematic discovery of mutational spectra in cancer samples. Here, we used whole genome sequencing to characterize somatic mutations and structural variation in a primary acral melanoma and its lymph node metastasis. Our data show that the somatic mutational rates in this acral melanoma sample pair were more comparable to the rates reported in cancer genomes not associated with mutagenic exposure than in the genome of a melanoma cell line or the transcriptome of melanoma short-term cultures. Despite the perception that acral skin is sun-protected, the dominant mutational signature in these samples is compatible with damage due to ultraviolet light exposure. A nonsense mutation in ERCC5 discovered in both the primary and metastatic tumors could also have contributed to the mutational signature through accumulation of unrepaired dipyrimidine lesions. However, evidence of transcription-coupled repair was suggested by the lower mutational rate in the transcribed regions and expressed genes. The primary and the metastasis are highly similar at the level of global gene copy number alterations, loss of heterozygosity and single nucleotide variation (SNV). Furthermore, the majority of the SNVs in the primary tumor were propagated in the metastasis and one nonsynonymous coding SNV and one splice site mutation appeared to arise de novo in the metastatic lesion.
Melanocytes are specialized pigmented cells that are primarily located in the epidermis in humans. However, they have also been found in lower numbers in the brain, the meninges, the eyes, the ears, and some mucosal surfaces. These cells are the precursors of melanoma, a potentially deadly cancer that, in advanced form, has a poor prognosis with a median survival of 6–10 mo and five-yr survival of ∼10% (Lee et al. 1995). Worldwide, there are ∼140,000 cases and over 40,000 deaths from melanoma each year (Parkin et al. 2005; Lucas et al. 2006). The commonest form of melanoma (∼90% of cases) is cutaneous melanoma, which largely affects Caucasian populations, generally occurs on habitually sun-exposed hairy skin, and is associated with intermittent or chronic ultraviolet (UV) light exposure.
Acral melanoma is a rare subtype of cutaneous melanoma, so-called because it occurs on nonhairy or acral skin, which includes palms, soles, and nailbeds. Several lines of clinical and epidemiological evidence suggest that acral and common cutaneous melanomas are distinct entities. Thus, unlike common cutaneous melanoma, acral melanoma is not associated with naevi, family history, or known melanoma-susceptibility genes (Phan et al. 2007). Acral melanoma is the predominant melanoma subtype in non-Caucasians and has a notably worse prognosis than common cutaneous melanoma (Bradford et al. 2009). In recent years, it has also become clear that acral and common cutaneous melanomas are genetically distinct. Whereas acral melanomas are characterized by a high frequency of focal amplifications (particularly involving CCND1, CDK4, and GAB2) and losses, common cutaneous melanoma exhibits fewer gene copy number changes. Similarly, whereas the protein kinase BRAF gene and the small G-protein NRAS gene are mutated in ∼50% and ∼20% of common cutaneous melanomas, respectively, and mutations in the receptor tyrosine kinase KIT gene appear to be absent, in acral melanoma BRAF mutations account for ∼16% of cases, and KIT mutations for a further ∼20% (Curtin et al. 2005, 2006).
Next generation sequencing (NGS), and especially whole genome sequencing (WGS), has enabled systematic discovery of mutational spectra in many cancer samples, and recently the whole genome sequencing of COLO829 melanoma cells was reported (Pleasance et al. 2010a). That study provided the first insight into the UV light-induced DNA damage and consequent mutational processes in melanoma cells, as well as evidence of transcription-coupled repair in this particular sample. Transcriptome sequencing of melanoma short-term cultures has revealed several novel gene fusions (Berger et al. 2010), and furthermore, sequencing of melanoma exomes demonstrated that TRRAP is recurrently mutated, suggesting that it is a new melanoma oncogene (Wei et al. 2011). These sequencing efforts have focused on the commonest melanoma subtype, and of the rare subtypes, only uveal melanoma has been sequenced (Harbour et al. 2010). Thus, at the single nucleotide level, our understanding of the genome-wide changes that underpin the biology of acral melanomas remains limited.
Recently, a patient presented at the Royal Marsden Hospital Melanoma Unit with a primary acral melanoma and concurrent metastatic involvement of the regional lymph nodes but no systemic metastasis. The patient had not received neoadjuvant or adjuvant chemotherapy and underwent surgery to remove both the primary and metastatic lesions. These samples provided a rare opportunity to perform WGS on an acral melanoma and simultaneously on its corresponding metastatic lesion. Comparisons of this kind have been performed in breast cancer, but this is the first comparison of a melanoma primary-metastasis pair by WGS. In addition to providing single nucleotide level sequence information, this analysis also gives an insight into the mutational processes and the relationship to UV light and DNA repair in a pair of primary and metastatic acral melanoma samples.
