
Sex-biased genetic effects on gene regulation in humans
- Antigone S. Dimas1,2,3,
- Alexandra C. Nica1,
- Stephen B. Montgomery1,
- Barbara E. Stranger4,5,6,
- Towfique Raj4,5,6,
- Alfonso Buil1,
- Thomas Giger1,
- Tuuli Lappalainen1,
- Maria Gutierrez-Arcelus1,
- MuTHER Consortium,
- Mark I. McCarthy2,7,8 and
- Emmanouil T. Dermitzakis1,9
- 1Department of Genetic Medicine and Development, University of Geneva Medical School, Geneva 1211, Switzerland;
- 2Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford OX3 7BN, United Kingdom;
- 3Biomedical Sciences Research Center Al. Fleming, 16672 Vari, Greece;
- 4Department of Medicine, Division of Genetics, Brigham and Women's Hospital, Boston, Massachusetts 02115, USA;
- 5Harvard Medical School, Boston, Massachusetts 02115, USA;
- 6Program in Medical and Population Genetics, Broad Institute of Harvard and Massachusetts Institute of Technology, Cambridge, Massachusetts 02142, USA;
- 7Oxford Centre for Diabetes, Endocrinology and Metabolism, University of Oxford, Churchill Hospital, Headington, Oxford OX3 7LJ, United Kingdom;
- 8Oxford NIHR Biomedical Research Centre, Churchill Hospital, Headington, Oxford OX3 7LJ, United Kingdom
Abstract
Human regulatory variation, reported as expression quantitative trait loci (eQTLs), contributes to differences between populations and tissues. The contribution of eQTLs to differences between sexes, however, has not been investigated to date. Here we explore regulatory variation in females and males and demonstrate that 12%–15% of autosomal eQTLs function in a sex-biased manner. We show that genes possessing sex-biased eQTLs are expressed at similar levels across the sexes and highlight cases of genes controlling sexually dimorphic and shared traits that are under the control of distinct regulatory elements in females and males. This study illustrates that sex provides important context that can modify the effects of functional genetic variants.
The majority of traits that distinguish the two sexes develop secondarily to the development of the ovaries and testes (Williams and Carroll 2009). Most studies of sexual dimorphism have focused on the impact of hormones or on the genetic contribution of sex chromosomes. However, there is growing evidence that genetic variation on the autosomes contributes to sexual dimorphism (Ober et al. 2008; Heid et al. 2010). Sex-specific QTLs for sexually dimorphic traits such as life span and HDL-cholesterol have been detected, respectively, in Drosophila (Nuzhdin et al. 1997) and mouse (Korstanje et al. 2004). Sex-specific eQTLs have also been detected in mice (Yang et al. 2006), but whether such effects on expression regulation are also seen in humans has not been explored to date.
To address the above question, we used gene expression levels quantified in EBV-transformed B cells (lymphoblastoid cell lines or LCLs) from four HapMap populations (CEU: 54 females, F; 55 males, M; CHB: 42 F, 38 M; JPT: 40 F, 42 M; YRI: 53 F, 55 M) (Stranger et al. 2012). A recent study examining the impact of EBV transformation on LCL expression and methylation profiles has shown that LCLs recapitulate naturally occurring expression variation in primary B cells (Caliskan et al. 2011). A large fraction of sex-specific epigenetic and gene expression effects are therefore likely to be maintained in LCLs. Sex-specific effects due to hormones, however, cannot be captured by this study since LCLs are grown in the absence of hormones. We stratified each population sample by sex and performed association of SNP genotype with mRNA levels using Spearman rank correlation (SRC) in each sex separately (sex-stratified analysis). Differences between LCLs derived from females and males are expected because: (1) LCLs retain an important fraction of their heritable epigenetic and methylation profiles (McDaniell et al. 2010), and (2) cell lines derived from females and males differ genotypically due to the presence of sex chromosomes (XX vs. XY).
