
Temporal uncoupling of the DNA methylome and transcriptional repression during embryogenesis
- Ozren Bogdanović1,
- Steven W. Long2,
- Simon J. van Heeringen1,
- Arie B. Brinkman1,
- Jose Luis Gómez-Skarmeta3,
- Hendrik G. Stunnenberg1,
- Peter L. Jones2 and
- Gert Jan C. Veenstra1,4
- 1Department of Molecular Biology, Faculty of Science, Nijmegen Centre for Molecular Life Sciences, Radboud University Nijmegen, 6500 Nijmegen, The Netherlands;
- 2Department of Cell and Developmental Biology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, USA;
- 3Centro Andaluz de Biología del Desarrollo, Consejo Superior de Investigaciones Científicas and Universidad Pablo de Olavide, Carretera de Utrera Km1, 41013 Sevilla, Spain
Abstract
DNA methylation is a tightly regulated epigenetic mark associated with transcriptional repression. Next-generation sequencing of purified methylated DNA obtained from early Xenopus tropicalis embryos demonstrates that this genome is heavily methylated during blastula and gastrula stages. Although DNA methylation is largely absent from transcriptional start sites marked with histone H3 lysine 4 trimethylation (H3K4me3), we find both promoters and gene bodies of active genes robustly methylated. In contrast, DNA methylation is absent in large H3K27me3 domains, indicating that these two repression pathways have different roles. Comparison with chromatin state maps of human ES cells reveals strong conservation of epigenetic makeup and gene regulation between the two systems. Strikingly, genes that are highly expressed in pluripotent cells and in Xenopus embryos but not in differentiated cells exhibit relatively high DNA methylation. Therefore, we tested the repressive potential of DNA methylation using transient and transgenic approaches and show that methylated promoters are robustly transcribed in blastula- and gastrula-stage embryos, but not in oocytes or late embryos. These findings have implications for reprogramming and the epigenetic regulation of pluripotency and differentiation and suggest a relatively open, pliable chromatin state in early embryos followed by reestablished methylation-dependent transcriptional repression during organogenesis and differentiation.
During early embryonic development, both transcriptional repression and activation play an important role in maintaining correct gene expression patterns. Cytosine methylation within CpG dinucleotides is one of the predominant gene-silencing mechanisms in vertebrates, which have the majority of their genomic CpG sequences methylated (Hendrich and Tweedie 2003). The exceptions are CpG islands, GC-rich regions that often coincide with gene promoters and that are mostly methylation-free (Bird 1986). Apart from CpG methylation, a number of recent studies on mammalian ES cells have provided experimental evidence for the existence of non-CpG (mainly CpA) methylation (Ramsahoye et al. 2000; Lister et al. 2009; Laurent et al. 2010). DNA methylation has so far been described to play an important role in a variety of biological processes like X-chromosome inactivation, genomic imprinting, and silencing of intra-genomic sequences of parasitic origin (Bird 2002; Weber and Schubeler 2007). Developmental changes in DNA methylation have been comprehensively studied in different organisms (Bogdanović and Veenstra 2009). In contrast to the global wave of demethylation and subsequent gradual remethylation of the mouse zygote (Mayer et al. 2000; Oswald et al. 2000), the genome of the amphibian Xenopus laevis maintains high DNA methylation levels throughout early development (Veenstra and Wolffe 2001), although some changes in the DNA methylation content have been reported for a number of developmentally regulated promoters (Stancheva et al. 2002). Depletion of the maintenance DNA methyltransferase (DNMT1) in Xenopus results in an early apoptotic phenotype (Stancheva and Meehan 2000; Stancheva et al. 2001), and mouse knockouts for Dnmt1 and the de novo DNA methyltransferase (Dnmt3b) are embryonically lethal (Li et al. 1992; Okano et al. 1999). However, DNA methylation itself may not be essential for early embryogenesis because ablation of DNMT1 could be rescued by a catalytically inactive DNMT1 mutant (Dunican et al. 2008). DNA methylation in animals can repress transcription via methyl-CpG binding domain (MBD) proteins (Hendrich and Bird 1998), which recruit HDAC-containing complexes and promote formation of inactive chromatin on targeted loci (Jones et al. 1998; Nan et al. 1998; Wade et al. 1999; Zhang et al. 1999). MECP2, however, the Rett Syndrome MBD protein (Amir et al. 1999; Shahbazian and Zoghbi 2001), may also act as an activator of transcription (Chahrour et al. 2008). In addition to DNA methylation, modifications of N-terminal histone tails are known to influence chromatin structure by serving as docking stations for transcriptional modulators (Bannister et al. 2001; Lindroth et al. 2004; Wysocka et al. 2005; Vermeulen et al. 2007). The tri-methyl at histone H3 lysine 4 (H3K4me3) deposited by the Set1/Tritorax group proteins marks promoter regions of active genes (Santos-Rosa et al. 2002), whereas the tri-methyl H3K27 (H3K27me3) mark deposited and read by the Polycomb group proteins is known to be involved in developmentally regulated gene repression (Czermin et al. 2002; Muller et al. 2002). Even though DNA methylation and histone modifications influence gene regulation by different molecular entities, it is clear that they act in a highly orchestrated way. For example, methylation of H3K4 inhibits the binding of DNA methyltransferase DNMT3L, which is thought to associate with DNMT3A and DNMT3B to promote de novo DNA methylation (Ooi et al. 2007; Hu et al. 2009). In contrast, the Polycomb group protein EZH2 has been shown to interact with both DNMT1 and DNMT3 to deliver DNA methylation at the sites of its recruitment (Vire et al. 2006). Surprisingly, studies carried out in different models demonstrated that DNA methylation and H3K27me3 probably take part in different regulatory pathways as they usually mark different gene populations (Tanay et al. 2007; Fouse et al. 2008; Komashko et al. 2008; Kondo et al. 2008; Rush et al. 2009). DNA methylation is not essential for pluripotency as ES cells can proliferate but not differentiate in the absence of DNMT1 (Chen et al. 2003; Jackson et al. 2004; Tsumura et al. 2006). A number of recent studies provided valuable insights into the dynamics of DNA methylation within the context of cellular differentiation (Meissner et al. 2008; Laurent et al. 2010). Nevertheless, our understanding of how DNA methylation contributes to normal embryonic development has remained far from complete. Not much is known about the DNA methylome in nonmammalian vertebrate species, and a key question is how DNA methylation affects developmental gene regulation in early vertebrate embryogenesis. To address this issue, we carried out a DNA methylome survey of Xenopus tropicalis embryos and studied how DNA methylation relates to H3K4me3 and H3K27me3 marks and influences embryonic transcription. We observe relatively high levels of DNA methylation on promoters and gene bodies of actively expressed genes, whereas the actual transcriptional start sites (TSS) bearing the active H3K4me3 mark are hypomethylated. Likewise, we find DNA methylation absent from broad H3K27me3 domains, indicating that Polycomb-mediated repression and DNA methylation-dependent repression do not cooperate during early embryogenesis. Comparison with human ES cell data demonstrates that these patterns of DNA methylation and histone modifications, including high promoter-proximal methylation on active genes, are conserved. Injection and transgenesis experiments show an early embryonic uncoupling of DNA methylation and repression of transcription initiation. These data indicate that DNA methylation and transcription are compatible during early development, whereas repression is restored during organogenesis and differentiation. This type of regulation bypasses the need for active reprogramming of the methylome during the dynamic early phase of development.