Results
Case description
A 65-yr-old male was diagnosed with a primary melanoma of the left great toe nail bed with concurrent metastatic involvement of the ipsilateral inguinal lymph nodes but no systemic metastasis. He was treated with surgical amputation of the toe and groin lymph node clearance. No neoadjuvant or adjuvant chemotherapy was administered. Histopathological examination revealed an ulcerated pigmented primary lesion of the nail bed involving the epidermis and dermis and extending down to subcutaneous fat with focal perineural and perivascular invasion. An acral lentiginous growth pattern was evident only in a small minority of the cells in the primary lesion. Breslow thickness was 5 mm. Immunohistochemistry confirmed the melanocytic origin of the tumor with strong staining for Melan A and HMB45. All but one of the lymph nodes contained metastatic deposits which, like the primary tumor, were composed of an admixture of atypical and pleomorphic epithelioid and spindle melanocytes. Informed consent for full genome sequencing was obtained, and DNA was prepared from the patient's peripheral blood. Primary tumor and a lymph node metastasis were microdissected as previously described (Geyer et al. 2010) to ensure tumor cell enrichment in the samples, and subjected to DNA and RNA extraction. Sanger sequencing did not reveal somatic mutations in either KIT (exons 9, 11, 13, 17) or BRAF (exons 11 and 15).
Whole genome sequencing
Sequencing of the matched normal and tumor samples was performed using unchained combinatorial probe anchor ligation chemistry on arrays of self-assembling DNA nanoballs (Drmanac et al. 2010). The gross mapping yields were 169.28, 156.96, and 150.20 Gb for the normal, primary tumor, and metastatic deposit samples, resulting in an average coverage of ∼59×, 55×, and 53× haploids, respectively. The reads were aligned to the NCBI build 37 reference genome, and the percentages of fully called bases for the normal, primary, and metastasis samples were 94%, 92.5%, and 91.6% (Supplemental Table S1A). Variations between the reference genome and each of the samples were called and scored using a local de novo assembly approach (Drmanac et al. 2010). High genotype call accuracies for each sample were confirmed by genome-wide SNP genotyping arrays (Supplemental Table S1B). We identified widespread somatic alterations throughout the genome on all scales, from single nucleotide variants (SNVs) and small insertions or deletions (indels) to structural variations in both primary and metastatic tumors (Fig. 1A,B).
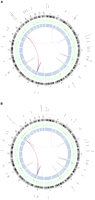
Summary of lesions found in acral melanoma. Shown are primary and metastasis Circos plots (Krzywinski et al. 2009) of somatic mutations in the primary (A) and metastatic (B) tumors. The outer circle contains whole genome high-confidence SNVs (black dots) and nonsynonymous SNVs (orange dots; annotated with HGNC/Ensembl gene symbols). Copy number alterations are shown in the inner two plots (green circle shows gains and blue shows losses). Validated structural variations are depicted as links in the interior of the plot.
Mutational analysis
Relative to the germline sequence, we initially identified 12,661 somatic SNVs in the primary sample and 11,711 in the metastasis. Each SNV was assigned a “somatic score” (see Methods), which is an estimate of the probability of the SNV being a true positive. Since this list is likely to contain false positives (Lee et al. 2010), we selected at random 50 SNVs from each of the primary and metastasis candidate lists. These SNVs were analyzed by conventional Sanger sequencing in the normal, primary, and metastasis to determine a somatic score threshold. Receiver operating characteristic (ROC) curves were then constructed using the somatic scores (Supplemental Figs. S1, S2). A somatic score threshold of −10 was selected, which equates to a true positive rate (TPR) of 0.93 and a false positive rate (FPR) of 0.10 in the primary-normal comparison and 0.93 and 0.13, respectively, in the metastasis-normal comparison. Applying this threshold resulted in a total of 6164 high-confidence somatic SNVs in the primary tumor and 5582 high-confidence somatic SNVs in the metastasis (Table 1). To validate the somatic SNVs in the coding regions, we also performed exome capture and paired-end short read sequencing of the matched normal and tumor samples (Table 2). Irrespective of their somatic score, all putative nonsynonymous coding SNVs and indels detected by either whole genome or exome sequencing were analyzed by conventional Sanger sequencing.
Sequencing coverage summary for normal primary and metastasis whole genomes
Exome sequencing summary for the primary and metastatic tumors
Mutation rates and spectrum
Using the threshold described, we calculated a genome-wide point mutation rate of 2.16 per Mb in the primary tumor and 1.95 per Mb in the metastasis. The mutation rate per Mb in the exonic regions was 1.12 in the primary tumor and 0.99 in the metastasis. The mutation rate in the intronic regions was 1.63 in the primary tumor and 1.48 in the metastasis. Thus, the rate of mutations was significantly lower in the exonic regions compared to the intergenic (P < 1 × 10−15) and intronic (P < 1.4 × 10−4) regions. In addition, the mutation rate in the intronic regions was lower than the intergenic regions in both samples (P < 1 × 10−15). To investigate mutation rates in the genic regions, we performed expression microarrays of the primary and metastatic tumors to identify all expressed genes. We found that the mutation rate in the genic sequences was significantly lower in the expressed genes compared to nonexpressed genes in both tumor samples (P < 1 × 10−15) (Fig. 2A). Taken together, these data provide circumstantial evidence to suggest that transcription-coupled repair (TCR) is active in the samples under study. Finally, we observed that 60% of the high-confidence somatic mutations in the primary and metastasis genomes were C>T (G>A) transitions (Fig. 2B), and of these, 84% occurred at the 3′ base of pyrimidine di-nucleotides (TpC or CpC) (Fig. 2C,D).