Results and Discussion
We tested all SNPs mapping in a 2-Mb window centered on the transcription start site (TSS) of genes, defined cis eQTLs as SNPs with SRC p < 10−5, and eQTL-genes as genes with at least one cis eQTL. We subsequently increased our stringency by ensuring equal levels of estimated false discovery rate (FDR) for eQTLs that were detected in both sexes (shared) and eQTLs that were detected in one sex only (threshold-based sex discordant or TBSD). We found that at an estimated FDR of 13%–17%, approximately one-third of eQTL-genes were TBSD (Table 1; Supplemental Fig. S1). In CEU, for example, from a total of 178 and 151 eQTL-genes detected in females and males, respectively, 68 and 41 were TBSD. The remaining two-thirds of eQTL-genes were shared across sexes for each population.
We also carried out expression association testing for the whole population sample (whole sample study), not stratifying by sex, to enable comparison of findings. In this analysis, we detected almost all shared, and 60%–78% of TBSD eQTL-genes identified in the sex-stratified study (Table 1). In CEU, for example, we detected 109 of the 110 shared eQTL-genes, as well as 41 out of 68, and 26 out of 41 TBSD eQTL-genes that were identified in females and males, respectively. Notably, 22%–40% of TBSD eQTL-genes (corresponding to 10%–15% of the sex-stratified study total discoveries) were not discovered in the whole sample analysis. This fraction of eQTLs is likely to harbor the true variants that exert effects in a sex-dependent manner, and pooling of both sexes achieves a greater sample size but dilutes rather than strengthens the statistical signal (Supplemental Fig. S2).
Autosomal eQTL-genes detected in the whole population sample study (A) and in the sex-stratified study (B)
To evaluate the strength of TBSD signals, we applied a continuous (vs. threshold-based) measure of significance. We considered eQTLs detected in one sex only (discovery sex) and explored their SRC nominal P-values in the other sex (non-discovery sex). Applying the methodology (Q-values) described in Storey and Tibshirani (2003), we estimated the fraction of the distribution that is enriched for statistically significant effects (π1) and found that this corresponds, on average, to 72%. Therefore, we estimate that 28% (standard deviation 1%) of TBSD associations had negligible effects in the non-discovery sex, and we define this subset of eQTLs as sex-biased. In CEU, for example, 28% of the 109 TBSD discoveries corresponds to 30.52 eQTL-genes. At an estimated FDR of 17%, we expect 25.33 true discoveries. This brings the fraction of truly sex-biased discoveries to ∼12% of the total (219). Similarly, truly sex-biased eQTL-genes range from 12% to 14% in CHB, JPT, and YRI.
As an independent means to evaluate sex-biased eQTLs, we tested TBSD SNP-expression probes using ANOVA with a SNP × sex interaction term to capture sex-biased effect size patterns. We observed enrichment of low P-values (≤0.05) for the interaction term in overwhelming excess compared with what is expected by chance (Supplemental Table S1) and estimate that 44%–50% of TBSD associations are sex biased. Taken together with estimated FDR, ANOVA results put the fraction of truly sex-biased eQTLs at ∼15%. This is consistent with our previous estimate of 12%–14%. Furthermore, we highlight that the variance in gene expression explained by the SNP × sex interaction term is significantly higher for sex-biased genes (Supplemental Fig. S3). It is worth noting that ANOVA highlights both differences in statistical significance and effect size (fold change or slope). This fold change difference constitutes an additional measure of sex bias (Nica et al. 2011).
Sampling effects arising from small sample size and population specificity of eQTLs (Stranger et al. 2012) hamper cross-population replication. Indeed, we observed low levels of replication for both shared and TBSD eQTLs. To overcome this limitation, we endeavored to replicate CEU eQTLs in a population-based cohort (MuTHER) of female twins from the United Kingdom (Nica et al. 2011). Our expectation was that CEU female-TBSD eQTLs would replicate at higher levels than CEU male-TBSD eQTLs in MuTHER twins. Indeed, we observed higher levels of replication of female-TBSD eQTLs with low P-value enrichment (Storey and Tibshirani 2003), π1 = 0.81, than male-TBSD eQTLs, where π1 = 0.53.
