
Abstract
Second-generation sequencing technologies allow surveys of sequence variation on an unprecedented scale. However, despite the rapid decrease in sequencing costs, collecting whole-genome sequence data on a population scale is still prohibitive for many laboratories. We have implemented an inexpensive, reduced representation protocol for preparing resequencing targets, and we have developed the analytical tools necessary for making population genetic inferences. This approach can be applied to any species for which a draft or complete reference genome sequence is available. The new tools we have developed include methods for aligning reads, calling genotypes, and incorporating sample-specific sequencing error rates in the estimate of evolutionary parameters. When applied to 19 individuals from a total of 18 human populations, our approach allowed sampling regions that are largely overlapping across individuals and that are representative of the entire genome. The resequencing data were used to test the serial founder model of human dispersal and to estimate the time of the Out of Africa migration. Our results also represent the first attempt to provide a time frame for the colonization of Australia based on large-scale resequencing data.
Owing to rapidly declining costs, second-generation sequencing has become an affordable means to perform surveys of sequence variation on a genome-wide scale. Several complete genome sequences have already been obtained for humans (and recently also for Neanderthals) (Bentley et al. 2008; Wheeler et al. 2008; Abdulla et al. 2009; Ahn et al. 2009; McKernan et al. 2009; Pushkarev et al. 2009; Green et al. 2010; Schuster et al. 2010) and for model organisms (Doniger et al. 2008; Ossowski et al. 2008; Daines et al. 2009; Hillier et al. 2009). In addition, de novo whole-genome sequencing has become feasible for nonmodel organism species, thus extending the power of genomic approaches to species important for ecological studies or for conservation biology.
Despite the lower sequencing costs, collecting whole-genome sequence data for many individuals is still unaffordable for many laboratories. Surveying a large and representative set of unlinked loci, rather than the entire genome, can provide a valuable alternative for many types of studies, especially if the cost of preparing the sequencing target is low. For example, this is the case when the goal is to develop markers for genetic mapping in species for which genetic tools are unavailable. It is also the case if the goal is to estimate demographic parameters; this is because, in the presence of recombination, the variance of the estimators decreases as the surveyed length increases due to the sampling of a larger number of independent realizations of the evolutionary process (Pluzhnikov and Donnelly 1996). Therefore, as long as many independent loci are sampled, resequencing data can provide sufficient information to estimate population parameters accurately even if only a portion of the genome is surveyed and even if obtained from a single sampled individual (Felsenstein 2006). Owing to their efficiency, surveys of variation in single individuals have been used to estimate interpopulation and interspecies divergence (Sun et al. 2009) and, together with ascertained variation data, to reconstruct the allele frequency spectrum (Keinan et al. 2007). Until recently, most variation surveys used Sanger sequencing of PCR products in many individuals per population at relatively few loci (Voight et al. 2005; Wall et al. 2008; Laval et al. 2010). Second-generation sequencing has overcome many of the limitations of Sanger sequencing, but specific protocols for the preparation of the resequencing target are required.
Two main approaches have been developed to select a subset of a genome to be analyzed by second-generation sequencing. The first approach enriches the resequencing target for specific regions of interest, which are selected either by PCR amplification or by hybridization to complementary oligonucleotides (Albert et al. 2007; Okou et al. 2007); a disadvantage of this approach is that the oligonucleotide libraries significantly add to the overall costs. An alternative approach is to produce a reduced representation of the genome by restriction digestion followed by direct sequencing the ends of either a subset of the size-selected restriction fragments (Van Tassell et al. 2008) or all the restriction fragments in the genome (Baird et al. 2008). To increase cost efficiency, samples subjected to reduced representation can be pooled prior to sequencing.
Many previous studies employing reduced representation protocols aimed to develop markers for subsequent genotyping studies and did not attempt to estimate evolutionary parameters. In general, obtaining unbiased estimates of evolutionary parameters based on shotgun sequence data presents several challenges, which include sequencing and alignment errors as well as missing data (for review, see Pool et al. 2010). Deep sequencing (e.g., in the order of 30–40×) may overcome these challenges by increasing the probability of sampling both alleles at heterozygous sites and the ability to distinguish true heterozygous sites from sequencing and alignment errors. An alternative approach consists in sequencing multiple individuals from the same population at low depth (e.g., average 4× as in the case of the 1000 Genomes project; www.1000genomes.org/), thus reaching higher confidence in the genotype calls for each individual by leveraging the sequence information gathered for other individuals in the same population.
Here, we have developed a reduced representation approach for second-generation sequencing that is suitable for population genetic analyses. Because our main goal was to collect good-quality genotype data for population genetic inferences, we modified the reduced representation protocol developed by Van Tassell et al. (2008) to obtain an appropriate sequencing depth for single individual samples. Our protocol retains the features of being inexpensive and applicable to any species for which a draft or complete reference genome sequence is available. Because of the wealth of information available on human sequence variation, we applied it to 19 individuals from a total of 18 different human populations, and where available, we compared genotype calls to the HapMap calls. We showed this method to be reproducible across individuals, resulting in a large overlap in surveyed regions. In addition to the experimental protocol for preparing the sequencing target, we have developed new tools and methods for aligning reads, calling genotypes, and incorporating sample-specific sequencing error rates in the estimate of population genetic parameters. When applied to human population samples, this approach provided, for the first time, a test of the serial founder model of human dispersal and a time frame for the colonization of Australia based on unascertained sequence variation data.
Results
A simple and inexpensive reduced representation protocol
We implemented a simple and inexpensive protocol to prepare sequencing targets that contain many independent genomic regions, can be reproducibly generated across samples, and are representative of the entire genome (see Methods). We applied this protocol to human samples, but we note that it can be easily applied anytime a draft or complete reference genome sequence is available for the species of interest or for a closely related one.
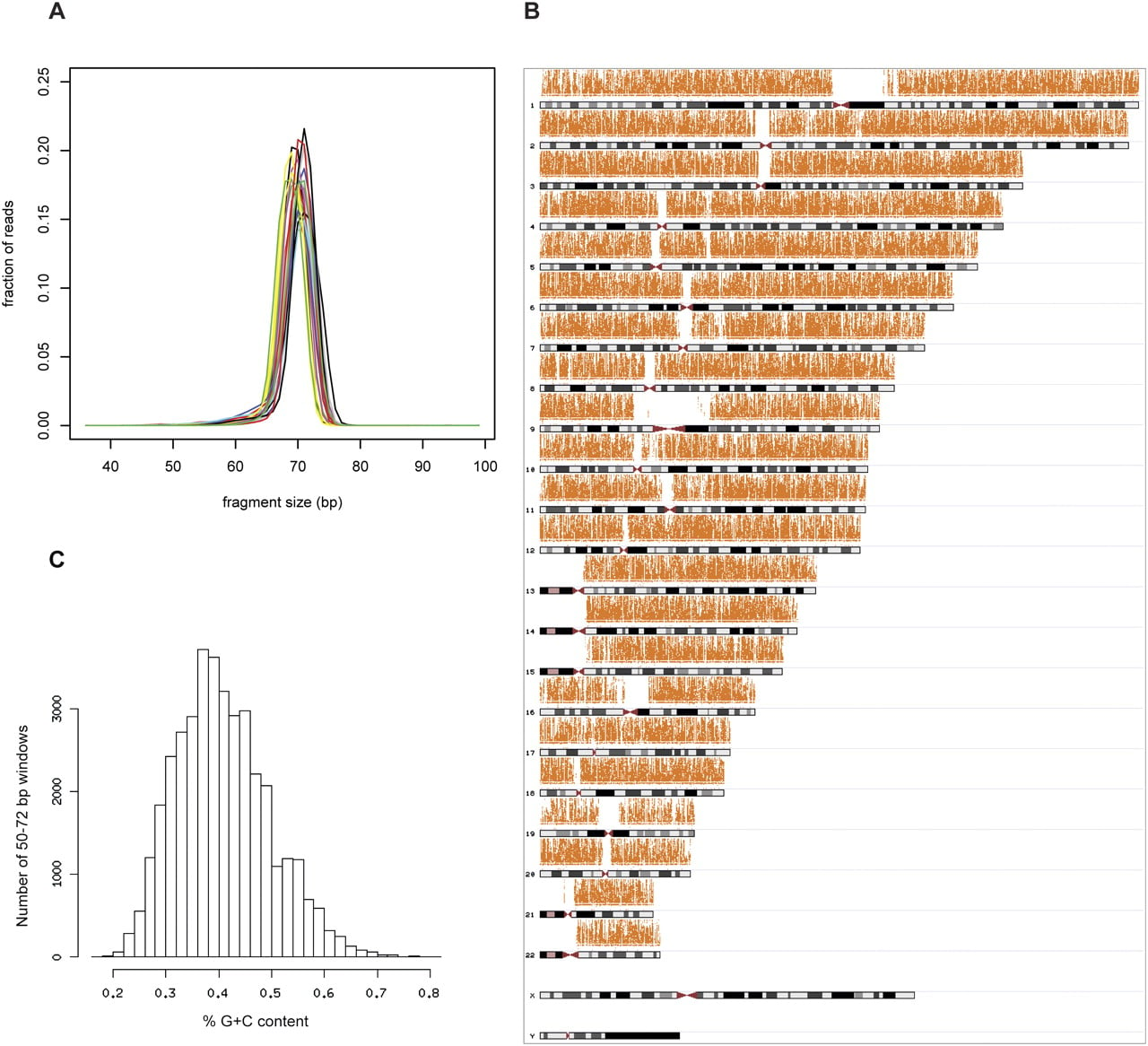
Human genomic DNA was digested to completion using the enzyme RsaI. The restriction fragments were size-selected from a polyacrilamide gel. Fragments in the 70- to 75-bp size range were purified and sequenced on an Illumina GAII sequencer. This protocol was applied to 19 individual samples from 18 human populations (Fig. 1A). Each population is represented by a single male individual except for the Berber population, for which only a female individual was included, and the Native Australian population, which is represented by a male and a female. The reads for each individual were mapped against the in silico restriction digest of the human genome by means of a new alignment strategy that limits the redundancy in the reference sequence, therefore reducing the likelihood that reads with polymorphic sites and sequencing errors are misaligned (see Methods). As shown in Table 1, on average 61.7% of reads were uniquely aligned in each individual, resulting in 92–159 Mb of usable sequence data for subsequent analyses. To assess the reproducibility of the gel excision, we calculated the frequency distribution of the proportion of reads by restriction fragment size. As shown in Figure 2A, in all cases the mode of the distribution is located within the targeted 70- to 75-bp interval, and little between-sample variation is observed in the span of the distribution, leading to the expectation that the surveyed segments largely overlap across individuals. Accordingly, after genotype calling, any two samples share on average 1.11 Mb of surveyed sequence (corresponding to ∼68% of the sites called in each sample), while 0.55 Mb of sequence was shared in at least 80% of the samples. This large overlap in surveyed segments across individuals allows the assessment of several aspects of genetic variation, including the allele frequency spectrum and the divergence between populations, which require data for orthologous segments for more than one individual. On average, 97.4% of the surveyed fragments are in the size range 50–100 bp. This range is wider than the fragment size interval that was excised from gel, probably due to diffusion during the electrophoretic migration of DNA fragments (Altshuler et al. 2000).