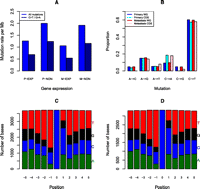
Somatic SNVs analysis in acral melanoma samples. (A) Somatic SNV mutation rates of all SNVs and C>T/G>A transitions for expressed genes in the primary (P-EXP) and metastatic tumors (M-EXP) and nonexpressed genes (P-NON and M-NON). (B) Proportion of somatic SNVs by class in the primary and metastatic tumors for the whole genome (WG) and coding regions (CDS). (C) Frequency of bases ±5 bp of whole genome C>T/G>A transitions in the primary tumors. (D) Frequency of bases ±5 bp of whole genome C>T/G>A transitions in the metastatic tumors.
Somatic coding mutations in the primary and metastatic tumors
We combined the whole genome, exome, and Sanger sequencing data to identify the somatic nonsynonymous SNVs in the coding regions of both tumors. The SNVs that were identified as somatic variants by NGS but were present in the germline by Sanger sequencing were classified as germline SNVs. We identified a total of 40 somatic nonsynonymous SNVs in the primary tumor and 41 somatic nonsynonymous SNVs and one splice site variant in the metastasis. The range of mutant allele frequencies was 21%–82% in the primary and 11%–84% in the metastasis (Table 3). Overall, 39 nonsynonymous SNVs were confirmed in both the primary and metastatic tumors, most of which were present at comparable frequencies in the two samples (Table 3). No somatic mutations were detected in miRNAs in either sample.
Summary of confirmed nonsynonymous mutations in the primary and metastatic tumors
To determine whether the metastatic deposit stemmed from a nonmodal clone from the primary tumor, we sought to define the differences in the repertoire of somatic mutations found in the metastatic deposit and in the primary tumor. Where there was discordance between the primary and metastasis calls, we visually inspected the binary sequence alignment/map (BAM) files to determine if the mutant allele was present at low frequencies in one of the samples even when it had been designated as wild type by our calling algorithms. Variants were considered exclusive to one of the samples (i.e., primary or metastasis) if they were determined as mutant by at least two of the orthogonal methods (i.e., whole genome, exome, or Sanger sequencing) and there was no evidence of that mutation in the other sample by any of the sequencing methods. A nonsynonymous coding mutation in SCAF1 was present in the primary but not the metastasis, and a nonsynonymous coding mutation in WNT1 and a point mutation in a splice site of SUPT5H were present in the metastasis but not the primary tumor (Table 3; Supplemental Fig. S3A,B).
Short insertions and deletions
We detected 4193 putative somatic small indels in the whole genome of the primary tumor and 3905 in the metastasis. Twenty-seven small indels were identified as somatic in the coding regions in the primary tumor and 32 in the metastasis. Only one indel was common to both primary and the metastasis. We successfully amplified and sequenced 47 coding regions containing putative indels. Three deletions were validated, of which one was also present in the germline. The two somatic deletions in PLCH2 and PDS5A validated in both the primary and metastasic tumors (Supplemental Fig. S4A,B). Based on this validation rate, we would predict 178 of the genome-wide indels to be true positives in the primary tumor and 166 in the metastatic lesion. We note that the only published cancer genome sequencing study using the same sequencing platform did not present data on indel validation (Lee et al. 2010).
Germline single nucleotide variants
Acral melanoma has not been linked to specific melanoma-susceptibility genes, but some individuals with acral melanoma have a higher risk of nonskin malignancies (Nagore et al. 2009), suggesting that acral melanoma might occur in a nonmelanoma cancer-susceptibility setting. Therefore, we analyzed the germline variants in the normal DNA from this patient to determine if it contained known cancer- susceptibility gene variants. We identified 8080 nonsense and missense SNVs compared to the reference genome. Additional germline variants were documented through invalidation of potential somatic variants. Most (97%) of the germline SNVs in this patient were not novel and are reported in dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/). No variants were found in melanoma-susceptibility genes such as MC1R and CDKN2A, but several variants mapped to IFNA16, which is within the melanoma-susceptibility locus on 9p21 (Yang et al. 2010), while others are in the Cancer Gene Census (www.sanger.ac.uk/genetics/CGP/Census) and so are known to be susceptibility genes in other cancers (Supplemental Table S2A). For example, known variants of MSH2, APC, MEN1, and MSH2 and novel variants of BRCA1 and ERCC2 were present in the germline of this patient (Supplemental Table S2B). The functional effects of each of the germline variants was assessed by SIFT (Kumar et al. 2009) (Supplemental Table S2A). The variants in the genes involved in the DNA repair pathway are of particular interest, but larger numbers of acral melanoma patients will need to be interrogated to ascertain the significance of these findings.
Copy number alteration and loss of heterozygosity
We detected regions of copy number alterations (CNAs) in the primary and metastatic tumors relative to the matched normal. Sequence depth-based CNA analysis was consistent with the findings of the other platforms, including array CGH and genome-wide SNP arrays (Fig. 3; Supplemental Figs. S5–S7). Amplifications of 4q12, 11q13, 11q14, 17p11, and 20q11 were present in both the primary and the metastatic tumors. Using fluorescence in situ hybridization (FISH), we confirmed the amplification of KIT within the 4q12 amplicon (Supplemental Fig. S8).