To address whether TBSD and (ANOVA-defined) sex-biased eQTL discovery is driven by differences in gene expression levels between females and males, we compared log2 expression medians across the sexes for (1) all autosomal genes tested (Fig. 1A), (2) eQTL-genes (Fig. 1B), (3) TBSD eQTL-genes (Fig. 1C), and (4) sex-biased eQTL-genes (Fig. 1D). In all cases, we detected very high correlation between female and male expression medians (r2 = 0.98–0.99, p < 10−4), and in line with other studies (Zhang et al. 2007, 2009; Idaghdour et al. 2008), we did not detect significant differences between females and males. This demonstrates that TBSD and sex-biased eQTLs do not arise as a consequence of expression level differences between the sexes. Therefore, sex bias in genetic effects of gene regulation is not of the same nature as differential expression of genes between sexes.
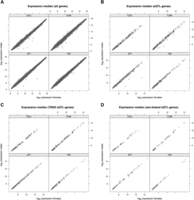
Comparison of female (x-axis) versus male (y-axis) expression medians. Log2 expression medians were compared across the sexes for (A) all genes, (B) eQTL-genes, (C) TBSD eQTL-genes, and (D) sex-biased eQTL-genes. In all cases, the correlation between log2 expression levels in females and males was found to be very high (r2 = 0.99 and p < 10−4), suggesting that expression level differences between sexes are not the primary driver of eQTL discovery.
Although almost all shared eQTL-genes detected in the sex-stratified study were also detected in the whole sample analysis, we identified five cases of female–male shared eQTL-genes that were not discovered when pooling the two sexes into a single analysis (Fig. 2A–E; Supplemental Table S2). In these cases, although the eQTL-gene is shared, there are independent regulatory elements in each sex. These eQTL-SNPs have negligible significance in the non-discovery sex (Supplemental Figs. S4–S8; Supplemental Table S2), explaining why such signals are likely to be diluted when both sexes are analyzed simultaneously. These cases include genes (see below, Fig. 2A–E, created using the UCSC Genome Browser; Kent et al. 2002; http://genome.ucsc.edu) with a role in gamete formation, fertility, and sexual dimorphism, but also genes involved in processes that are not linked to perceived sex-related traits. This suggests that there may be a sex-biased dimension for traits that, to date, are considered to have similar biology across sexes. SPO11 (CEU) (Fig. 2A; Supplemental Fig. S4) is involved in meiotic recombination (Bellani et al. 2010) and spermatocyte formation, it is expressed in oocytes, and both female and male knockout mice are infertile (Bellani et al. 2010). CKLF (JPT) (Fig. 2B; Supplemental Fig. S5) encodes a chemokine with a role in muscle development and neuronal migration (Wang et al. 2010). Its expression is increased in systemic lupus erythematosus (SLE) and in rheumatoid arthritis (RA), diseases that are nine and three times more common in women, respectively. MRFAP1L1 (JPT) (Fig. 2C; Supplemental Fig. S6) is thought to have a role in spermatogenesis through its interaction with TSNAX (Rual et al. 2005), a gene involved in spermatogenesis, neuronal regulation, and genome stability (Jaendling and McFarlane 2010). ODF2L (YRI) (Fig. 2D; Supplemental Fig. S7) interacts with PRSS23 (Stelzl et al. 2005), a serine protease involved in proteolytic degradation of extracellular matrix components, an essential process for ovulation (Wahlberg et al. 2008). Finally, PSAP (YRI) (Fig. 2E; Supplemental Fig. S8) codes for a conserved glycoprotein involved in the development of the reproductive and nervous systems (Hu et al. 2010). It has a developmental role in prostate cancer, its inactivation in mice leads to atrophy of the male reproductive system, and its down-regulation decreases metastatic prostate cancer cell adhesion, migration, and invasion (Hu et al. 2010).