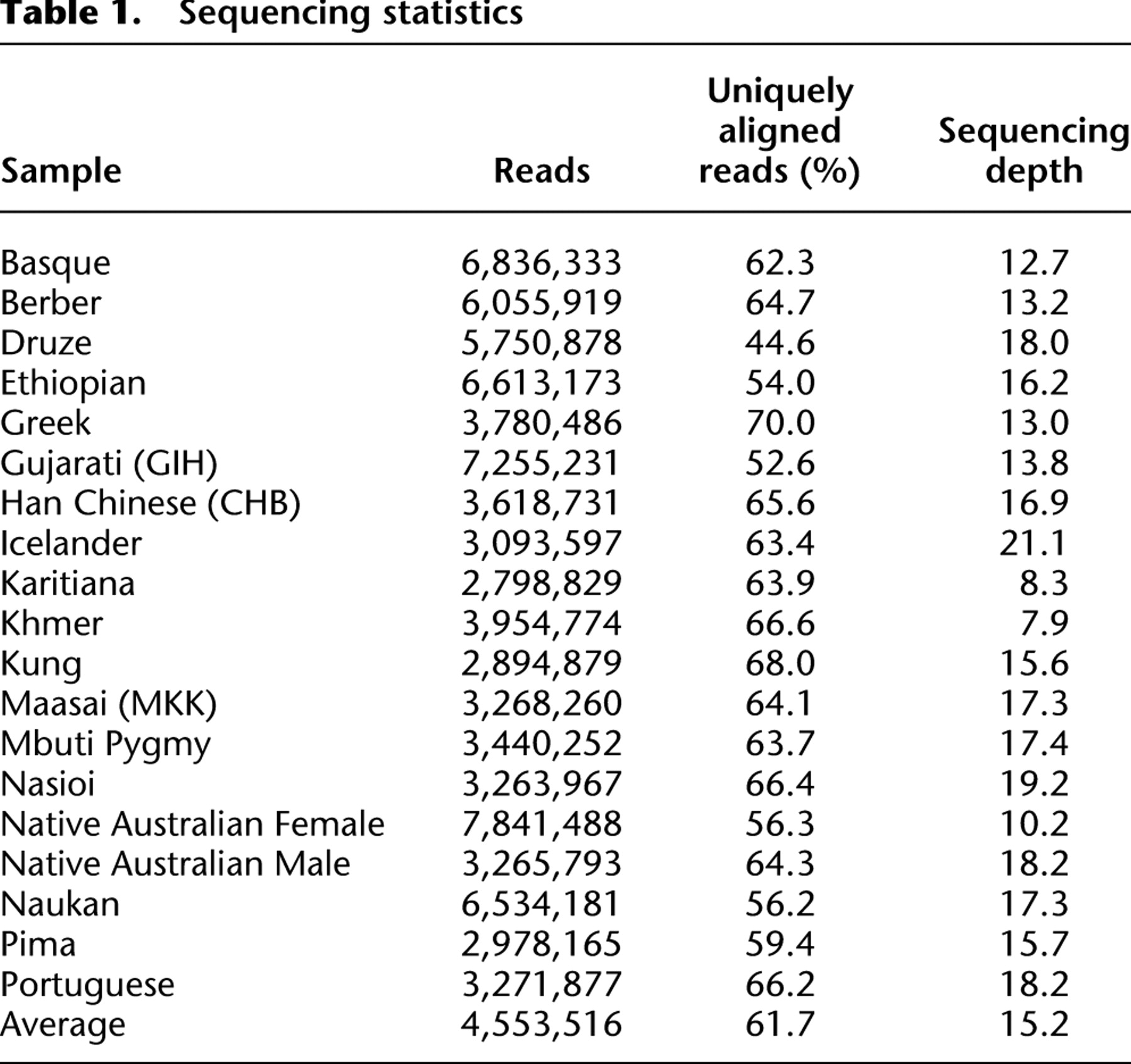
(A) Map of the approximate geographic locations for the populations sampled in this study. (B) Nucleotide diversity decreases with distance from a location in Eastern Africa (Addis Ababa). Geographic distance is calculated through waypoints to account for large masses of water.
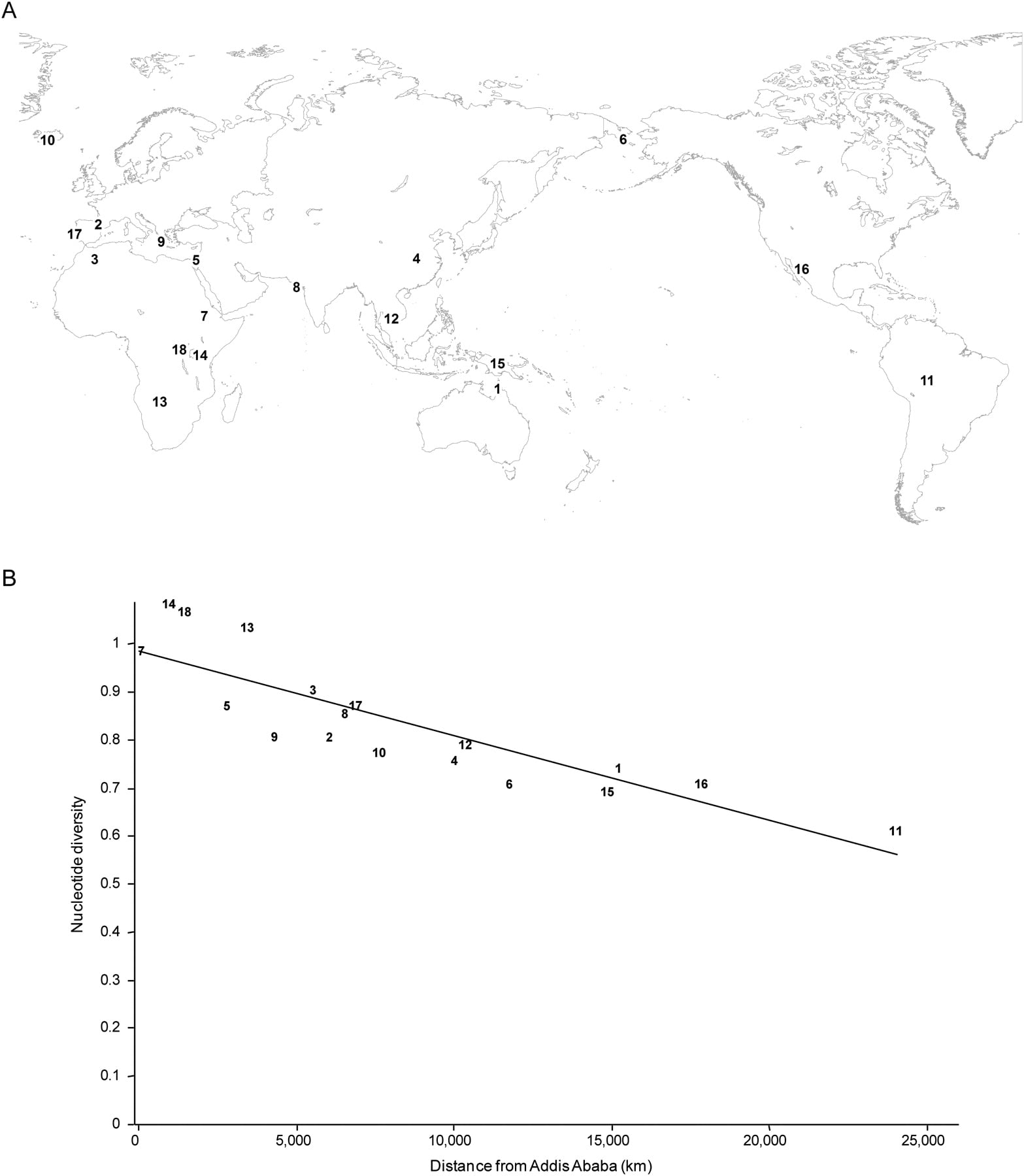
Features of the sequencing target prepared by our reduced representation protocol. (A) The plot shows the distributions of the proportion of reads aligned to restriction fragments in the range 40–100 bp for the samples in this study. (B) Autosomal regions are evenly represented in the resequencing target and no apparent bias is detected in the genomic distribution of coverage depth (vertical axis). (C) The plot shows the distribution of %G + C content for the sequencing target, which is very similar to the distribution reported for the entire human genome (Lander et al. 2001).