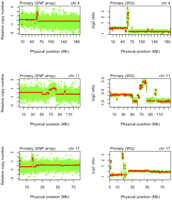
Copy number alterations in the primary tumor. SNP array plots showing somatic copy number alteration (CNA) in the primary tumor detected by genome-wide SNP arrays (left column) and whole-genome (WG) sequencing normalized read-depth per 10 kb (right column) for chromosomes 4, 11, and 17.
The zygosity of each of ∼924,000 SNP positions was determined in the normal, primary, and metastasis DNA by genome-wide SNP array. Regions of loss of heterozygosity (LOH) (in which SNPs were heterozygous in the normal but homozygous in the tumor) were identified (see Methods; Supplemental Figs. S9, S10). These regions largely overlap with the potential LOH regions detected by whole genome sequencing (i.e., where SNVs are called as heterozygous in the normal but homozygous in the tumor) (Fig. 4).
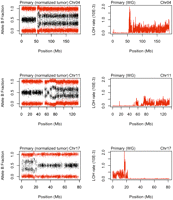
Loss of heterozygosity in the primary tumor. SNP array plots showing loss of heterozygosity (LOH) in the primary tumor as detected by allele B frequency from SNP array (left column) and whole-genome sequencing LOH rate in 100-kb windows (right column) for chromosomes 4, 11, and 17.
Structural variation
We utilized the mate-paired nature of the sequence library to identify putative large-scale structural variations (SVs). De novo assembly around putative SV breakpoints was conducted (Drmanac et al. 2010), and SVs present in the patient's germline genome were excluded. We applied a number of criteria to eliminate false positives. SV events were discarded unless there were more than ten mate-pairs in a cluster, de novo assembly of the junction was successful, there was a high mapping diversity, and specific repeat sequences on the left and right side of the junction were absent. In addition, we imposed a stringent criterion that the breakpoints must be identical in both the primary and metastatic tumors so as to identify a set of high- confidence somatic SV candidates. This resulted in the detection of 121 somatic SV candidates in the sequenced genomes of both the primary and metastatic lesions. To validate these predicted SVs, we used PCR to amplify the breakpoints and then Sanger-sequenced these products. The sequences were mapped to the reference genome to confirm breakpoints at single nucleotiode resolution. Of the 121 putative SVs, 57 (47%) and 71 (58%) were found to be somatic changes in the primary and metastasis, respectively; 55 of these were present in both (Supplemental Tables S3, S4). Of the validated rearrangements, 38 in the primary tumor and 42 in the metastasis have at least one breakpoint that maps to a genic region. Furthermore, seven gene fusions were predicted from the validated somatic rearrangements, but reverse-transcription PCR (RT-PCR) analysis failed to detect expression of the putative chimeric transcripts. However, this may be due to the complex nature of the rearrangements in which these events occur or the fact that these genes possess multiple or novel transcript isoforms (Berger et al. 2010), making it difficult to predict all possible fusion transcripts.
Discussion
Whole genome sequencing of a primary acral melanoma and its matched metastasis has revealed a range of somatic single nucleotide variants, copy number alterations, and structural variations, the combination of which are likely to have contributed to the pathogenesis of the tumors. Importantly, as this particular patient did not receive prior treatment, this case presented a rare opportunity to obtain a baseline mutation rate and spectrum in a sample that had not been exposed to therapeutic DNA-damaging agents.
Unlike some acral melanomas that do bear resemblance to the conventional cutaneous subtype by exhibiting nonacral growth patterns or harboring mutations in the BRAF gene, this case had the bona fide histopathological and clinical features of an acral melanoma. Furthermore, our NGS and aCGH analyses revealed amplifications in 4q12, 11q13, 11q14, 17p11, and 20q11 and a prominent amplification of KIT in both the primary and the metastatic tumors. These observations are consistent with the results of previous reports on the patterns of CNAs in acral melanomas (Bastian et al. 2000; Curtin et al. 2005, 2006) and indicate that the lesions we studied have the cardinal genetic features currently ascribed to acral melanomas. In addition to these general features, we also identified a number of novel genetic alterations, including 41 nonsynonymous SNVs. Many of the genes mutated in our samples are mutated in other cancers, but only PLCB4 has previously been reported to be mutated in melanoma (Wei et al. 2011), and CNTN5 and PLCH2 have been reported to be mutated in melanoma cell lines (http://www.sanger.ac.uk/genetics/CGP/cosmic/). Notably, none of the specific SNVs that we report has been described previously, and thus, they all appear to be private or low-frequency events, although this remains speculative until these genes are sequenced in multiple samples.