Results
DNA methylation profiles of X. tropicalis embryos
Genomic DNA isolated from blastula and gastrula (Nieuwkoop-Faber stages 9 and 12.5) embryos was purified by affinity capture (MethylCap) using the methyl-CpG-binding domain (MBD) of human MECP2 protein (Cross et al. 1994; Kangaspeska et al. 2008; Brinkman et al. 2010; Martens et al. 2010). MethylCap has been benchmarked as a robust, accurate, and effective approach to map DNA methylation (Bock et al. 2010). Subsequent to binding to MBD-beads, methylated DNA fragments were retrieved by two elution steps (500 and 700 mM NaCl) and subjected to massive parallel sequencing. The resulting reads were aligned to the X. tropicalis genome (Joint Genome Institute genome assembly version 4.1). Visualization of DNA methylation profiles in the UCSC Genome Browser (Kent et al. 2002) revealed a dense methylation landscape, similar between both developmental stages and salt elutions (Fig. 1A). Enriched regions (peaks) were identified using MACS (Model-based Analysis of ChIP-Seq data) (Zhang et al. 2008), resulting in between 72,000 and 104,000 methylated regions for each of the four tracks (Supplemental Tables S1–S5). These numbers are comparable to those observed in human cells with a similar enrichment and sequencing approach (Serre et al. 2009). The identified DNA methylation peaks are enriched for CpG dinucleotides when compared to the genome average, whereas their GC content is only marginally higher than that of the Xenopus genome (Fig. 1B).

(A) Genome Browser view of sequenced DNA methylation tracks: late blastula (stage 9) and late gastrula (stage 12.5) tracks of both salt elutions (500 mM and 600 mM/700 mM). The X. tropicalis genome is robustly methylated in both intergenic and intragenic regions during blastula and gastrula stages. (B) Boxplots showing the distribution of the CpG observed/expected ratio (CpG O/E) and the GC content (GC%) of DNA methylation peaks. Peaks of DNA methylation are enriched for CpG dinucleotides, whereas their GC content is similar to the genome average. JGI 4.1 refers to the genome assembly used (Joint Genome Institute genome assembly version 4.1). (C) Bisulfite sequencing of randomly selected MethylCap peaks (see also Supplemental Fig. S2B). (Right) Embryonic stages and MethylCap salt elutions of the DNA methylation tracks; (black boxes) PCR amplicons; (below) bisulfite sequencing profiles of the amplicons. (Black circles) Methylated CpGs; (white circles) unmethylated ones.
The methylation status of these genomic regions was verified and validated. Randomly selected MethylCap peak regions were verified by quantitative PCR (experimental FDR ≤ 0.067) (Supplemental Fig. S1). Two validation approaches were employed. First, the results obtained with a different enrichment strategy, using a methyl-CpG-specific antibody (MeDIP) (Weber et al. 2005; Mohn et al. 2009), were highly congruous (stage 9, R2 =0.7859; stage 12.5, R2 = 0.8356) (Supplemental Fig. S2). Second, a number of randomly selected DNA methylation peaks were subjected to bisulfite sequencing for further validation (Fig. 1C; Supplemental Fig. S2B,C). As shown previously (Bock et al. 2010; Brinkman et al. 2010), genomic regions identified by MethylCap-sequencing can be validated using MeDIP and bisulfite sequencing.
In the 500 mM MethylCap fraction, we observed a modest increase in the number of peaks corresponding to the gastrula stage, whereas the number of regions identified in the 700 mM fraction is similar between the two stages examined (Supplemental Table S1). Also, the analysis revealed a small number of sequences that were recovered from the MBD affinity purification but did not contain any CpG dinucleotides. These outliers were all simple sequences featuring repeats of the CA dinucleotide, which can be found methylated in mammalian cells and, in particular, embryonic stem cells (Ramsahoye et al. 2000; Lister et al. 2009; Laurent et al. 2010). To identify the genomic locations of DNA methylation peaks, their genomic distribution was determined relative to gene position (Supplemental Fig. S3). No differences were observed in the overall distribution of DNA methylation between the developmental stages or salt elutions. Approximately 50% of the peaks are located in gene-distant regions (>5000 bp upstream of the gene start), while the remaining half is distributed over six locations: 5′ far (500–5000 bp to 5′ end), 5′ near (1–500 bp to 5′ end), exon, intron, 3′ near, and 3′ far (Supplemental Fig. S3). When normalized for the genomic coverage of these locations, exons appear slightly enriched for DNA methylation. DNA methylation of exons has been described and proposed to serve as a splicing and RNA elongation regulatory mechanism (Choi et al. 2009; Laurent et al. 2010). The data demonstrate that the X. tropicalis genome is methylated in both intergenic and intragenic regions in late blastula and gastrula stages.