We compared a number of features of the resequenced target to those for the entire human genome in order to assess whether the surveyed restriction fragments represent a random subset of the human genome. We found that the surveyed fragments are evenly distributed across chromosomes and across genomic regions (Fig. 2B), and across genic and nongenic regions (48% genic regions compared with 50% in the entire genome). Likewise, the average recombination rate (1.25 cM/Mb) and %G + C content (40.9%) are very similar to the genome-wide averages (1.3 cM/Mb [Yu et al. 2001], 41% GC content [Lander et al. 2001]) (Fig. 2C).
False negatives represent the largest fraction of errors in the data set
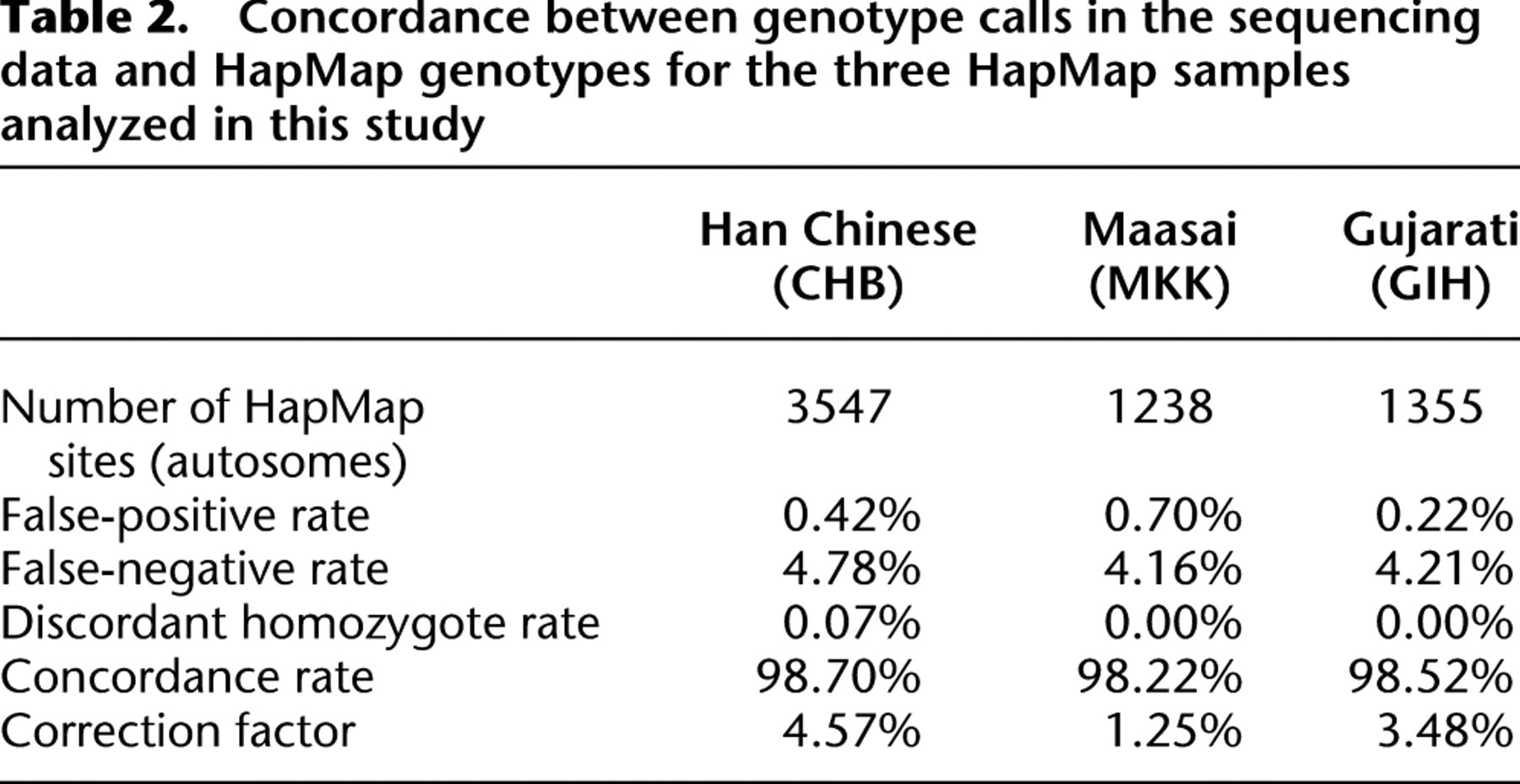
After applying position and base quality filters (see Supplementary Methods), we called individual genotypes at the remaining sites. If the two alleles across all true heterozygous sites are sampled independently and with equal probability, the per allele sequencing depth distribution is expected to be binomial conditional on the total coverage at the site. In this case, a critical value threshold could be used to discriminate between a true heterozygous site and a site with sequencing errors. We assessed the accuracy in genotype calls based on the binomial distribution by comparing the genotype calls to the genotype data available for the three HapMap samples included in the study. Because of our experimental design, a fragment is sampled only if the restriction site is present; therefore, heterozygotes at a restriction fragment length polymorphism (RFLP) will be hemizygous in the sequence data. As expected, we found that our data deviated from the binomial expectations to the extent that using such a binomial filter for heterozygous sites proves overly conservative; that is, a substantial number of true heterozygous sites are called homozygous (Supplemental Table 1). As an alternative to this model-based approach, we have implemented an empirical genotype calling scheme that relies on sample-specific allele coverage matrices (see Methods; for an example, see Supplemental Fig. 1). Again, we assessed the accuracy of genotype calls by comparison to the HapMap genotype data; a site heterozygous in the resequencing data and homozygous in the HapMap data is referred to as a false positive, while a site homozygous in the resequencing data set and heterozygous in the HapMap data is referred to as a false negative. Table 2 presents the concordance rate for each HapMap individual in terms of false positives and false negatives. The proportion of false negatives is 9.9-fold higher than the proportion of false positives. Additionally, we identified a small subset of discordant sites that were called as homozygous in both data sets, but for different alleles. These are likely to represent HapMap genotyping errors. On average, the concordance rate between our and the HapMap genotype calls is 98.5%.
Concordance between genotype calls in the sequencing data and HapMap genotypes for the three HapMap samples analyzed in this study
Because all but two samples in our data set are males, we were also able to estimate the false-positive error rate in the data as the proportion of heterozygous sites called on the X chromosome (Supplemental Table 2). The false heterozygous sites detected on the X chromosomes probably result from misincorporation events that occur and are propagated by PCR amplification during the library preparation. This problem is usually solved by removing reads that start at the same position; however, in our study design, all reads for a given restriction fragment have the same start site, thus making it impossible to identify and omit duplicate reads. We estimated a false-positive rate of 3.4 × 10−6 over 894 kb of surveyed sequence in 17 individuals. This false-positive rate is three orders of magnitude lower than the one estimated through comparison to the HapMap data (average 0.45%) (Table 2); because the genotyping error rate in the HapMap data is ∼0.5% (http://hapmap.ncbi.nlm.nih.gov/), it is plausible that the discrepancy between our two estimates of the false-positive rate is due to HapMap genotyping errors.
Overall, these results indicate that the largest fraction of errors in our data is represented by false negatives rather than false positives. To take into account allele drop-out (defined as the probability that one of the alleles at a heterozygous site is not detected) due to RFLPs, we calculate an approximate correction factor based on equilibrium neutral theory (see Methods and Supplementary Methods); this correction factor is generally estimated to be 3%. This estimate is in good agreement with simulated data (see Supplementary Methods) and with an average correction factor estimate based on the comparison between the HapMap and the resequencing data generated in this study (3.1%) (Table 2).