We detected considerable genetic heterogeneity in both the primary tumor and the metastasis as evidenced by the wide range of mutant allele frequencies (11%–84%). The comparison of the mutational repertoires in the two tumors shows that the majority of the coding SNVs and the CNAs are present in both, only one mutation is present in the primary and absent in the metastasis, and two mutations appeared to arise de novo in the metastasis. These findings are akin to the results of similar studies in breast cancer (Ding et al. 2010) and pancreatic cancer (Yachida et al. 2010), in which most of the original mutations present in the primary were propagated in the metastasis. Previous studies have suggested that in melanoma the metastatic gene expression pattern emerges during the vertical growth phase (Jaeger et al. 2007; Soikkeli et al. 2007), and, considering that the primary tumor was diagnosed at an advanced stage (i.e., in vertical growth phase) and the lymph node metastasis was detected synchronously, it is, perhaps, not surprising that they are highly similar at single nucleotide resolution.
Possible drivers of melanomagenesis
The consequences of different mutations cannot be determined from these data alone. The majority of events are expected to be passenger mutations, but among the putative driver oncogenes are genes that have previously been linked to other cancers, including ASB9 (Tokuoka et al. 2010), FAT2 (Matsui et al. 2008), PTRF (Aung et al. 2011), RHOB (Mazieres et al. 2004), and CNDP2 (Zhang et al. 2006), and those in pathways known to be important in melanoma, including KSR1 (Takata et al. 2005), PREX2 (Muchemwa et al. 2008), and MFI2 (Bertrand et al. 2007).
Putative cooperating genetic lesions may be associated with nonsense mutations or deletions leading to tumor suppressor gene inactivation. Nonsense mutations were observed in both the primary and metastatic tumors in DROSHA (Q1087*), ERCC5 (E418*), LRRK1 (E880*), and LRRFIP1 (R380*). DROSHA encodes the critical nuclease in microRNA (miRNA) processing (Lee et al. 2006), a function that is linked to cancer (Lu et al. 2005), and reduced DROSHA expression is associated with poor prognosis in neuroblastoma (Lin et al. 2010) and ovarian cancer (Merritt et al. 2008). The ERCC5 gene product is a component of the nucleotide excision repair (NER) pathway, and, although somatic mutations in ERCC5 are not reported in melanoma (www.sanger.ac.uk/genetics/CGP/cosmic/), germline mutations are associated with xeroderma pigmentosum, which carries a 1000-fold increased melanoma risk (Wang et al. 2009). The LRRK1 gene product regulates EGFR trafficking (Hanafusa et al. 2011) which is associated with a wide range of human cancers. Intriguingly, germline mutations in LRRK genes are linked to Parkinson's disease and appear to confer a higher risk of melanoma (Pan et al. 2011), which is congruent with studies linking melanoma to neurodegenerative diseases (Albert 2010). Finally, we note that we also observed a somatic 8-Mb deletion spanning CDKN2A and CDKN2B, which are melanoma tumor suppressor genes (Maelandsmo et al. 1996).
Potential drivers of metastasis
Only two single nucleotide variants were detected in the metastasis and not the primary tumor. One affected the coding region of WNT1 (C369F), and the second disrupted a splice site in SUPT5H. While our data suggest that these mutations arose de novo in the metastatic tumor, it is also possible that they were present in a small subclone in the primary that evaded detection by all of our sequencing methods. Wnt-β-catenin signaling is a key driver of melanoma metastasis (Chien and Moon 2007), but mutations in the components of this pathway have not previously been reported in melanoma. This is the first report of a nonsynonymous mutation in WNT1 in any cancer, but functional studies will be required to determine its role in the metastatic process. In contrast, SUPT5H, a regulator of transcription elongation, has not previously been linked to cancer. Mutations in SCAF1 have not been reported in cancer, and the mutation detected in our study (T694S) was present only in the primary and not the metastasis, although we cannot discount the possibility of its presence in a subclone in the metastatic lesion.
Genomic landscape
As part of this study, we analyzed somatic mutation rates and signatures, which have previously been used to elucidate the factors that underlie tumorigenesis. The mutational rates in the acral melanoma tumors in this study are lower than those reported for COLO829 melanoma cells (Pleasance et al. 2010a) and the mutation rates estimated from RNA sequencing of short-term melanoma cultures (Berger et al. 2010) and exome sequencing of melanoma tumors (Wei et al. 2011). Whole genome sequencing of a common cutaneous melanoma tumor is not available at this stage, but several other solid malignancies have been sequenced. We observe that the mutational rates in our tumors are 10-fold lower than is reported in a non-small cell lung cancer sample that was sequenced by the same method (Lee et al. 2010). Notwithstanding the differences in sequencing methods and analysis, the mutational burden of the tumors under study is similar to that observed in breast cancer (Shah et al. 2009; Ding et al. 2010) and in prostate cancers (Berger et al. 2011) and fourfold higher than was reported in an acute myeloid leukemia (Ley et al. 2008).
Thus, the overall mutation rate in these tumors is comparable to solid tumors that are not associated with obvious carcinogens such as UV light or cigarette smoke. Nevertheless, the predominant mutations (60%) in the acral tumors were C>T transitions, a type of lesion that is linked to UV exposure (Pfeifer et al. 2005). Importantly, the majority of the C>T transitions (84%) were at the 3′ base of pyrimidine di-nucleotides (TpC or CpC), another feature associated with UV-induced damage (Daya-Grosjean and Sarasin 2005). Previous studies (Greenman et al. 2007) have shown that C>T transitions predominate in melanoma samples, and in the whole genome sequencing studies, they accounted for ∼70% of mutations in the COLO829 melanoma cells but only ∼20% of mutations in lung cancer samples (Lee et al. 2010; Pleasance et al. 2010b). Clearly, the differences between the mutational rates and spectra of the COLO829 cells and the samples under study may stem from the fact that the former is a cell line and the latter comprises a primary tumor and its metastatic deposit. However, taken at face value, our data indicate that the mutational burden in our acral melanoma samples is considerably lower than that seen in a tumor from a sun-exposed site but that their mutational signatures appear to be similar.