Methylation of repetitive elements
The distribution profiles of DNA methylation unveiled that ∼50% of the genomic DNA methylation lies within the gene-distant regions (Supplemental Fig. S3A). DNA methylation of repetitive sequences is a common phenomenon observed in plants, fungi, and mammals (Selker 2004; Schulz et al. 2006; Zilberman et al. 2007). Almost 35% of the Xenopus genome corresponds to transposable elements, mostly DNA transposons (72% of all transposable elements), which constitute 25% of the genome (Hellsten et al. 2010). To gain insight into the nature of DNA methylation in these gene-distant regions and repetitive DNA, the repetitive DNA methylation content was mapped to the genome by assigning non-unique sequence reads randomly to matching sequences. DNA methylation in three different repeat collections was analyzed: (1) The UCSC RepeatMasker collection of interspersed repeats and low-complexity DNA sequences (http://www.repeatmasker.org); (2) Simple Repeats identified by the Tandem repeats finder program (Benson 1999); and (3) Microsatellite DNA, a subset of the Simple Repeats track representing sequences of 15 or more dinucleotide and trinucleotide repeats. DNA methylation is enriched over the repeating elements included in the Simple Repeats and RepeatMasker groups to a varying extent, with one of the blastula-stage fractions showing less DNA methylation compared to the other fractions (Fig. 2A). The Microsatellite Repeat group is depleted of DNA methylation, in line with the absence of CpG dinucleotides in these microsatellites. To investigate in more detail the methylation status of specific repeats, the UCSC RepeatMasker track was intersected with a merged DNA methylation peak file. Repeats displaying >80% overlap with DNA methylation peaks were called “methylated” and were used for subsequent analysis. Within the RepeatMasker group, different types of repeating elements show a differing extent of DNA methylation as shown by the percentage of methylated repeats (Fig. 2B; Supplemental Table S6). For example, >70% of the 1723 and Harbinger DNA transposons (Kay and Dawid 1983; Kay et al. 1984; Jurka et al. 2007) are robustly methylated. The methylation status of RepeatMasker elements (Fig. 2B) positively correlates with CpG density, which is not surprising knowing that MethylCap is an affinity-based technique that selects CpG-dense methylated sequences. However, some repeat groups such as Sat1 with low CpG density are frequently found in methylated portions of the genome. Among elements with similar CpG density, the Gypsy retrotransposon and the 1723 DNA transposons are two to three times more frequently methylated than the OCR DNA transposons. The results demonstrate strong methylation of specific repetitive DNA elements.
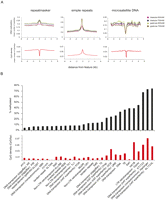
DNA methylation of repetitive elements. (A) Distribution profiles of DNA methylation over three distinct repeat tracks demonstrate an enrichment of repetitive elements for DNA methylation. Microsatellite DNA lacks CpG dinucleotides and therefore is not methylated. However, the RepeatMasker track is depleted for CpG dinucleotides but enriched for DNA methylation, while in the case of the Simple Repeats track DNA, methylation positively correlates with CpG density. (B) Extent of overlap between repeat types and DNA methylation (merged MethylCap peaks). (Top panel) Percentage of methylated genomic copies. (Bottom panel) CpG density of different repeat types. There is an overall correlation of DNA methylation and CpG density. However, some repeats such as Sat1 are highly methylated in spite of their low CpG density.
DNA methylation and gene regulation
Mammalian genes frequently contain methylation-free patches of CpG dinucleotides called “CpG islands” (CpGis) in their promoter regions (Cooper et al. 1983; Gardiner-Garden and Frommer 1987). To investigate whether 5′ ends of Xenopus genes are also enriched for CpGis, and to determine their methylation status, the overlap between these features was determined. As a first step, CpGis were called using a sliding-window approach with the relatively stringent CpGi criteria proposed by Takai and Jones (2002): Length > 500 bp, G+C content > 55%, CpG observed over expected (O/E) > 0.65. This results in 24,283 CpGis. All DNA methylation peaks of blastula and gastrula stages merged together were intersected with the CpGis and the 5′ ends of two gene collections (Fig. 3A): the FilteredModels (FM) gene collection generated by the Joint Genome Institute (JGI) and the XTEV_VV gene models, which are the subset of experimentally validated (Xenopus tropicalis experimentally validated) gene models that have an H3K4me3 peak at the 5′ end in gastrula-stage embryos, in addition to evidence of expression (Akkers et al. 2009). The intersections show that relatively few 5′ ends of the actively expressed XTEV_VV gene models overlap with methylated CpGis. Interestingly, as many as 10,943 (45%) of CpGis in the Xenopus genome are methylated; however, this overlap constitutes only 8% of the 141,246 MethylCap peaks (Fig. 3A).
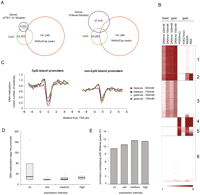
DNA methylation and transcriptional regulation. (A) Venn diagrams showing a direct overlap of merged DNA methylation peaks (141,246 regions, all four peak sets combined), the 5′ ends of genes (left: 8625 XTEV_VV gene models; right: 27,916 Joint Genome Institute, JGI, FilteredModels), and genomic CpGis (24,283 Takai-Jones CpGis). DNA methylation is largely absent from 5′ ends of genes. A large portion of the genomic CpGis are methylated (overlap between CpGis and MethylCap peaks). (B) K-means clustering of DNA methylation, histone methylation, and active transcription within CpGis. Clusters 1 and 3 correspond to methylated CpGis with different methylation profiles. Cluster 2 represents exons of genes expressed during gastrulation. Clusters 4 and 5 consist of active CpGi promoters. Cluster 6 corresponds to CpGis that are neither associated with active transcription units nor enriched for either histone modification or RNAPII. (C) Distribution of DNA methylation over CpG and non-CpGi promoters. Both CpG and non-CpG promoters show a dip in DNA methylation around the TSS. To define CpGi and non-CpGi promoter subsets, the TSS regions of XTEV_VV gene models were intersected with Takai-Jones CpGis. (D) Boxplots showing the abundance of DNA methylation within promoter regions of genes with distinct expression intensities. (E) Percentage of promoters (region 1 kb upstream of TSS) overlapping (≥500 bp) with peaks of DNA methylation.
To examine the relationship between CpGis, DNA methylation, and expression, we integrated our data with published profiles of histone H3 methylation, RNA polymerase II (RNAPII) occupancy, and poly(A)+ RNA (RNA-seq) (Akkers et al. 2009), the number of sequence reads corresponding to these data was determined for all CpGis, and k-means clustering was performed (Fig. 3B). RNAPII is found at promoters, introns, and exons of expressed genes, whereas RNA-seq reads are only found at exons of expressed genes. Histone H3 lysine 4 trimethylation (H3K4me3) is enriched at the 5′ ends of active genes. Clusters 1 and 3 correspond to methylated CpGis with different methylation profiles between stages. A few of these are located in introns of actively transcribed genes (RNAPII-positive, RNA-seq-negative), but the majority are located outside gene bodies or in inactive genes. The second (2) cluster apparently represents CpGis that are colocated with exons of genes expressed during gastrulation (presence of both RNA-seq and RNAPII ChIP-seq reads) and that are strongly methylated. Clusters 4 and 5 feature CpGi promoters that are characterized by a lack of DNA methylation and enrichment for H3K4me3 and RNAPII. These promoters are expressed as indicated by the enrichment of RNA-seq and RNAPII ChIP-seq reads. The majority of CpGis in the sixth (6) cluster are not associated with active transcription units, nor are they enriched for either histone modification or RNAPII. These data demonstrate a strong anticorrelation between DNA methylation and H3K4me3-enriched CpGi core promoters, consistent with mechanisms that link H3K4 trimethylation to unmethylated CpGis by recruitment of CFP1 (Thomson et al. 2010). In contrast, CpGis in exons or introns of expressed genes are mostly methylated. CpGis enriched for H3K27me3 do not form a separate cluster but are found among CpGis that are also enriched for H3K4me3 and RNAPII (Clusters 4 and 5).