Estimating nucleotide diversity
Because sequencing targets prepared by reduced representation contain thousands of random fragments across the genome, the proportion of heterozygous sites in one individual is a reasonable estimate of the average nucleotide diversity in the population. To this end, we used a maximum likelihood method to estimate nucleotide diversity from sites with coverage 10× and above in each individual (see Methods and Supplementary Methods). This method is similar to the maximum likelihood method of Lynch (2008) in having binomially distributed allele counts conditional on the coverage. For our data the approach had to be adapted to take account of restriction site polymorphisms. In addition, we ignore data from very low coverage sites (less than 10×), and when there are more than two alleles at a site, we ignore the third and fourth alleles. Unlike the method of Lynch (2008), we do not assume that sequencing errors are equally likely between all bases, nor do we assume that the pair of bases at a heterozygous site is found in proportion to the nucleotide frequencies in the genome.
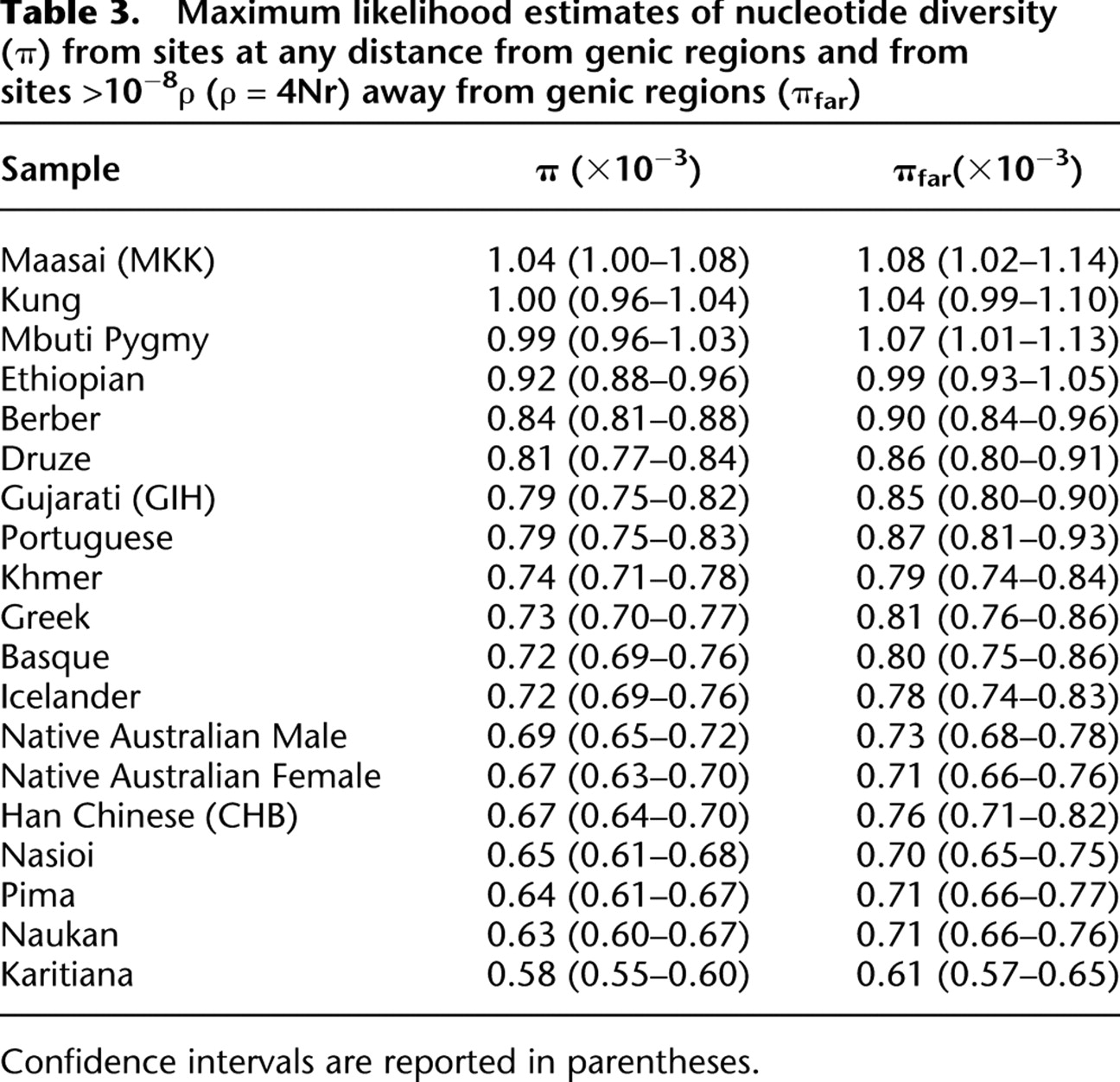
Maximum likelihood estimates of nucleotide diversity for each sample are reported in Table 3. We compared these estimates to published estimates available for noncoding regions surveyed in five population samples overlapping or closely related to the Mbuti Pygmy, Kung, Basque, Nasioi, and Han Chinese samples used in this survey (Wall et al. 2008). As shown in Supplemental Table 3, our estimates of nucleotide diversity for these samples are consistently lower than the published ones (by 22%, on average). However, the published estimates originate from a resequencing survey of genomic regions distant from genes (at least 100 kb) and with a low recombination rate. When we include only sites >10−08 ρ units (ρ = 4Nr, where N is the effective population size and r is the recombination rate between adjacent sites) away from genes, the estimates of nucleotide diversity in our data are closer to the published ones (12% lower, on average). Therefore, we included only sites outside genic regions (on average 50% of all surveyed sites) in all subsequent analyses; the amount of surveyed noncoding sequence in this subset of the data is 1.04 Mb on average per individual.
Maximum likelihood estimates of nucleotide diversity (π) from sites at any distance from genic regions and from sites >10−8ρ (ρ = 4Nr) away from genic regions (πfar)
We also used a method of moments to estimate nucleotide diversity on the genotype calls from the allele coverage matrices. In contrast to the maximum likelihood method, the allele coverage matrices allowed us to include only sites for which we had high confidence of performing accurate genotype calls in any given sample. As a consequence, the minimum coverage considered in each sample is different and varies from 12–22×. Although the genotype calls obtained with this method have low error rates (as reported above), the uncertainty around the estimate of nucleotide diversity is greater because fewer sites are called using the allele coverage matrices. Despite this, nucleotide diversity estimates were highly concordant between the two methods (Pearson's r = 0.98) (Supplemental Table 4).
Testing the serial founder model
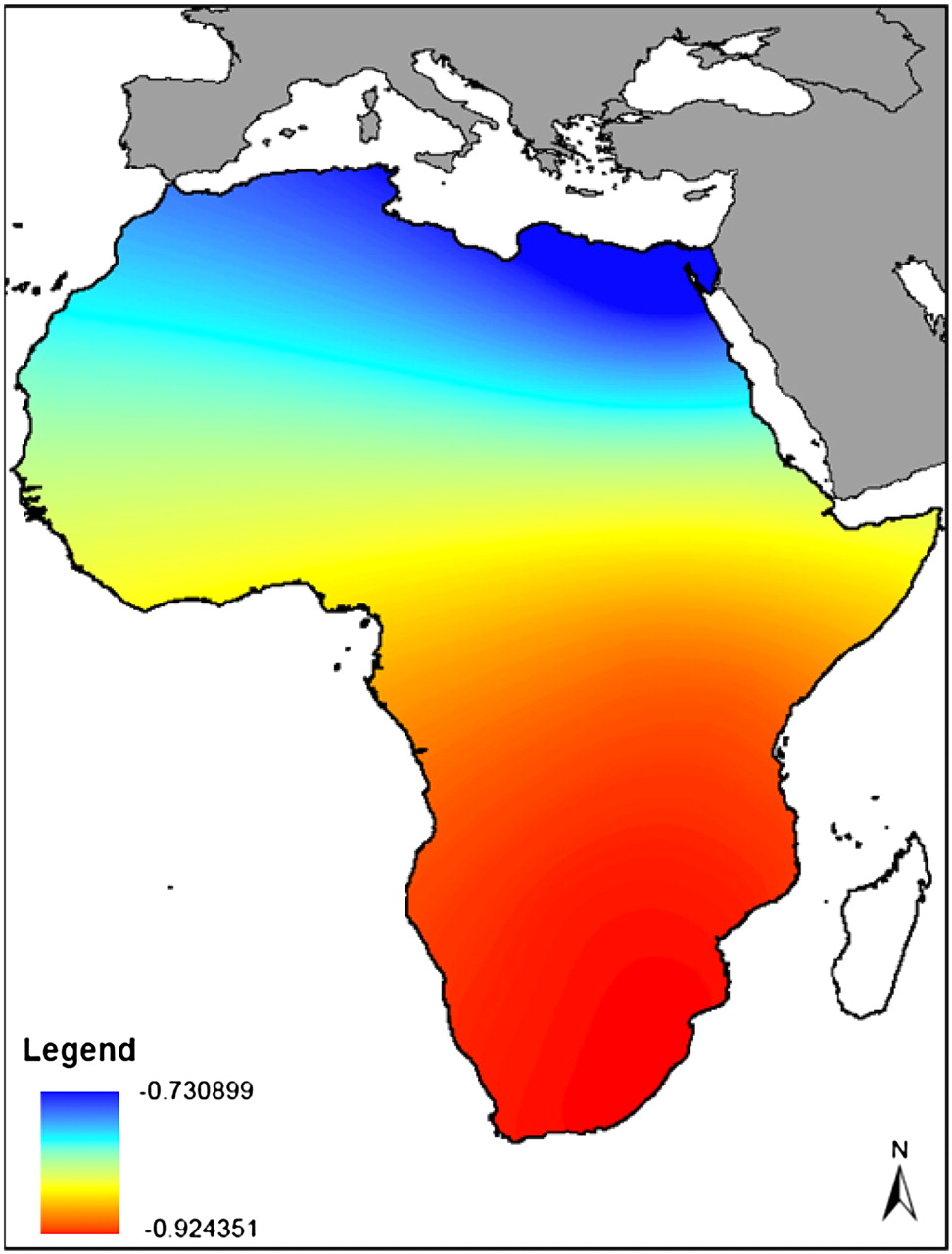
Previous studies showed that microsatellite and haplotype diversities decrease in worldwide population samples as a function of distance from Ethiopia (Ramachandran et al. 2005; Li et al. 2008). These findings were interpreted as evidence for a serial founder model of dispersal of human populations out of Africa. However, this model has not been tested based on unascertained sequence variation data. Therefore, we used our estimates of nucleotide diversity to ask this question. Figure 1B shows the correlation between nucleotide diversity and the great circle distances from Addis Ababa for each sampling location corrected for large masses of water (waypoints). Our estimates return an R2 of 0.7643, which is very similar to what was previously reported (0.7630). However, Ramachandran et al. (2005) observed the strongest correlation between microsatellite diversity and a location in central-western Africa, while we observe the strongest correlation between nucleotide diversity and geographic distance for a location in southern Africa (Fig. 3). This is in agreement with the location of highest correlation between distance and diversity determined in two studies that sampled a larger set of African populations (Tishkoff et al. 2009; Henn et al. 2011).