Furthermore, our data suggest that the nail matrix does not provide complete UV protection as previously suggested (Parker and Diffey 1983) and that UVB light can penetrate the human nail. Nevertheless, there appear to be quantitative and/or qualitative differences between the UV exposure experienced by this acral case and the melanoma tumors in previous studies (Greenman et al. 2007; Pleasance et al. 2010a). It is also possible that the UV mutation signature, in part, occurred due to inefficient cyclobutane pyrimidine dimers repair by NER (Gaddameedhi et al. 2010) due to the presence of the ERCC5 mutation. Although somatic mutations have not previously been reported in NER genes in melanoma (www.sanger.ac.uk/genetics/CGP/cosmic/), and some nonacral melanoma cell lines retain NER activity (Bowden et al. 2010; Gaddameedhi et al. 2010), ERCC5 mutations are associated with microsatellite instability in colorectal and gastric cancers (Park et al. 2002).
Comparison of primary and metastasis
Although fewer SNVs were called in the metastasis than in the primary, this is likely a consequence of lower coverage and yield in the metastasis rather than a difference in tumor cell content. Comparison of the SNP array data suggests that the primary and metastasis have comparable levels of stromal contamination (Supplemental Fig. S11A,B) which excludes a difference in tumor cellularity as a likely confounder in our analyses.
Notably, we observed a wide spectrum of mutations ranging from nucleotide-level to copy number alterations that were present in both the primary tumor and in its metastatic deposit. These observations could be consistent with different models of metastatic dissemination. First, a metastatic subclone emerging within a primary tumor became the fittest and dominated the primary tumor bulk (“clonal dominance”) (Kerbel et al. 1988; Talmadge 2007). Second, the metastatic clone could have stemmed from a nonmodal subclone of the primary tumor that emerged at a late stage of development of the primary lesion and comprised a small percentage of the tumor bulk. In this case, the metastatic clone would harbor specific mutations in addition to the mutations found in the modal population of the primary lesion. Third, it is possible that both the primary tumor and the lymph node metastasis are composed of mosaics of nonmodal populations and that an enrichment for a minor subclone from the primary tumor happened in the metastatic deposit. Fourth, it is possible that the subset of the mutant alleles we observe in both primary and metastasis was acquired by incipient tumor cells early in tumorigenesis (Bernards and Weinberg 2002) and that the genetic changes involved exclusively in metastasis do not exist. In this case, the role, if any, of the additional genetic lesions we detected in the metastasis is ambiguous. It is clear, however, that to truly interrogate the differences between the primary and metastasis at the single nucleotide level and investigate the “metastasis genes,” if they, indeed, exist, a substantial number of matched primary-metastasis pairs will need to be sequenced.
Conclusion
The systematic comparison of the acral melanoma samples described here provides a view of mutational processes, DNA repair, and selection during this particular tumor's evolution. It also provides evidence of UV exposure in an acral melanoma. Our findings offer further support to the hypothesis that acral melanoma is a genetically distinct melanoma subtype. The rate of SNVs is more comparable to that detected in solid malignancies not associated with a particular carcinogen and is substantially lower relative to COLO829 cells (Pleasance et al. 2010a). The presence of structural variants suggests that only whole genome sequencing, or RNA sequencing coupled with exome sequencing, will provide a comprehensive compendium of the genetic changes that occur in this melanoma subtype. We note also that the heterogeneity inherent in these tumors could present a challenge in the era of targeted therapy.
Methods
Tumor tissue
Following patient consent, tumor tissue was obtained at surgery and snap-frozen in liquid nitrogen. Samples were anonymized and access to tissue and clinical data was in accordance with the Human Tissue Act and MREC Guidelines. All the studies presented here were conducted in accordance with the study protocol CCR3097, approved by the Royal Marsden Hospital Research Ethics Committee on Oct. 21, 2008.
Extraction and quality assessment of nucleic acids
Both tumors were microdissected to ensure >90% purity of neoplastic cells. Microdissection was performed with a sterile needle under a stereomicroscope (Olympus SZ61) from 50 consecutive 8-μm-thick representative sections of the tumors, stained with nuclear fast red as previously described (Geyer et al. 2010). Tumor DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen Ltd.) according to the manufacturer's recommendations. RNA was extracted using TRIzol (Invitrogen) and eluted in RNAse-free water. Germline DNA was extracted from peripheral blood mononuclear cells using the DNeasy Blood and Tissue Kit (Qiagen). To ensure that the tumor and germline DNA were derived from the same patient, STR analysis using PowerPlex kit (Promega) was carried out on all three samples and confirmed that all were derived from the same individual. All DNA samples were quantified using PicoGreen dsDNA Quantification Reagent according to manufacturer's recommendations (Invitrogen). Structural integrity of DNA was checked by gel electropheresis. RNA quantity and quality were assessed using Agilent's 2100 Bioanalyzer.