These results were obtained with a collection of 24,283 CpGis that meet the criteria proposed by Takai and Jones (2002). For comparison, two more inclusive CpGi collections were examined: (1) CpGis determined according to criteria proposed by Gardiner-Garden and Frommer (1987) and (2) The UCSC Genome Browser CpGis. These collections differ in number, CpG density, GC content, and overlap with transcription start sites (Supplemental Fig. S4). The Takai-Jones CpGi collection shows the highest enrichment for both 5′ ends of genes and TSS-seq transcription start sites (van Heeringen et al. 2011). K-means clustering performed on all three CpGi collections demonstrates that similar clusters of DNA methylation, histone modifications, RNAPII, and RNA are found independently of the CpG island collection used (Supplemental Fig. S5).
To extend our analysis to non-CpGi promoters, the distribution of DNA methylation around transcription start sites (TSS) of genes was examined (XTEV_VV gene models). The genes were divided into two groups depending on the presence or absence of a CpGi at the TSS. Both CpGi and non-CpGi promoters are similar with respect to a general lack of DNA methylation at the TSS, regardless of whether the Takai-Jones CpGi (Fig. 3C) or the Gardiner-Garden CpGi definition is used (data not shown). We then wanted to interrogate the relationship of promoter DNA methylation and gene expression. The promoters (TSS-1 kb) were grouped depending on the number of RNAPII reads located in the gene body normalized for gene length. All the genes marked by RNAPII reads were divided into three equally sized expression bins (high, medium, low RNAPII recruitment). Nonexpressed genes (“no”) were defined as a subgroup of XTEV gene models with a verified 5′ end (H3K4me3 peak) but no evidence for expression as judged by RNA-seq. Average values of the four tracks were plotted to show the distribution of DNA methylation in promoter regions (Fig. 3D). Promoters (1 kb upstream) belonging to nonexpressed genes are enriched for DNA methylation; however, there is no inverse correlation between gene expression and promoter methylation; the genes from the high expression bin actually have more DNA methylation in their promoter regions than the weakly expressed genes (Fig. 3D). Furthermore, ∼10% of promoters from all four expression bins show a significant (≥500 bp) direct overlap with DNA methylation peaks (Fig. 3E). A similar observation has been made in mouse ES cells, in which promoter DNA methylation is compatible with expression (Fouse et al. 2008).
To investigate in more detail the relationship of DNA methylation and transcription, DNA methylation reads from all four tracks were mapped to H3K4me3 peaks and their surrounding regions. Consistent with the k-means clustering (Fig. 3B) and the genomic intersections (Fig. 3A), there is strongly reduced DNA methylation inside H3K4me3-positive regions; however, these regions are flanked by above-average DNA methylation (Fig. 4A,B). This finding is consistent with reports that the N terminus of histone H3 must remain unmethylated to provide a binding site for DNA methyltransferases (Ooi et al. 2007; Hu et al. 2009), and that DNA must be unmethylated for H3K4me3 to be deposited (Hashimshony et al. 2003). Nevertheless, a few genes have an H3K4me3 peak, RNAPII enrichment, and a DNA methylation peak overlapping with their TSS (Supplemental Table S7). To test whether these core promoters are, indeed, fully methylated, bisulfite sequencing was performed on two of these loci (Supplemental Fig. S7). Bisulfite sequencing indicated that the expressed bet1 and c20orf151 transcription start sites are, indeed, fully methylated, rendering it less likely that mixed cell populations are the explanation of this phenomenon. While these expressed-methylated core promoter examples should be regarded as rare exceptions, as many as 3722 genes display DNA methylation within 1000 bp upstream of their TSS, 865 of which are expressed (Supplemental Fig. S6; Supplemental Table S8). Together, these results do not strongly support a dominant repressive role of DNA methylation during blastula and gastrula stages (Fig. 3D,E; Supplemental Figs. S6, S7).
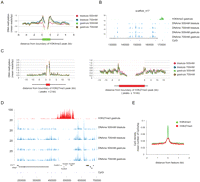
DNA methylation and H3K4me3/H3K27me3 are mutually exclusive on the majority of genomic locations. (A) Distribution of DNA methylation over boundaries of H3K4me3 peaks. DNA methylation is excluded from the bulk of genomic H3K4me3 sites even though above average DNA methylation can be observed on sequences flanking the H3K4me3 peaks. (B) Genomic example of the exclusion of DNA methylation and H3K4me3. (C) Broad H3K27me3 domains are mostly DNA methylation-free, even though smaller H3K27me3 peaks can be methylated. (D) hoxb cluster as an example of the mutual exclusion of H3K27me3 and DNA methylation. (E) H3K4me3 and H3K27me3 peaks are enriched for CpG dinucleotides but depleted of DNA methylation.
To identify differentially methylated regions (DMRs) between blastula and gastrula stages, Fisher's exact test was applied to determine statistical significance of differences in peak read counts between the two stages (multiple testing q-value cutoff 0.05) followed by filtering for DMRs with a substantial (greater than fourfold) difference in read counts between the two stages. This resulted in 3549 DMRs, 3197 of which are more strongly methylated at stage 9, whereas 352 DMRs are more strongly methylated at stage 12.5 (Supplemental Tables S9, S10). Out of 3197 DMRs displaying higher DNA methylation at stage 9, only 187 show a direct overlap with promoter regions (Supplemental Table S9). Similarly, only 12 promoter regions intersect the DMRs that are more abundantly methylated at stage 12.5 (Supplemental Table S10). However, neither of these gene groups represents specific Gene Ontology categories, nor does their differential DNA methylation status correlate with expression data (data not shown).
The H3K27me3 mark is deposited after the onset of embryonic transcription and is mostly associated with spatial restriction of transcription (Akkers et al. 2009). In contrast to the H3K4me3 peaks, which are more uniform in size and generally mark gene promoter regions, H3K27me3 peaks range from ∼2 kb to >100 kb and can spread through gene clusters. Therefore, H3K27me3-enriched regions were divided into four groups depending on the peak size, and DNA methylation profiles were determined for each of these groups (Fig. 4C; Supplemental Fig. S8). Whereas DNA methylation is absent from large H3K27me3 domains, smaller H3K27me3-marked regions (≤2 kb), which are often located close to the borders of larger H3K27me3 domains, are enriched for DNA methylation, as illustrated by the hoxb gene cluster (Fig. 4C,D). The absence of DNA methylation from H3K4me3 and H3K27me3 peaks is not due to the low CpG density of these regions as both features are CpG-rich (Fig. 4E). In conclusion, DNA methylation is generally not found at the TSS of expressed genes that are enriched for histone H3K4 trimethylation, however many genes with proximal promoter methylation are robustly expressed. Also, DNA methylation is excluded from large H3K27me3-decorated regions.