Heat map of the correlation between nucleotide diversity and geographic distance from locations within Africa; correlation coefficients for each location are color-coded according to the legend in the figure. The African continent was subdivided into regions 100 km square, and the distance from each region's center point to each of the samples' locations was calculated analogously to what had been done previously (Ramachandran et al. 2005). Distances from geographic locations in Southeast Africa are the most strongly correlated with nucleotide diversity.
Estimating population divergence and split times
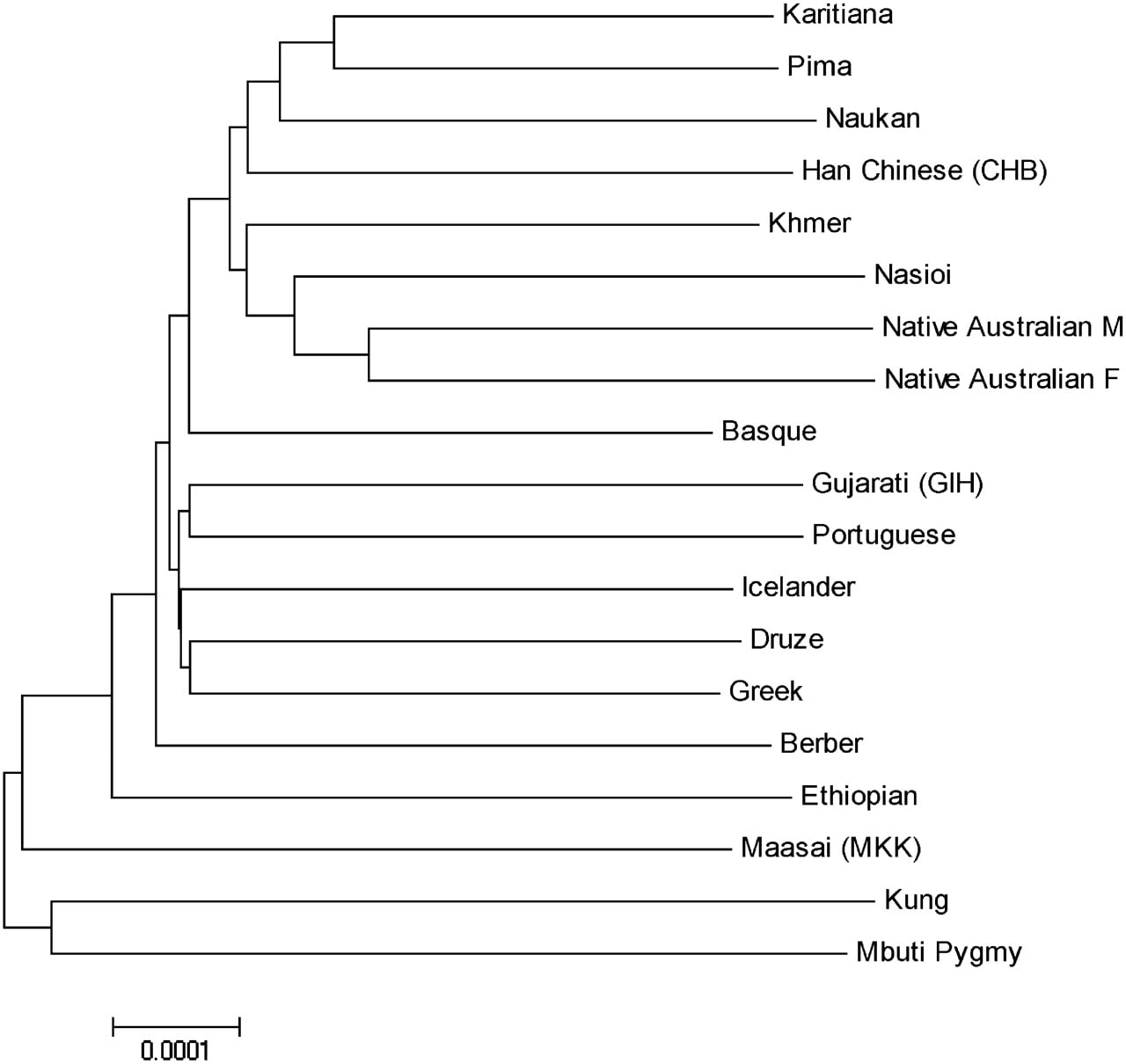
The sequence data for the reduced representation targets can also be used to calculate the sequence divergence between pairs of individuals as an estimate of the divergence between pairs of populations. We calculated pairwise divergences between the 19 individual samples and used them to construct a neighbor joining tree (Fig. 4; Tamura et al. 2007). Consistent with previous genetic variation studies (for a review of the results from uniparentally inherited markers, see, e.g., Cavalli-Sforza and Feldman 2003; Li et al. 2008), the deepest lineages lead to African individuals, with the Kung and Pygmy individuals having the most basal branches. The Middle Eastern, European (except for Basque), and Gujarati samples form a separate clade. As expected, Native Australians are most closely related to the Melanesian Nasioi and the Native American samples branch together with the Naukan Yupik from north eastern Siberia. The tree is characterized by long terminal branches, reflecting the fact that our estimate of divergence is not corrected for intrapopulation polymorphism, which accounts for the majority of variation in humans.
Neighbor joining tree calculated using the sequence divergence between individuals; M and F stand for male and female, respectively.
Given that the neighbor joining tree recapitulates the relationships between populations supported by genetic and archeological data, we used divergence and nucleotide diversity to estimate the split times between populations and the severity of the bottlenecks associated with the dispersal; the bottleneck is assumed to be followed by an instantaneous recovery to the ancestral population size. The ancestral and daughter populations are assumed not to exchange genes after the split.
Divergence estimates were calculated from a smaller subset of sites (0.58 Mb on average) relative to nucleotide diversity estimates (1.05 Mb on average); therefore, they are associated with greater uncertainty. To achieve more reliable estimates of split times, we averaged divergence estimates across samples, when appropriate. To calculate the split time associated with the Out of Africa migration, we considered the Kung and Pygmy samples as representative of the founder population and each of the remaining populations as the daughter population and then averaged across daughter populations. Under the assumption of a single migration wave out of the ancestral population, this average value represents an estimate of the time of the migration; the estimated value (50,776 yr ago [standard deviation: 19,265 yr; standard error: 4673 yr]) is compatible with the archeological record and with previous estimates based on genetic data for the Out of Africa migration (Stringer and Andrews 1988; Quintana-Murci et al. 1999; Underhill et al. 2000; Macaulay et al. 2005; Mellars 2006a,b; Fagundes et al. 2007; Laval et al. 2010). Previous studies inferred a more severe bottleneck associated with the dispersal of modern humans into Asia compared with the one associated with the dispersal into Europe (Marth et al. 2004; Voight et al. 2005; Laval et al. 2010). Consistent with these findings, we estimated the severity of the bottleneck associated with the split from the African founder population (Kung and Pygmy) to be lower for western Eurasian (0.25) than for east Asian populations (0.32) (Severity is defined here to be the fractional reduction in nucleotide diversity due to the bottleneck.). Interestingly, the highest bottleneck severity is estimated for the Karitiana, who live at the end of the geographic range expansion out of Africa.