Shared eQTL-genes regulated by independent regulatory variants in females (F) and males (M). (A) In CEU, rs6025625 (CEU F) and rs3787152 (CEU M), mapping within 760 kb of each other, are associated with SPO11 expression levels. SPO11 is essential for meiotic recombination, and both F and M knockout mice are infertile. (B) In JPT, rs2271025 (JPT F) and rs3826161 (JPT M) (r2 = 0.26, D′ = 0.64) are eQTLs for CKLF, a gene encoding a chemokine, with a role in muscle development and neuronal migration. (C) In JPT, rs11734984 (JPT F) and rs10020189 (JPT M), mapping within 580 kb of each other, are eQTLs for MRFAP1L1, a gene that is likely to have a role in spermatogenesis through its interaction with TSNAX. (D) In YRI, rs506733 (YRI F) and rs12097932 (YRI M) (r2 = 0.00, D′ = 0.09) are eQTLs for ODF2L, a gene thought to have a role in ovulation through its interaction with PRSS23. (E) In YRI, rs877663 (YRI F) and rs730722 (YRI M) (r2 = 0.04, D′ = 0.43) are eQTLs for PSAP, a gene encoding a conserved glycoprotein involved in the development of the reproductive and nervous systems that has been linked to prostate cancer. Boxes indicate eQTL-genes. r2 and D′ calculated for SNPs within 500 kb of each other. Figures were made using the UCSC Genome Browser (http://genome.ucsc.edu) (Kent et al. 2002).
To gain a better understanding of the biology behind eQTLs in females and males, we explored the properties of TBSD and sex-biased eQTLs. We found that the direction of allelic effects was consistent across the sexes (Supplemental Fig. S9), suggesting that if an eQTL allele increases/decreases expression in one sex, it will have the same direction of effect in the other sex. Similarly to cell-type-specific (Dimas et al. 2009) and population-specific (Stranger et al. 2012) eQTLs, we found that TBSD and sex-biased eQTLs (at similar levels of FDR to shared eQTLs) have lower effects and span a range of distances from the TSS, whereas shared eQTLs have higher effects and cluster around the TSS (Supplemental Fig. S10). This trend is also revealed when plotting the significance differential across the sexes for eQTLs that are sex biased and those with ANOVA interaction p > 0.05 versus distance to TSS (Supplemental Fig. S11).
To quantify the impact of regulatory variants on gene expression, we measured the expression fold change between the two homozygote classes (Fig. 3; Supplemental Fig. S12). As expected, we found that sex-biased eQTLs exert a higher fold change in expression in the discovery sex, whereas shared eQTLs tend to result in similar fold changes in females and males. Notably, however, there are cases of shared eQTLs in which large differences in fold change across the sexes are observed. This implies that distinct regulatory effects are exerted in each sex, even in cases in which the eQTL has been designated as shared using threshold-based significance criteria. In CEU, for example, a shared eQTL for PNMAL1 (Fig. 3A) shows a higher fold change in females compared with males (2.7 vs. 1.3, respectively). Similarly, WBSCR27 (Fig. 3B) shows a higher fold change in males compared with females (1.1 vs. 2.9, respectively). Currently, very little is known about these genes, but the above observation may help elucidate their role. Furthermore, this observation highlights the value of considering parameters of sex bias beyond statistical significance (e.g., effect size).
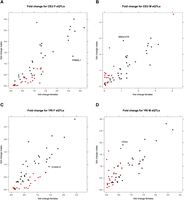
Expression fold change between major and minor allele homozygotes for shared and sex-biased eQTLs. Sex-biased eQTLs exert greater expression fold change in the discovery sex. Expression fold change in females (x-axis) and males (y-axis) for 65 SNP-expression probes in CEU females (A) and for 59 SNP-expression probes in CEU males (B). Although sex-biased eQTLs exert greater fold change differences in the discovery sex, there are cases of shared eQTLs that also display prominent fold change differences between females and males, e.g., (A) PNMAL1 (fold change: CEU F 2.7; CEU M 1.3), (B) WBSCR27 (fold change: CEU F 1.1; CEU M 2.9). Expression fold change in females and males for 62 SNP-expression probes in YRI females (C) and for 62 SNP-expression probes in YRI males (D). Cases of shared eQTLs that display prominent fold change differences between females and males include: (C) S100A13 (fold change: YRI F 1.4; YRI M 0.9), (D) CPA4 (fold change: YRI F 0.7; YRI M 1.9). (Black) Shared eQTLs; (red) sex-biased eQTLs.