Conservation of gene regulation in human ES cells and Xenopus embryos
The dynamics of histone and DNA methylation in pluripotency and differentiation has been under extensive investigation over the last couple of years (Boyer et al. 2005; Lee et al. 2006; Mikkelsen et al. 2007; Fouse et al. 2008; Mohn et al. 2008). Lister et al. (2009) generated methylome maps corresponding to human embryonic stem (ES) cells and fetal fibroblasts and identified two groups of genes with distinct transcriptional activity and epigenetic makeup in the two cell types. To investigate if these patterns of gene regulation are conserved in Xenopus, orthologs of these two groups were identified, and reads corresponding to DNA methylation, H3K27me3, H3K4me3, and RNAPII were mapped from −20 to +20 kb relative to their TSS. Strikingly, the differences observed between the two groups of genes in human ES cells are by and large also observed between the orthologous groups of genes in Xenopus embryos. The first group of orthologs, which are more highly expressed in H1 ES cells (H1-exp), was found to be more highly expressed in Xenopus embryos than the second group, which are orthologs of genes with stronger transcriptional activity in fibroblasts (Fib-exp) (Fig. 5A). Moreover, the H1-exp genes have a higher DNA methylation content (−10 kb to +10 kb, P-value = 0.002, Wilcoxon rank test) and much less H3K27me3 than the Fib-exp genes (Fig. 5A), consistent with their epigenetic makeup in ES cells (Lister et al. 2009). In contrast, both gene groups feature a similar enrichment for H3K4me3 around their TSS. These results were analyzed in more detail by hierarchical clustering of DNA methylation and histone methylation around the TSS for both groups of genes (Fig. 5B; Supplemental Fig. S9). We find that, although the average patterns are conserved between human ES cells and Xenopus embryos (Fig. 5A), several subgroups of genes can be identified (Fig. 5B). This may reflect differences in developmental stage or divergent gene regulation. However, even within distinct groups there is an overall positive correlation of DNA methylation and transcription and a strong anticorrelation of H3K27me3 and DNA methylation. These results confirm that the methylated genes are generally more highly expressed, whereas the H3K27me3-decorated genes are generally methylation-free and show no or much lower expression. Moreover, there is a strong mutual exclusion of DNA methylation and H3K27me3 around the TSS (Figs. 4D, 5B), consistent with the hypothesis that the two epigenetic marks represent two independent pathways; of these, H3K27me3 is more tightly correlated with transcriptional repression than DNA methylation. H3K27me3-enriched genes are often expressed in a spatially regulated fashion in embryos (Akkers et al. 2009); however, DNA methylation is not enriched over spatially regulated genes (data not shown). These observations raise the question how effective DNA methylation in promoter-proximal regions is in transcriptional repression.
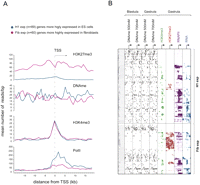
Conserved patterns of epigenetic regulation in human ES cells and Xenopus embryos. (A) Distribution of RNAPII, H3K4me3, H3K27me3, and DNA methylation (average values of all four tracks) around TSSs corresponding to Xenopus orthologs of two gene groups differing in their expression status among fetal fibroblasts (Fib) and ES cells (H1). The genes with higher expression in ES cells (H1-exp) are also more highly expressed in Xenopus embryos and show increased DNA methylation both upstream of and downstream from the TSS. These orthologs also have a significantly higher DNA methylation in the region spanning 10 kb upstream of the TSS (P-value = 0.031, Wilcoxon rank test), the region 10 kb downstream from the TSS (P-value = 0.0026), and the total region from −10 kb to +10 kb relative to the TSS (P-value = 0.0023). (B) Hierarchical clustering performed on Xenopus orthologs of H1-exp and Fib-exp genes shows an inverse relationship between DNA methylation and H3K27me3. It also shows a high level of DNA methylation on promoters of robustly expressed H1-exp orthologs.
Transcription from methylated promoters in early embryos
To directly assess the ability of DNA methylation to repress transcription initiation in early embryos, we injected unmethylated and SssI-methylated promoter reporter constructs into oocytes and embryos. The oocyte is known to repress methylated DNA very effectively (Kass et al. 1997; Jones et al. 1998). We used a plasmid containing the hsp70 promoter driving the expression of the CAT reporter gene, the expression of which was determined by quantitative RT-PCR. In the oocyte, the unmethylated plasmid was highly transcribed, whereas the transcript from the methylated template was only present at background levels (Fig. 6A), confirming that DNA methylation acts as a potent signal for transcriptional repression in the oocyte. Strikingly, when we measured the expression from these templates in the embryo, we observed significant transcription from the fully methylated hsp70 promoter (Fig. 6A). A similar effect was observed for the Xenopus histone H2B promoter (Supplemental Fig. S10A). Both methylated and unmethylated templates can be recovered from oocytes and embryos with equal efficiency and with the same methylation status they had before injection (Fig. 6A; Supplemental Figs. S10B, S11).

Methylated promoters in early Xenopus embryos are able to initiate transcription. (A) Unlike Xenopus oocytes, gastrula embryos fail to repress transcription from the injected hsp70 methylated (SssI) promoter. Levels of injected plasmid DNA recovered from embryos were similar for all samples. (B) Effects of DNA methylation on stably integrated transgenes. A fully methylated CMV-luciferase transgene is robustly expressed compared to its unmethylated counterpart during blastula and gastrula stages. Strong DNA methylation-dependent repression observed in the oocyte is restored gradually at neurula and tailbud stages. The results represent three independent experiments and were normalized for the number of transgene insertions. (C) When targeted to the injected hsp70 promoter by the heterologous Gal4 DNA-binding domain in oocytes, the transcription repression domain of MECP2 shuts down reporter construct transcription. Such an effect is not observed in gastrula embryos where the Gal4-TRD fusion is expressed at equivalent levels (see Supplemental Fig. S15). Error bars represent the SEM of three experiments.