The allele frequency spectrum
Resequencing data such as those collected here are suitable for the analysis of the allele frequency spectrum, which in turn is informative about the demographic history of the population. We used our data to calculate Tajima's D (Tajima 1989), which is a summary statistic of the frequency spectrum based on the difference between two estimators of the population mutation rate parameter. To this end, we pooled individuals by major geographic areas, thereby identifying three groups: sub-Saharan Africa (Kung, Pygmy, Maasai, Ethiopian), Europe (Greek, Portuguese, Basque, Icelandic), and Asia (Gujarati, Khmer, Han Chinese, Naukan Yupik). Only sites >1 kb away from genes and that were called in all individuals within a geographic region were used (488, 406, and 299 kb for the sub-Saharan Africa, Europe, and Asia groups, respectively). Consistent with previous studies (Akey et al. 2004; Voight et al. 2005; Wall et al. 2008), Tajima's D was negative in the African populations group (−0.29), consistent with a model of population expansion, and positive in the European (0.24) and Asian (0.32) population groups.
Genome-wide patterns of diversity reduction
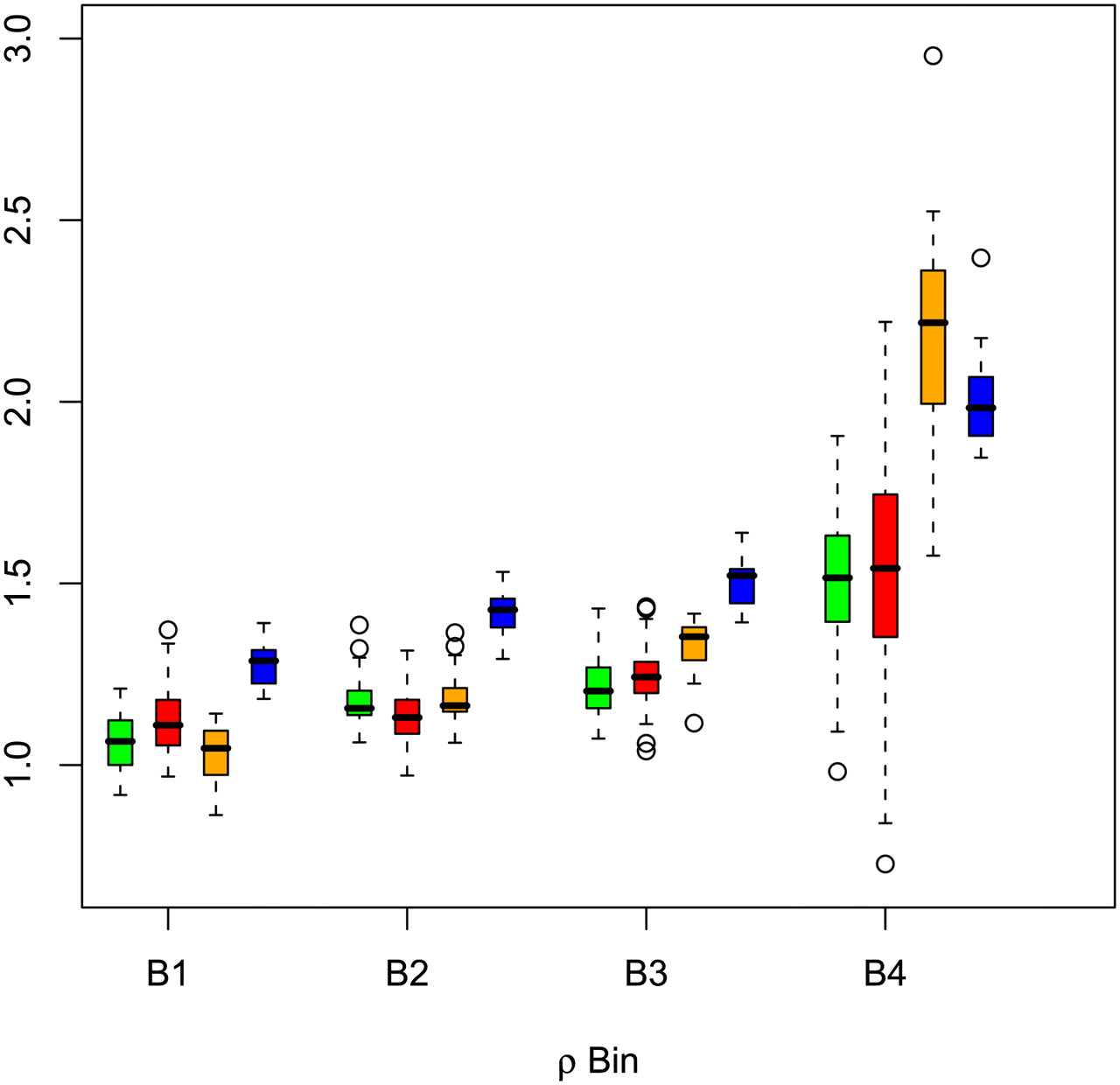
A reduction of neutral sequence diversity attributable to the effect of natural selection has been observed near functional elements in the genome of human and ancestral hominids (McVicker et al. 2009; Hammer et al. 2010). To determine whether the same pattern was detected in our data, we examined nucleotide diversity and sequence divergence between human and chimpanzee in genomic segments at increasing distances from genes. We also calculated the sequence divergence between all pairs of individuals for the same segments. To this end, we subdivided the sites with genotype calls into equally populated bins based on their genetic distance from the closest gene (Fig. 5). Our results are consistent with previous findings by showing a clear pattern of increasing diversity with increasing distance from genes. A similar pattern is evident when comparing measures of genetic diversity with sequence conservation across distantly related species. We observe a 32% reduction in nucleotide diversity for genic regions (distance from genes: ρ < 10−5) compared with regions far from genes (ρ > 1). This reduction is within the previously reported range (McVicker et al. 2009) when the 10% of neutral sites nearest to exons are compared to the 50% of neutral sites farthest from exons.
Patterns of diversity reduction are related to distance from genes and sequence conservation. Genetic distance is reported in units of ρ = 4Nr; using the estimates of r from the HapMap project (http://hapmap.ncbi.nlm.nih.gov/). B1: 5 × 10−6 < ρ ≤ 10−2; B2: 10−2 < ρ ≤ 10−1; B3: 10−1 < ρ ≤ 1; B4: ρ > 1. Green indicates nucleotide diversity; red, sequence divergence between populations; orange, sequence divergence between human and chimpanzee; and blue, phascons score. In each bin, the parameters have been scaled by their corresponding values for ρ ≤ 5 × 10−6.
Discussion
We have optimized a protocol to prepare reproducible sequencing targets containing thousands of random genomic fragments that are representative of the entire genome. The resequencing data sets generated with this protocol are of a size intermediate between those obtained by PCR-resequencing approaches and whole-genome sequencing projects. This protocol is easily applicable in the presence of a reference genome sequence for the species of interest or a closely related one, although de novo assembly is also possible (J.D. Wall, pers. comm.). Importantly, our protocol is highly cost effective (less than $5/sample) compared with methods based on sequence capture. By applying this protocol to samples from human populations, we have been able to assess and correct for errors and establish that data collected through this protocol recapitulate known features of human variation. We illustrate the utility of our approach by showing a strong correlation between unascertained diversity levels and geographic distance and by providing an estimate for the split time from the ancestral founder population and for the severity of the bottleneck associated with this event.
Accounting for different sources of error in second-generation sequencing data
Different sources of errors in second-generation sequencing data have been previously identified (for a review, see Rokas and Abbot 2009; Pool et al. 2010); however, a full understanding of the error structure has not been reached yet. Additionally, many of these biases/errors are a function of the specific study design, including sequencing depth, sequencing platform and chemistry, library preparation, and post-sequence processing. Most of the efforts dealing with errors in second-generation sequencing data have focused on minimizing the false-positive rate, for example, by requiring high sequencing depth and quality to call SNP genotypes. However, as Johnson and Slatkin (2006, 2008) pointed out, stringent SNP-calling criteria may result in a high false-negative rate and in underestimating nucleotide diversity. Therefore, they have suggested incorporating quality scores directly into the procedure for estimating nucleotide diversity; however, the exact relationship between base quality scores and error probabilities is not well established for second-generation sequencing data. Until a better understanding of this relationship is achieved, it remains difficult to account for error probabilities (Hellmann et al. 2008). However, in our data set, it is clear that our scheme for genotype calling based on coverage matrices is able to reduce the false-positive rate to negligible levels (3.4 × 10−6 as estimated from the X chromosome data of male samples), thus leaving false negatives as main contributors to the error. False negatives may result from different aspects of the data collection. Alignment errors have been previously reported to introduce biases in the allelic composition of genotype calls, mainly by favoring the alignment of reads containing the reference over the nonreference allele, so that heterozygotes are misclassified as homozygotes for the reference allele (Degner et al. 2009). As proposed by Pool et al. (2010), this can be considered a missing data problem, where one allele is sampled more often or exclusively compared to the alternative allele. In these cases, because the reads carrying the alternative allele also carry sequencing errors, they may be discarded by the alignment strategy used or alternatively they could be mapped with higher confidence to alternative locations in the genome. To deal with this type of errors, we have developed an ad hoc alignment strategy, which limits redundancy in the reference sequence, therefore strongly reducing the likelihood that reads containing both polymorphic sites and sequencing errors are misaligned. RsaI RFLPs are another contributor to the false-negative rate because only one allele is sampled in heterozygous individuals (this is distinct from the effect of low coverage that results in randomly sampling one of the two alleles). To account for the false-negative errors due to polymorphisms in RsaI restriction sites, we have incorporated a correction factor in our estimates of nucleotide diversity and divergence that is calculated based on theoretical expectations and is very similar to the one calculated from the empirical comparison to HapMap genotype data. Analogously, when applying this protocol to any species for which a complete or draft reference sequence is available, it will be possible to introduce a correction factor that accounts for the predicted rate of RFLP.