To obtain an overview of eQTL-gene biological functions, we used the DAVID Functional Annotation tool (Huang da et al. 2009) and interrogated Gene Ontology biological processes for TBSD and sex-biased versus shared genes. We observed only weak enrichment for GO terms linked to reproduction and meiosis possibly because we are underpowered for this analysis. A more interesting hypothesis, however, is that sex-biased gene regulation affects perceived sexually dimorphic and other traits in a similar way. Given the role of regulatory variation in shaping complex traits and determining disease risk (Nica et al. 2010), we also interrogated the overlap between sex-biased eQTLs and GWAS SNPs (Hindorff et al. 2009) and found two cases of overlap, both for diseases with a well-established sex imbalance (Supplemental Fig. S13; Supplemental Table S3). In both cases, regulatory trait concordance (RTC) scores (Nica et al. 2010), an index integrating eQTL and GWAS data to detect disease-causing regulatory effects, were equal to one (RTC = 1), suggesting that the disease and regulatory signal are overlapping. rs167769 is associated with eosinophilic esophagitis (Rothenberg et al. 2010), a disease four times more common in young males. The eQTL-gene for this SNP is STAT6, a component of the JAK–STAT signaling cascade, which is implicated in other inflammatory diseases (Barrett et al. 2008). rs2872507 is associated with RA (Stahl et al. 2010) and Crohn's disease (Barrett et al. 2008). The GWAS-reported locus on 17q12-q21 is a region under complex gene regulation and harbors multiple disease signals (Verlaan et al. 2009). Most studies have focused on the potential role of IKZF3 and ORMDL3 in disease, but we report the eQTL-gene ZPBP2 and bring this gene to the fore as a potential mediator of the disease association.
In this study, we searched for eQTLs separately in females and males of four HapMap populations and found that ∼12%–15% of detected eQTLs, and their underlying regulatory variants, are sex biased. This bias extends to a subset of genes that possess the same eQTL in both sexes but that exerts a very distinct regulatory effect, measured as the resulting fold change in gene expression, in each sex. We highlight that a fraction of eQTLs that have been detected to date in eQTL studies where no distinction by sex is made, is driven by strong effects in one sex. Furthermore, we report that sex-related effects can affect traits where no sexual dimorphism has been observed to date. Given the prominence of sex-biased effects, this study emphasizes the importance of considering each sex separately in genomic studies to uncover new disease and trait variants.
Methods
RNA preparation
Total RNA was extracted from lymphoblastoid cell lines (LCLs) grown in a hormone-free environment, in the presence of phenol red•Na (0.0053 g/L; Sigma-Aldrich). LCLs were from 379 individuals of four HapMap populations. The numbers of individuals of each population include (CEU) 109 Caucasians living in Utah USA, of northern and western European ancestry; (CHB) 80 Han Chinese from Beijing, China; (JPT) 82 Japanese in Tokyo, Japan; and (YRI) 108 Yoruba in Ibadan, Nigeria (The International HapMap Consortium 2005) (Coriell). Two in vitro transcription (IVT) reactions were performed as one-quarter-scale Message Amp II reactions (Ambion) for each RNA extraction using 200 ng of total RNA as previously described (Stranger et al. 2005). One and one-half micrograms of the cRNA was hybridized to an array (Stranger et al. 2007b, 2012).
Gene expression quantification
To assay transcript levels in LCLs, we used Illumina's commercial whole-genome expression array, Sentrix Human-6 Expression BeadChip version 2 (Illumina) (Kuhn et al. 2004). These arrays use a bead pool with about 48,000 unique bead types (one for each of 47,294 transcripts, plus controls), each with several hundred thousand gene-specific 50-mer probes attached. On a single BeadChip, six arrays were run in parallel (Stranger et al. 2007b). Each of the two IVT reactions from the 379 samples was hybridized to one array each, so that each cell line had two replicate hybridizations. cRNA was hybridized to arrays and subsequently labeled with Cy3-streptavidin (Amersham Biosciences) and scanned with a Bead Station (Illumina) as previously described (Stranger et al. 2005). Samples were processed in an order randomized with respect to population of origin and IVT batch.