The injected promoter-reporter templates are largely maintained in an episomal state without efficient DNA replication (Marini and Benbow 1991). Therefore, we hypothesized that the lack of DNA methylation-dependent repression in early embryos could be due to the lack of genomic integration. Moreover, in the absence of genomic integration, the distribution of the plasmids in the embryo is mosaic and cannot be properly assayed in late embryonic stages. To further test our findings, fully methylated or unmethylated constructs containing a luciferase reporter placed under the control of the CMV promoter were integrated into the Xenopus genome by transgenesis. The activity of the CMV promoter was measured in F0 transgenic embryos (n = 260; three independent experiments with 20 or more transgenic embryos per sample) by quantitative RT-PCR for both methylated and unmethylated transgenes, and the results were normalized to their integrated copy numbers. The methylated transgenes were expressed in both blastula and gastrula embryos, whereas the expression was significantly reduced at the neurula stage and below the detection threshold at the tailbud stage (Fig. 6B). Similar results were obtained with transgenic embryos obtained with a different transgenesis method (SceI nuclease-mediated transgenesis instead of REMI transgenesis) (Supplemental Fig. S12A). Bisulfite sequencing of the stably integrated transgenes showed that the DNA methylation status of methylated and unmethylated transgenes does not change during the experiment (Supplemental Fig. S12B).
These results led us to examine the expression and activity of methyl-CpG-binding domain (MBD) proteins. In addition, to measure the ability of MBD proteins to bind to methylated DNA, an assay was used in which an in vitro methylated or unmethylated, concatamerized DNA probe was immobilized on magnetic beads and incubated with oocyte and embryo extracts (Supplemental Fig. S13A). The MBD proteins MECP2 and MBD3 are expressed in blastula and gastrula embryos (Stancheva et al. 2003; Iwano et al. 2004); however, both their expression and their ability to bind methylated DNA are very low in embryos compared to oocytes (Supplemental Fig. S13B; data not shown). To test whether repression could be restored by overexpression of MBD proteins, synthetic MECP2 mRNA was injected into fertilized eggs, and the effect on promoter activity was determined. Even though overexpressed MECP2 is able to bind methylated DNA in the early embryos, comparable to the endogenous activity of MECP2 in the oocyte, no transcriptional repression of the injected reporter plasmid was observed (Supplemental Fig. S14). To further test this issue, we examined whether such repression can be restored if the MECP2 repression domain is targeted to the promoter by a heterologous DNA-binding domain. For that purpose, the hsp70 promoter with five Gal4-binding sites was injected into oocytes and embryos together with a synthetic RNA coding for the fusion of the yeast Gal4 DNA-binding domain and the MECP2 transcription repression domain (Gal4–TRD) (Jones et al. 1998; Kaludov and Wolffe 2000). The Gal4–TRD fusion represses transcription very efficiently in oocytes; however, in gastrula embryos, Gal4–TRD did not repress transcription even though it was expressed at the same level as that obtained in oocytes (Fig. 6C; Supplemental Fig. S15). These results demonstrate that the strong MECP2-mediated repression observed in the oocyte is ineffective in the early embryo; transcriptional repression is largely uncoupled from DNA methylation in early embryos, but not in oocytes or late embryos.
Discussion
In this study, we describe the methylome of X. tropicalis and its implications in embryonic gene regulation. Using high-throughput genomic sequencing combined with DNA methylation affinity capture, we generated genomic DNA methylation maps of late-blastula and gastrula stages. The genome of X. tropicalis is heavily methylated at both intergenic and intragenic regions throughout gastrulation. Interestingly, a significant portion (45%) of genomic CpGis is methylated. A strong anticorrelation exists between DNA methylation and histone H3K4 trimethylation, in accordance with earlier reports (Ooi et al. 2007; Hu et al. 2009). Similarly, we find DNA methylation absent from large H3K27me3 domains. Finally, we detect cases of compatibility between DNA methylation and active transcription and a conserved pattern of epigenetic regulation in Xenopus blastula and gastrula embryos and mammalian ES cells. Functional assays performed in oocytes and embryos show a temporal relief of DNA methylation-dependent repression during gastrulation.
Using previously published epigenetic profiles of human ES cells (Lister et al. 2009), we found that Xenopus embryos and ES cells use the same type of epigenetic regulation on two distinct gene groups; one of them being more highly expressed in ES cells and the other being more highly expressed in fibroblasts. Interestingly, the genes with higher DNA methylation content were more active than genes enriched in H3K27me3, suggesting that Polycomb-mediated repression is more tightly correlated to gene silencing in both early embryos and ES cells. These findings are compatible with the fact that ES cells tolerate loss of both maintenance and de novo DNA methyltransferases. ES cells deficient for DNMT1 were able to proliferate and initiate differentiation (Jackson et al. 2004). Similarly, DNMT3A/3B depletion does not influence ES cell self-renewal but, rather, causes a failure in differentiation. Strikingly, a triple knockout (Dnmt1−/− Dnmt3a−/− Dnmt3b−/−) in ES cells demonstrated that in the absence of DNA methyltransferases, ES cells remain viable and preserve their genome integrity (Tsumura et al. 2006). A similar observation has been made in Dnmt1-depleted Xenopus tissue explants that were cultured for up to 48 h without signs of apoptosis (Stancheva et al. 2001). However, this is clearly not the case with other cell types such as mouse fibroblasts, where DNMT1 knockdown causes cell death (Jackson-Grusby et al. 2001). Similar to the case of DNA methylation, loss of Polycomb proteins Eed and Ring1b does not influence the self-renewal properties of ES cells. However, a loss of Polycomb destabilizes ES cells due to expression of lineage-specific markers (Leeb and Wutz 2007; Chamberlain et al. 2008; Leeb et al. 2010). Taken together, these findings suggest that transcriptional repression is not essential for pluripotency.
Moreover, ES cells do not repress methylated promoters very strongly. When ES cells differentiate to terminally differentiated neurons, methylation of CpG-poor promoters in ES cells is compatible with transcription as assayed by RNA Pol II occupancy (Mohn et al. 2008). Commitment to a lineage leads to an activation of a number of promoters of which only some lose DNA methylation. Genome-wide mapping of DNA methylation to promoter regions demonstrated that promoters in mouse ES cells are heavily methylated, including active promoters regulating pluripotency genes (Fouse et al. 2008). Actually, ∼75% of methylated genes bound by either NANOG or POU5F1 (also known as Oct4) were expressed compared to 93% of expressed, unmethylated POU5FI/NANOG bound targets (Fouse et al. 2008).