Whole exome capture
Exome capture was performed according to the Agilent SureSelect target enrichment sample preparation protocol using 5 μg of nonamplified genomic DNA (v.2.1 May 2010).
Sanger sequencing
A total of 100 regions were amplified by PCR to assess the mutational status of randomly chosen SNVs in normal primary and metastasis DNA. The products were directly sequenced using dye-terminator chemistry. A further 187 regions were amplified by PCR to assess the putative coding variants detected by whole genome or exome sequencing in the normal primary and metastasis DNA. Sequences were visualized using Sequencher software. Details of the PCR and sequencing primers are given in the Supplemental Material.
Validation of gene fusions by RT-PCR
Fusion candidates were validated by reverse-transcriptase PCR. RNA was reverse-transcribed to generate cDNA using M-MLV reverse Transcriptase (Sigma). PCR primers were designed to flank the fusion breakpoints, and following PCR, the products were detected by gel electropheresis. Details of PCR primers are given in the Supplemental Material.
Whole genome sequencing and somatic variant detection
Sequencing of the matched normal and tumor samples was carried out using unchained combinatorial probe anchor ligation chemistry on arrays of self-assembling DNA nanoballs (Drmanac et al. 2010). The reads were aligned to the NCBI build 37 reference genome, and the percentages of fully called bases for the normal, primary and metastasis samples were 94%, 92.5%, and 91.6%. The gross mapping yields were 169.28, 156.96, and 150.20 Gb for the normal, primary tumor, and metastatic deposit samples, resulting in an average coverage of ∼59×, 55×, and 53× haploids, respectively. The percentages of the genomes covered at 10× were 96.5%, 95.2%, and 94.8% for the normal, primary tumor, and metastatic deposit samples (Supplemental Table S1A). The intersection of all three genomes with coverage of 10× was 92.2%. Variations between the reference genome (NCBI build 37) and each of the samples were called and scored using a local de novo assembly algorithm (Drmanac et al. 2010). Typically, at least seven high-quality, well-mapped reads are required to make a homozygous SNP call, and at least three reads from each allele are required to call a heterozygous SNP.
In addition, a reference score is calculated for each called base in the genomes (Drmanac et al. 2010). Allele calls for each sample were compared to those determined by Affymetrix Genome-wide SNP6 genotyping arrays (Supplemental Table S1B). Somatic single nucleotide variants and indels between (1) the normal and primary tumor samples, and (2) the normal and metastasized samples were uncovered using the calldiff function of cgatools (http://cgatools.sourceforge.net/), which assigns a somatic score to each SNV and, additionally, a somatic score to short insertions and deletions. The somatic score is a measure of the confidence that each SNV/indel is a true somatic variant and is calculated using the reference scores (described above) of the base in the normal and tumor sample (Drmanac et al. 2010).
To assess the accuracy of the somatic SNVs, we selected 50 SNVs at random from each of the primary and metastasis somatic candidates. The genotypes of these SNVs were assayed using conventional Sanger sequencing so as to determine a somatic score threshold. ROC curves were constructed using the somatic scores of the SNVs genotyped by Sanger sequencing (Supplemental Figs. S1, S2). A somatic score threshold of −10 was selected, equating to a true positive rate of 0.93 and a false positive rate of 0.10 in the primary-normal comparison, and a TPR of 0.93 and FPR of 0.13 in the metastasis-normal comparison. In addition, we constructed ROC curves for whole genome exonic SNVs using exome sequencing data as the validation data (Supplemental Figs. S1, S2). Selecting the same somatic score threshold of −10, we obtained a TPR of 0.85 and an FPR of 0.16 for the primary, and 0.85 and 0.13 for the metastasis comparison.
Somatic structural variations in the primary and metastatic tumors were called using the junctiondiff function of cgatools. High-confidence SV junctions were those that had at least 10 mate-pairs in a cluster, in which de novo assembly of the junction was successful, had a high mapping diversity, and for which there was an absence of specific repeat sequences on the left and right side of the junction.
Exome sequencing
Exonic sequences were targeted using the Agilent SureSelect 37-Mb exome capture kit and sequenced using the Illumina GAIIx. Seventy-six bp paired-end reads were aligned to the NCBI build 37 reference genome using BWA (Li and Durbin 2009). Base quality score recalibration and local realignment around indels were performed using GATK (McKenna et al. 2010). Duplicate removal was conducted using samtools (Li et al. 2009). Somatic SNVs and indels were determined using a combination of Varscan (Koboldt et al. 2009) and SomaticSniper (Ding et al. 2010) and annotated using the Ensembl Variant effect predictor (McLaren et al. 2010).