Testing the serial founder model on resequencing data
According to the serial founder model, a source population is subsampled to generate founders that colonize a neighboring geographic region and the process is repeated for progressively more distant locations from the origin of migration. Because of the sampling associated with each colonization, genetic diversity is expected to decrease as a function of distance from the geographic location of the source population. Patterns of microsatellite and haplotype diversity in the Human Genome Diversity Project (HGDP) panel fit this expectation, with the strongest negative correlation being observed between diversity and distance from sub-Saharan Africa (Ramachandran et al. 2005; Li et al. 2008). Here, we report a similar pattern detected for the first time by using resequencing data.
A previous study assumed that the origin of the Out of Africa expansion was Addis Ababa in Ethiopia (Ramachandran et al. 2005); however, they observed the strongest correlation between microsatellite diversity and distance from a location in central-western Africa. It is to be noted that the HGDP panel does not include an Ethiopian population sample; therefore, this result may be explained by incomplete sampling of genetic diversity within Africa. Two more recent studies (Tishkoff et al. 2009; Henn et al. 2011) identified south-western Africa as the origin of human migration and the region close to the midpoint of the Red Sea as the most likely waypoint for the Out of Africa. In agreement with these studies, when we considered all possible locations in Africa over a grid of geographic coordinates, we identified southern Africa as the location with the strongest correlation with genetic diversity. Because the current location of the founder population may be different from the location of the ancestral population, inferences about the geographic origin of the migration based on this approach should be interpreted with caution.
Reconstructing past demographic scenarios
The application of the reduced representation approach to human populations indicates that it is a useful method for generating informative data to make inferences about population history. Even though we only sampled genetic variation in one individual per population, we gathered enough information across loci to be able to infer a population tree that is consistent with a large body of genetic variation data (Cavalli-Sforza and Feldman 2003; Li et al. 2008). Understanding the mode and timing of the dispersal of natural populations over a geographic area may help interpret the interplay between genetic and environmental variation and may contribute to understanding the mechanism of speciation. In human populations, understanding the mode and timing of dispersal across the globe has implications for anthropological, medical, and epidemiological studies. Our split time estimate for the migration from the ancestral African population is consistent with both archeological and molecular data (Stringer and Andrews 1988; Quintana-Murci et al. 1999; Underhill et al. 2000; Macaulay et al. 2005; Mellars 2006a,b; Fagundes et al. 2007; Laval et al. 2010). These estimates assume a single migration event. However, the archeological record supports an early presence of modern humans in Australia (probably as early as 60,000 yr ago), suggesting that an independent migratory flux out of Africa reached Oceania (Roberts et al. 1990; O'Connell and Allen 2004; Balme et al. 2009; Oppenheimer 2009). Interestingly, both Native Australians and the Nasioi have three out of the four oldest divergence times from the African ancestral population (Table 4); under the assumption of an independent migration out of Africa into Oceania, these divergence times provide a time frame for this migration based on genetic data. Moreover, under the assumption of a single out of Africa migration followed by a split between the western Eurasians and east Asians, the Oceanic, east Asian, and Native American samples are expected to be equally divergent from the Europeans. However, the Oceanic samples are more divergent from European samples (0.920 × 10−3 on average) than the East Asian (0.866 × 10−3 on average) or Native American samples (0.848 × 10−3 on average) (Supplemental Fig. 2). Although this is a qualitative observation, it is consistent with an independent migration into Oceania.
Estimates of split time from the African founder population and bottleneck severity
The concordance of our time estimate with current models of human dispersal suggests that the data collected using our reduced representation approach is suitable to make broad inferences about population history in humans and other species. It should be noted that because our method assumes no gene flow, we probably underestimate the true split times. Additionally, our split time estimates are associated with large confidence intervals. Although this is a common problem when using genetic data to estimate the time of historical events, in the case of our study, this problem is aggravated by the fact that divergence was calculated by using a smaller number of sites, that is, sites with coverage greater than 20× and that overlap across pairs of samples. In contrast, nucleotide diversity is estimated from a larger subset of sites, and it is, therefore, associated with less uncertainty. More precise estimates of divergence could be achieved by sequencing reduced representation targets to greater depth or by increasing the range of fragment sizes included in the target.
In conclusion, we developed an effective approach to generate second-generation sequencing data sets suitable for population genetic analyses. By applying this protocol to human samples, we have demonstrated that the results obtained in terms of nucleotide diversity, allele frequency spectrum, and population split time estimates are consistent with previously reported ones. Additionally, we have provided new insights into the timing of human dispersal into Oceania.
Methods
The samples
Genomic DNA aliquots from 19 males (Icelander, Basque, Portuguese, Greek, Druze, Gujarati [GIH], Han Chinese [CHB], Khmer, Nasioi, Native Australian, Maori, Ethiopian, Maasai [MKK], Mbuti Pygmy, Kung, Karitiana, Pima) and two females (Native Australian, Berber) were purchased from ECACC (Berber and Native Australians) and Coriell Cell repository; a genomic DNA aliquot of a Naukan Yupik male collected in the Chukchi peninsula was also included (Volodko et al. 2008). To minimize the probability of including inbred individuals, we chose only individuals sampled from the general population or unaffected individuals from families with autosomal dominant Mendelian diseases as reported in the Coriell Cell Repository. A map of the approximate geographic location for each sample's population is provided in Figure 1A. To assess whether the individuals included in the study are representative of the population they were selected from, we used the genotype calls obtained by sequencing for SNPs that overlap with SNPs genotyped in the HGDP panel or in the HapMap (see Supplementary Methods). All the samples, but the Maori, can be attributed to the population they were chosen to represent based on the proportion of the different components. The Maori sample appears to be admixed with individuals of European origin and therefore was excluded from all subsequent analyses (Supplemental Fig. 3).
Reduced representation protocol
To create a reduced representation of the human genome, we applied a protocol modified from the method of Van Tassell et al. (2008). We restriction digested 10 μg of genomic DNA with 120 U of Rsa I (NEB) overnight, followed by restriction digestion with 120 additional units for 3 h in a total volume of 300 μL. This enzyme was chosen because the sequence of the restriction site does not contain CpGs and because the cutting site is within the restriction site. We also performed in silico restriction digestion of the human genome to identify a size interval (70–75 bp) that contains about 51,000 fragments, which at the time of planning this project corresponded to a sequencing depth of about 50× on a single lane of Illumina GAII. The restriction digest also showed that no repetitive elements were predicted in the 50- to 100-bp size range.
We performed quantitative real-time PCR, targeting a restriction site in each DNA sample before and after the digestion to test whether each sample was digested to completion. Samples with a ratio of nondigested to digested DNA >0.2% were discarded and the restriction digestion was repeated. Restriction fragments were separated on a 20% polyacrilamide gel run at 100 V for 35 h. The gels were stained in a SYBR Gold (Invitrogen) solution, and fragments in the 70- to 75-bp range were excised on a blue light transilluminator. A O'RangeRuler 5-bp DNA ladder was used as a size marker. We then followed the protocol by Van Tassell et al. (2008) to isolate and purify the DNA fragments from the gel.
The Illumina library preparation and sequencing were performed at the High-Throughput Genome Analysis Core at the Institute for Genomics and Systems Biology (University of Chicago) for all samples except the Khmer, Kung, Mbuti Pygmy, and Karitiana, which were processed at the National Center for Genome Resources. In all cases, the Illumina protocol to prepare libraries for ChIP sequencing was used. Each sample was run on one or two lanes of the Illumina GAII in order to obtain a minimum of ∼100 Mb of sequence per sample.
Read alignment
A reference genome library for alignment was created, which included ∼9.3 × 106 36-bp sequences taken from the 5′ and 3′ ends of the DNA fragments created by in silico RsaI digestion of the complete human reference genome (NCBI36/hg18). Before alignment, this library was masked by making all pairwise comparisons of 36-mer sequences, in both forward and reverse directions, and then removing all sequences that had four or fewer mismatches with at least one other sequence in the library. Masking the reference library in this manner guarantees the unique alignment of reads that align with two or fewer mismatches. The 36-bp reads were then mapped against this masked reference library using in-house software. Reads that failed to map with two or fewer mismatches were discarded.
Base calling and filtering
Before genotype calling, we used both read position and the sequencing base quality to filter read bases. Bases at read positions 1 and 2 were removed from analysis because these positions fall within the RsaI restriction site and should almost always be an A or C, respectively, on the forward read strand. Bases at read position 3 were also removed because it was found that the base composition of these sites was strongly influenced by the presence of the neighboring C base in the restriction site due to the higher transition mutation rates of CpG sites. Read bases with a phred base quality score less than 20 were also discarded. We chose this base quality threshold based on the observed sequencing error rates found for reads mapping to the nonpseudo-autosomal region of the X chromosome of male samples. For sites at which more than two different alleles were found among the read bases passing position and quality filters, we kept only the read bases with the two alleles having greatest sequencing depth.