In Xenopus oocytes, non–promoter DNA methylation is transmissible in cis over hundreds of base pairs to repress transcription (Kass et al. 1997), but at what locations DNA methylation can repress transcription is not known for embryonic or somatic cells. In blastula- and gastrula-stage embryos, we observe a compatibility of DNA methylation and active transcription in Xenopus embryos. A significant portion of actively expressed genes was found to have high methylation within 1 kb of the transcription start site. Even though DNA methylation is generally absent from the TSS of expressed genes, probably due to the deposition of the H3K4me3 mark, we also identified a small number of expressed genes having a fully methylated 5′ end. Consistent with earlier reports (Kass et al. 1997; Jones et al. 1998), we find that Xenopus oocytes repress methylated DNA very efficiently, but we observe a severely reduced potential for DNA methylation-dependent repression during gastrulation. Moreover, the MECP2 transcription repression domain, which in oocytes acts as a potent gene repressor when targeted to the promoter (Jones et al. 1998), was not able to shut down reporter transcription in the gastrulating embryo, possibly due to the absence of co-repressors such as the Sin3 complex (Jones et al. 1998; Nan et al. 1998; Wade et al. 1999; Fuks et al. 2003). A recent global study performed in mouse neurons demonstrated the ability of MECP2 to interact with both the Sin3 complex and the transcriptional coactivator CREB1 (Chahrour et al. 2008). It is possible that such a differential recruitment of transcriptional repressors and activators is occurring in a developmental stage- or context-dependent fashion. It is important to note that the readout of DNA methylation is highly diverse. Although none of the tested promoters are dominantly repressed by DNA methylation in early embryos, some promoters are unaffected by DNA methylation, whereas other promoters are, though transcribed, less active when methylated. The effects may not only be determined by methyl CpG-binding proteins, but also by sequence-specific DNA-binding proteins, some of which are inhibited when their binding site is methylated (for review, see Bogdanović and Veenstra 2009).
Interestingly, Xenopus embryos and mammalian ES cells display similarities in terms of requirements for DNA methyltransferase function. Recent studies in Xenopus embryos showed that the strong phenotypes observed upon ablation of DNMT1 (Stancheva and Meehan 2000) are not caused by perturbations in DNA methylation but, rather, by the loss of transcriptional repressor function of the DNMT1 protein, independent of its catalytic activity as a DNA methyltransferase (Dunican et al. 2008). The phenotype caused by the loss of DNMT1 was rescued by a catalytically inactive form of human DNMT1 (hDNMT1C1226Y) (Jair et al. 2006; Dunican et al. 2008), suggesting that the DNA methyltransferase function is not required for pluripotency. All of these observations are compatible with the general inability of DNA methylation to dominantly repress transcription initiation in early development and differentiation. Even though Xenopus embryos and mammalian ES cells show similarities in epigenetic profiles, differences in chromatin structure between mammalian ES cells and Xenopus embryos have also been documented. Bivalent domains marked by both H3K4me3 and H3K27me3 are a reported hallmark of the chromatin state of mammalian ES cells (Azuara et al. 2006; Bernstein et al. 2006). It has been proposed that bivalently marked genes are resolved upon differentiation (Bernstein et al. 2006; Stock et al. 2007; Landeira et al. 2010). However, in X. tropicalis embryos, H3K27me3 deposition follows H3K4me3 deposition in time and is linked to spatial restriction of gene expression (Akkers et al. 2009). Furthermore, classical bivalency with the two opposing histone marks decorating the same nucleosomal population is not the predominant configuration in Xenopus embryos. Also, a recent study demonstrated that artificial CpG clusters, when integrated into the genome of mouse ES cells, are sufficient to deposit H3K4me3 via the recruitment of CFP1 (Thomson et al. 2010). cxxc1 (cfp1) is a highly conserved protein that is expressed during Xenopus development (data not shown). However, we identify a number of genomic CpG islands without H3K4me3 methylation. This could be due to differences in H3K4me3 deposition between Xenopus and mouse or due to different functions of CpGis in these two organisms. In contrast to mammals, Xenopus lacks a global demethylation program after fertilization and phenomena such as imprinting and X-inactivation (Yamada et al. 1999; Veenstra and Wolffe 2001). Development starts with a global transcriptional repression that in Xenopus is relieved at the mid-blastula transition (MBT) by a change in the cytoplasm-to-nucleus ratio (Newport and Kirschner 1982a,b). The MBT is characterized by changes in subcellular localization of transcription factors and regulation of various activating mechanisms (Veenstra 2002). Around the MBT the active H3K4me3 mark gets deposited on the TSS of active genes (Akkers et al. 2009). At this stage, methylated genes can be expressed, and the Polycomb H3K27me3 mark is largely absent. At late blastula and early gastrula stages, H3K27me3 is deposited on key developmental regulator genes to establish spatial domains of gene expression. Our transgenesis experiments demonstrate that it is only during organogenesis and terminal differentiation that DNA methylation-dependent repression is restored. Early embryogenesis may require a more relaxed interpretation of DNA methylation, which allows distinct transcriptional programs to operate without large changes in DNA methylation. Only later, during organogenesis and differentiation, is DNA methylation-dependent repression restored, locking cells in their fate by stable long-term repression.
Methods
Xenopus procedures
X. tropicalis embryos were collected after natural mating, which was induced by injection of females with human chorionic gonadotrophin (HCG). The embryos were then incubated in 3% cysteine to release the jelly coat. The staging was done according to Nieuwkoop and Faber (http://www.xenbase.org).
X. laevis oocytes and embryos were injected with 0.25–0.5 ng of hsp70 promoter templates. In the case of the Gal4-MRD RNA coinjection, an additional 0.2 ng of the RNA was injected. RNA injection into the oocytes was done 16 h prior to template injection, allowing the protein to be expressed at sufficient levels. Both the template plasmid and the synthetic RNA were coinjected at the same time in the embryos, and the embryos were left to develop until the gastrula stages.
X. laevis transgenesis was carried out essentially as described (Kroll and Amaya 1996); however, the sperm nuclei were not digested with restriction enzymes. Briefly, Xenopus high-speed egg extract (EXT) and sperm head nuclei were purified and stored at −80°C in aliquots until use. The pGL3-CMV plasmid or methyl pGL3-CMV was linearized with AlwNI, purified, and incubated along with linear pEGFP-N1 (100 ng each per experiment) and sperm nuclei for 5 min. EXT (10 μL) and 20 μL of Sperm dilution buffer (SDB = 250 mM sucrose, 75 mM KCl, 0.5 mM spermidine, 0.2 mM spermine) were mixed and incubated for 5 min at 65°C, then centrifuged at 16,000g for 3 min to remove precipitate. The soluble fraction (6 μL) was diluted to 22 μL with SDB plus 10 mM MgCl2. This activating solution was added to the nuclei/sperm mix and incubated for 15 min at room temperature. The nuclei were gently added to 170 μL of SDB and used for microinjection at a rate of 0.586 μL/min using a microliter syringe pump (Harvard Apparatus).