Raw expression data normalization
With the Illumina bead technology, a single hybridization of RNA from one cell line to an array produces on average ∼30 intensity values for each of 47,294 bead types. These background-corrected values for a single bead type are subsequently summarized by Illumina software and output to the user as a set of 47,294 intensity values for each individual hybridization. In our experiment, each cell line was hybridized to two arrays, thus resulting in two reported intensity values (as averages of the values from the 30 beads per probe) for each of the 47,294 bead types. Hybridization intensity values were normalized on a log2 scale quantile normalization method (Bolstad et al. 2003) across replicates of a single individual followed by a median normalization method across all individuals of the four populations. These normalized expression data were used as input for the expression analysis.
Selection of probes to analyze
Of the 47,294 probes for which we collected expression data, we selected a set of 22,744 probes to analyze (21,800 autosomal and 955 on chromosome X). We included in our analyses each probe that mapped to an Ensembl gene, but not to more than one Ensembl gene (Ensembl 49 NCBI Build 36), and we excluded probes mapping to the Y chromosome. The resulting sets of 21,800 autosomal and 955 chromosome-X probes were analyzed in the association analyses and correspond to 17,673 unique autosomal and 741 unique chromosome-X Ensembl genes, respectively (total 18,414 genes).
Genetic variation
Single nucleotide polymorphisms (SNPs) for the same 379 HapMap individuals of CEU, CHB, JPT, and YRI, were selected (Release version 2) for use in the association analyses. SNPs that were included in association analyses fulfilled three criteria: (a) were present in both females and males in each population, (b) had a MAF ≥5% in each population, and (c) had <20% missing data. This corresponds to between 1.1 million and 1.3 million SNPs per population.
Association testing
We performed association of SNP genotype with probe expression levels using Spearman rank correlation (SRC) as previously described (Stranger et al. 2007a,b; Dimas et al. 2009; Montgomery et al. 2010). All SNPs mapping in a 2-Mb window centered on genes' TSS were tested. We carried out two rounds of association testing. In the first round, we stratified each of the four population samples by sex (CEU: 54 F, 55 M; CHB: 42 F, 38 M; JPT: 40 F, 42 M; YRI: 53 F, 55 M) and for each population tested individuals of each sex separately (sex-stratified study). Expression association was carried out for autosomal genes. In the second round of association testing, we tested all individuals of the population sample irrespective of sex in the same association test (whole sample study). In both the sex stratified study and the whole sample study, we applied an empirical threshold of SRC P-value <10−5 to define eQTLs.
Permutations and FDR estimation
We performed 10,000 permutations of expression phenotypes relative to genotypes for females and males of the four populations (i.e., CEU_F, CEU_M, CHB_F, CHB_M, JPT_F, JPT_M, YRI_F, YRI_M). In each case, permutations were carried out for 2560 probes (2062 genes) chosen at random and resulted in a matrix of SRC permuted −log10 P-values of 2560 probes by 10,000 permutations. We calculated the average permuted −log10 P-value across probes and created a ranked distribution of permuted −log10 P-values that was subsequently used to estimate the false discovery rate (FDR) (Storey and Tibshirani 2003). This is sufficient since SRC permutation thresholds tend to be very tight around the mean so we did not require to perform permutations for all genes.
Determination of significant associations and designation of shared, threshold-based sex-discordant, and sex-biased eQTL-genes
We used an initial threshold-based significance filter to define cis eQTLs as those SNPs with a nominal SRC p < 10−5. eQTL-genes were defined as those genes with at least one cis eQTL, and for all analyses, we kept the most significant SNP association per gene. Empirically, we have found that most SNP-expression probe associations that pass the SRC p < 10−5 threshold correspond to a permutation threshold of 0.01–0.001 (Stranger et al. 2007b, 2012; Dimas et al. 2009). Applying this significance threshold to each population, we determined eQTL-genes that were (a) detected in both sexes (shared eQTL-genes) and (b) detected in one sex only. We subsequently filtered eQTL-genes detected in one sex only to FDR levels corresponding to shared eQTL-genes. These FDR-filtered eQTL-genes detected in one sex only comprise the threshold-based sex-discordant (TBSD) eQTL-genes detected in each population.