Gene expression arrays
RNA from the primary acral melanoma and its metastasis was extracted and assayed on Ilumina Human WG6v3 beadchips. Transcript expression was ascertained using the BioConductor package lumi (Du et al. 2008). Data were read into an expression set object using the lumiR function, and probes detected at a detection level threshold of 0.05 using the detectionCall function were considered as expressed. Probes were mapped to HGNC genes using the BioConductor (http://www.bioconductor.org/) packages illuminaHumanv3.db and annotated. Genes were designated as expressed if all probes mapping to the gene passed the detection level threshold and as nonexpressed if all probes mapping to the gene failed the threshold. Data were deposited in the Gene Expression Omnibus (accession number GSE28909).
Genome-wide genotyping
Genome-wide genotyping was conducted on the Affymetrix Genome-Wide Human SNP Array 6.0. Genotypes were called using the apt-probeset-genotype algorithm (birdseed-v2 method) of Affymetrix Power Tools. The cgatools function snpdiff (http://cgatools.sourceforge.net/) was used to compare the SNP array genotyping calls with confidence scores lower than 0.02 to the whole genome sequencing calls. Data and CEL files were deposited in the Gene Expression Omnibus (accession number GSE28910).
Estimation of tumor purity
We conducted an analysis of the purity of the tumors using the SNP array data with the ASCAT algorithm (Van Loo et al. 2010). This algorithm reports tumor cell fractions of 54% and 56% for the primary tumor and metastasis respectively.
Copy number alterations
SNP array
Regions of copy number alteration were determined from the Affymetrix SNP 6.0 array using the aroma.affymetrix and aroma.cn R packages (Bengtsson et al. 2009). The CRMA v2 method was implemented, which is a preprocessing and probe summarization method that provides full-resolution raw total copy-number estimates. The CbsModel method was used to identify somatic copy number alterations. Data and CEL files were deposited in the Gene Expression Omnibus (accession number GSE28910).
Whole genome sequencing
Read depth was corrected for GC content and normalized for average haploid genome coverage. Log2 ratios of the tumor to normal average read depths for windows of 10 kb were calculated. These values were smoothed and segmented as implemented in the BioConductor package DNAcopy using default values (Venkatraman and Olshen 2007).
Microarray comparative genomic hybridization
The microarray comparative genomic hybridization platform used for this study was constructed at the Breakthrough Breast Cancer Research Center and comprises ∼32,000 BAC clones tiled across the genome. DNA labeling, array hybridizations, and image acquisition were performed as previously described (Natrajan et al. 2010). No whole-genome DNA amplification was performed. aCGH data were preprocessed and analyzed using an in-house R script (BACE.R) in R version 2.8.0, as previously described (Natrajan et al. 2010). After filtering polymorphic BACs, a final data set of clones with unambiguous mapping information according to the NCBI 37 (hg19) of the human genome (http://www.ensembl.org) was smoothed using the circular binary segmentation (cbs) algorithm using default parameters. The cut-offs for gains, losses, and amplifications were defined as described previously (Natrajan et al. 2010).
Fluorescence in situ hybridization (FISH)
FISH was carried out for validation of the KIT amplicon detected by sequencing depth analysis and aCGH. An in-house biotin-labeled probe was used as previously described (Gomes et al. 2007). Tissue pretreatment, hybridizations, and washes were performed as previously described (Lambros et al. 2006). Signals were counted in nonoverlapping nuclei of morphologically unequivocal neoplastic cells. A given sample was considered to harbor KIT amplification if >50% of the neoplastic cells harbored (1) more than five signals per nuclei, or (2) large gene copy clusters (Gomes et al. 2007).
Loss of heterozygosity (LOH)
Regions of potential LOH were plotted from the Affymetrix SNP 6.0 array B allele frequency in the matched normal-normal samples using the TumorBoostNormalization function of the aroma.cn R package for normalizing allelic ratios from genotyping microarrays (Bengtsson et al. 2010), which is described in detail at http://aroma-project.org/vignettes/tumorboost-highlevel.
Regions of potential LOH from whole genome sequencing data were plotted along each chromosome by calculating the frequency of LOH sites (homozygous in the tumor and heterozygous in the normal sequence) in 100-kb windows. LOH rates were smoothed by the BioConductor package DNAcopy (Venkatraman and Olshen 2007).
Data access
SNP and mRNA arrays have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/), under Super Series accession number GSE28913. Sequencing data have been deposited at the European Genome-phenome Archive (EGA) under accession number EGA00001030866.
Acknowledgments
This work was supported by a Career Development Award from The Harry J. Lloyd Charitable Trust (S.T.), Cancer Research UK (ref: C107/A10433), and The Institute of Cancer Research. J.S.R.-F. is in part funded by Breakthrough Breast Cancer and is a recipient of the 2010 CR-UK Future Leaders Prize. We acknowledge NHS funding to the NIHR Biomedical Research Centre and thank Mr. Eric Ward, Dr. Kay Savage, and Ms. Annette Lane (ICR) for technical assistance with histological preparations, and Ms. Alice Smith for technical assistance with Sanger sequencing. We thank Jeff Almeida-King at the EGA/EBI for his help with the submission of sequencing data.
Footnotes
-
↵9 Corresponding author.
E-mail Richard.Marais{at}icr.ac.uk.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.125591.111.
- Received May 1, 2011.
- Accepted November 29, 2011.
- Copyright © 2012 by Cold Spring Harbor Laboratory Press
Freely available online through the Genome Research Open Access option.