Nucleic acid isolation and PCR
Genomic DNA was isolated using phenol–chloroform and precipitated with ethanol. Total RNA was extracted with TRIzol (Invitrogen) and RNAeasy (QIAGEN) columns. cDNA isolated from 10–20 gastrula embryos was synthesized using a Superscript III kit (Invitrogen). Quantitative RT-PCR reactions were carried out on 1:50 dilutions of cDNA using the iQ SYBR Green Supermix (Bio-Rad). Plasmid DNA was isolated from all the samples and subjected to PCR to control for the potential differences that might have occurred during embryo injections
MethylCap-seq
Genomic DNA was sheared by sonication to ∼500-bp fragments. Methylated DNA was affinity-purified using H6-GST-MBD fusion protein (Diagenode) following the manufacturer's protocol with one modification. The DNA was eluted in several steps with increasing NaCl concentrations: twice with 200 mM NaCl, once with 300 mM NaCl (washing steps 1–3), 500 mM and 700 mM (elutions 1 and 2). Sequencing samples were prepared using the manufacturer's protocols (Illumina). The DNA ends were repaired, and the adapters were ligated. The library was size-selected (300 bp) and amplified by PCR. The sequencing was carried out on a Genome Analyzer (Ilumina). MeDIP reactions were performed with 1 μg of sonicated DNA and following the MeDIP kit (Diagenode) protocol. DNA methylation reads were aligned to the X. tropicalis genome (JGI version 4.1) using either ELAND (GAPipeline version 1.0; Illumina) or MAQ (version 0.7.1) (Li et al. 2008) programs. The reads were extended to 300 bp after mapping. MAQ aligned reads were used for the analysis of repetitive DNA, while the rest of the data corresponds to uniquely mapped ELAND reads.
DNA methylation peaks
Genomic regions enriched for DNA methylation were identified by MACS (Model-based Analysis of ChIP-Seq data) (Zhang et al. 2008). The genomic distribution of DNA methylation peaks was calculated by PinkThing software (http://pinkthing.cmbi.ru.nl), using Ensembl gene models. The genomic intersections were performed by either the Galaxy bioinformatics tool (Blankenberg et al. 2007; Taylor et al. 2007) or the UCSC Table Browser. To validate peak finding settings experimentally, quantitative PCR reactions were carried out on 1:50 dilutions of MethylCap/MeDIP material using the iQ SYBR Green Supermix (Bio-Rad). All PCRs were performed on the MyIQ single-color real-time PCR detection system (Bio-Rad). For primer sequences, see the Supplemental Data.
CpG island identification
The locations of CpGis in the Xenopus genome were determined using a sliding-window operation applying either the Takai-Jones or Gardiner-Garden criteria for CpGis. The Takai-Jones CpGi criteria were as follows: Length ≥ 500 bp, % GC ≥ 55%, CpG observed/expected ratio ≥ 0.65. The Gardiner-Garden criteria were as follows: Length ≥ 200 bp, % GC ≥ 50%, CpG observed/expected ratio ≥ 0.6. After merging overlapping regions, a total of 259,903 CpGis were identified in the X. tropicalis genome (JGI version 4.1) by applying the Gardiner-Garden criteria. The Takai-Jones criteria produced a subset of 24,283 CpGis. The merged features do not always meet the criteria mentioned above for the total length of their sequence. This is the counterintuitive result of merging adjacent islands. Each 200-bp or 500-bp window contributing to the merged CpGi feature does meet these criteria, and therefore each CpGi feature contains one or more qualifying CpGis.
Data analysis
To generate average profiles, the mean number of DNA methylation reads was calculated for 100-bp bins surrounding the feature of interest (TSS, H3K4me3 and H3K27me3 peak borders, repetitive DNA elements). The y-axis is represented as the mean number of reads per base pair. K-means clustering (k = 6, default settings for TMEV 4.5) was performed using the TM4 software suite (http://www.tm4.org). The results are represented as log2 values of the number of mapped reads. To compare the results with human data sets (Lister et al. 2009), X. tropicalis orthologs of genes expressed in ES (H1-exp) cells and fetal fibroblasts (Fib-exp) were identified using BioMart (http://www.biomart.org). Reads corresponding to DNA methylation, H3K4me3, H3K27me3, RNAPII, and RNA-seq were mapped to these two groups of genes and their surrounding regions (TSS ± 20 kb). Gene orientation was taken into account. Hierarchical clustering was performed by the TM4 software suite (http://www.tm4.org). The values represent the log2 numbers of mapped reads. Boxplots were generated in R using default settings. The boxes show the interquartile range (IQR) around the median; the whiskers extend from the minimum value to the maximum value unless the distance to the first and third quartiles is more than 1.5 times the IQR.
Constructs
The constructs used in this study have been described (Landsberger et al. 1995; Jones et al. 1998; Veenstra et al. 1999; Kaludov and Wolffe 2000; Carro et al. 2004). The mRNA from the Gal4-MRD and xMECP2pSP64polyA vectors was transcribed using an in vitro synthesis kit (mMessage mMachine Kit; Ambion). Promoter templates were in vitro methylated by overnight incubation with the bacterial SssI methylase using a standard manufacturer's protocol (Invitrogen). HpaII restriction followed by agarose gel electrophoresis was subsequently performed to control the extent of the in vitro methylation reaction.
Data access
The data have been deposited in the NCBI Gene Expression Omnibus (GEO) (Edgar and Barrett 2006) and are accessible through GEO Series accession number GSE23913 (http://www.ncbi.nlm.nih.gov/ geo/query/acc.cgi?acc=GSE23913). Visualization tracks are available at the authors’ website (http://www.ncmls.nl/gertjanveenstra/resources.htm).
Acknowledgments
This work was funded by grants of the Netherlands Organization of Scientific Research (NWO-ALW VIDI grant 864.03.002) and the U.S. National Institutes of Health (grant R01 HD054356) to G.J.C.V. We thank the Spanish and Andalusian Governments for grants to J.L.G.-S. (BFU2010-14839, Proyecto de Excelencia CVI-3488 and CSD2007-00008). We thank A. Salcedo and G.W. van der Heijden for helpful suggestions and critical reading of the manuscript, R.C. Akkers and E.M. Jansen-Megens for MethylCap-seq assistance, and Ron Engels for help with frogs and animal care. We also thank Juan Ramón Martínez Morales for letting O.B. perform some experiments in his laboratory.
Footnotes
-
↵4 Corresponding author.
E-mail g.veenstra{at}ncmls.ru.nl.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.114843.110.
- Received September 3, 2010.
- Accepted April 14, 2011.
- Copyright © 2011 by Cold Spring Harbor Laboratory Press