To evaluate the threshold-based sex results, we used a continuous measure of significance. For each population, we took all TBSD SNP-expression probes and extracted their SRC nominal P-values in the non-discovery sex. For these SNP-expression probes, we quantified enrichment for low P-values using q-value criteria (Storey and Tibshirani 2003).
We estimated the number of truly sex-biased eQTLs genes as follows: From the Q-value analysis, we expect 28% of discoveries to be true nulls. For CEU, 28% of the 109 TBSD eQTLs (68 in females + 41 in males) correspond to 30.52 genes. Given that the estimated FDR is at 17%, 17% of 30.52 equals 25.33 true discoveries. 25.33 true discoveries corresponds to ∼12% of the total number of 219 eQTL-genes (110 shared + 68 in females + 41 in males). Similarly, we estimated numbers of sex-biased eQTL-genes for CHB, JPT, and YRI.
As a further means of evaluating TBSD eQTLs, we carried out ANOVA with an interaction term for SNP × sex, which captures sex-biased patterns of associations. For each population, we took all TBSD SNP-expression probes and registered those with an ANOVA interaction term p ≤ 0.05. This subset of TBSD SNP-expression probes constitutes the sex-biased eQTL-genes used in further analyses.
To assess levels of replication of TBSD associations, we used the MuTHER data set (Nica et al. 2011). MuTHER is a population-based cohort of female twins from the United Kingdom with expression levels quantified in LCLs, fat, and skin. In this study, only unrelated individuals were considered at a time by separating twins from the same pair and by performing two independent eQTL analyses. We sought to replicate CEU TBSD associations in LCLs from each twin population by testing for low P-value enrichment in MuTHER of CEU female-TBSD and CEU male-TBSD SNP-expression probes using q-values. Reported π1 values are the average of the two twin populations.
Properties of eQTLs and eQTL-genes
For shared and sex-biased SNP-expression probes in each population_sex, we calculated the fold change in median −log2 expression values between major and minor allele homozygotes. To compare direction of the allelic effects for shared and TBSD eQTLs (and as a consequence of sex-biased eQTLs since these are a subset of TBSD eQTLs), we explored the direction of the Spearman correlation coefficient (rho) for the union of significant SNP-expression probes across the sexes, for each population. To obtain an overview of eQTL-gene biological functions, we used the DAVID Functional Annotation tool (Huang da et al. 2009) and interrogated GO Biological Processes (FAT) for shared and TBSD eQTL-genes versus all 18,414 genes tested.
Overlap of sex-biased eQTLs with GWAS disease/trait SNPs
The NHGRI GWAS catalog (Hindorff et al. 2009) (http://www.genome.gov/gwastudies accessed 23 November 2010) was queried for GWAS SNPs that overlapped with the eQTLs detected in this study.
Data access
The expression data reported in this paper have been deposited in the Array Express (http://www.ebi.ac.uk/arrayexpress/) database (Series Accession Number E-MTAB-264 and E-MTAB-198). Furthermore, all TBSD eQTL results are provided as Supplemental Material.
Acknowledgments
This study was supported by funds from the Louis-Jeantet Foundation, the Swiss National Foundation and “NCCR Frontiers in Genetics,” and the Wellcome Trust to E.T.D. Financial support was also provided by NIH (R01 MH090941) and the Wellcome Trust (MuTHER 081917/Z/07/Z) to E.T.D and M.I.M. A.S.D. was funded by the Wellcome Trust (083270/Z/07/Z), ENGAGE (HEALTH-F4-2007-201413), and Marie Curie FP7-PEOPLE-2010-IEF. We thank Vital-IT for the computational resources to perform the analysis and Anastasia Lafargue Komnou for her help in figure preparation.
Author contributions: The study was designed by A.S.D. and E.T.D. Analyses were carried out by A.S.D. Additional analyses were performed by A.C.N., S.B.M., B.E.S., A.B., T.R., T.G., T.L., and M.G.A. The manuscript was written by A.S.D. and E.T.D. with contributions from M.I.M. and from the other authors.
Footnotes
-
↵9 Corresponding author
E-mail emmanouil.dermitzakis{at}unige.ch
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.134981.111.
Freely available online through the Genome Research Open Access option.
- Received November 17, 2011.
- Accepted August 13, 2012.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