Identification of genic regions in the human genome
We identified genic regions based on the annotation in the UCSC human genome assembly NCBI36/hg18. To compare the fraction of genic regions represented in our data to the same proportion for the entire genome, 10 kb of flanking sequence was included on both sides of each gene. In all other analyses, no flanking region was added to each genic region.
Estimating the recombination rate in the surveyed restriction fragments
To estimate the recombination rate in the surveyed restriction fragments, we used the genetic distance (r) between the closest HapMap sites on either sides of each restriction fragment (http://hapmap.ncbi.nlm.nih.gov/). The recombination rate ρ = 4Nr, was estimated assuming N = 10,000.
Genotype calling
For each sample, we created a matrix A = [aij ], where matrix element aij represents the count of autosomal sites having sequencing depth i (1 ≤ i ≤ 100) for both alleles and sequencing depth j (0 ≤ j ≤ i/2) for the allele with lower coverage (Supplemental Fig. 1). Thus, the counts across each matrix row represent the sequencing depth frequency distribution for the allele of lower coverage for sites having a total sequencing depth given by that row. Above some minimum total coverage i, these distributions are typically bimodal, with the mass of the distribution centered near j = 0 representing the sequencing depth distribution for the error base at putative homozygous sites, and the mass centered near j = i/2 representing the sequencing depth distribution for the lower coverage allele at putative heterozygous sites. For each row i, we find the smallest j for which aij = 0 or 1 and let this cell define the boundary between regions where genotypes can be called unambiguously, namely, cells to the left of this cell are taken to be counts of true homozygous sites, while cells to the right are taken to be true heterozygous sites. We then call each site's genotype depending on where in the matrix it falls, removing from analysis all sites that have a total sequencing depth i for which this mass separation does not occur (typically at low coverage). Additionally, to reject spurious heterozygous calls made with the matrix (typically occurring at high total coverage) that are more likely the result of sequencing error, we assume P = 0.5 as the probability for sampling either allele at heterozygous sites and apply a binomial filter with the low critical value of 1 × 10−10.
Calculating a correction factor
In this section, we calculate the expected fraction of “called” sites that are called heterozygous. This fraction will be less than the actual fraction of sites that are heterozygous because of restriction site polymorphisms that result in allele drop out. Drop out occurs when one of the alleles at a polymorphic site is part of a restriction fragment of the appropriate size, but the other allele is not, and hence we only observe one allele. The “expected fraction of sites called heterozygous” can be thought of as a conditional probability associated with a random focal site,
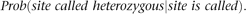
The conditioning event in this case, “site is called,” is the event that a site is located in a restriction fragment of the right size, and the coverage of the site is larger than a threshold value (t), so that a reliable genotype call can be made. The other event, “site called heterozygous,” means that the site is called but also that both alleles must be located in a restriction fragment of the right size, namely, that there is no allelic drop out. Thus we aim to calculate
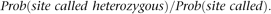
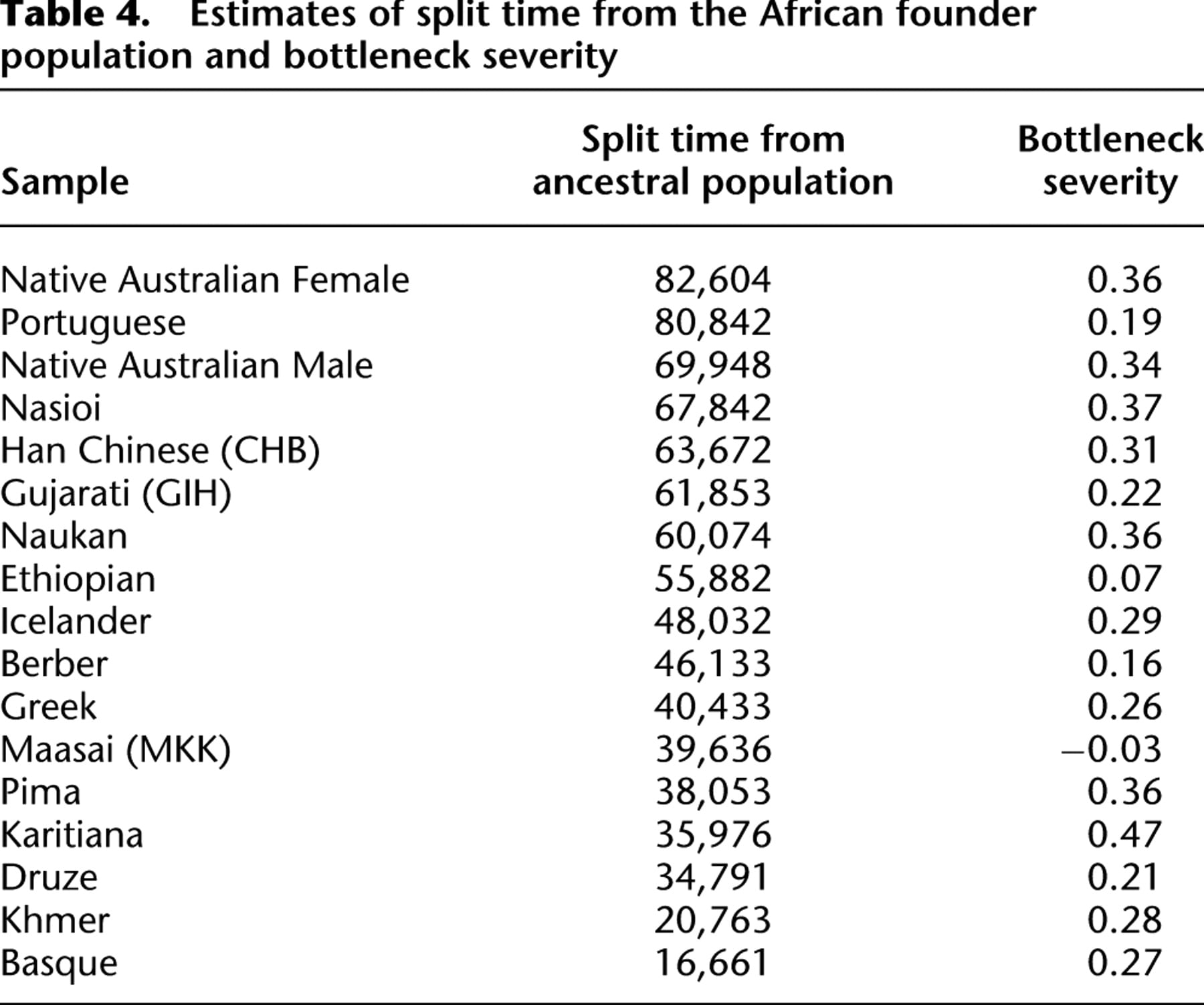
In this section, we ignore sequencing error, and thus the numerator is the probability that a focal nucleotide site is heterozygous and that there are nonheterozygous restriction sites at appropriate positions to generate fragments of the right size and that coverage is larger than t. Following the derivations provided in the Supplementary Methods, we obtain the following:
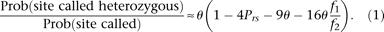
Where the symbol θ is the scaled per base pair neutral mutation rate (4Neμ), Prs is the probability of a restriction site at any position, f2 is the probability a site without dropout has coverage greater than t, and f1 is the same probability for a site with dropout. The quantity f1/f2 is estimated by
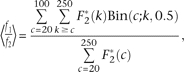
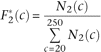
The expected fraction in Equation 1 can then be used to correct our observed fraction of sites, which appear to be heterozygous, to obtain a less biased, “corrected π”:
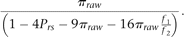
The theoretical expectation is that the raw π is ∼3.4% lower than the true π. Complete derivations for the above formulas are provided in Supplementary Methods.
Estimating nucleotide diversity by a maximum likelihood method
To estimate the fraction of sites with minimum coverage of 10 that are heterozygous in an individual (p), we used a maximum likelihood method that assumes that each site is independent. With linkage, this is not strictly true but may give a good approximation. We model the probability of configurations when a site is homozygous by a binomial distribution with an error rate parameter. When the site is heterozygous, we assume the configuration is binomially distributed. The different frequencies of nucleotide pairs when sites are heterozygous, as opposed to when they are homozygous with sequencing errors, are incorporated in the estimate. Similarly, our estimated correction factor is incorporated in the estimation method.
We assume the probability of an observed configuration (iA1, jA2), meaning i copies of the A1 allele and j copies of A2 is
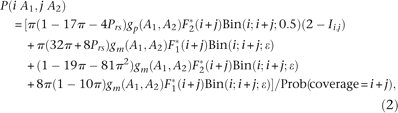
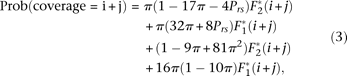
Estimating sequence divergence
For each pair of samples, we identified sites with called genotypes in both samples and calculated the proportion of genotypes falling in the following three categories: fixed differences, defined as sites homozygous for alternative alleles in the two samples; shared differences, defined as sites heterozygous in one or both samples; and invariant sites, defined as sites homozygous for the same allele in both samples. Raw pairwise divergence was then calculated as follows: proportion of fixed differences + (proportion of shared differences × 0.5). For each population pair, the raw divergence estimate was multiplied by the average of the correction factors estimated for each population in the pair.
Estimating population split times
Population split times were estimated assuming a serial founder model, where each population originated by splitting from an ancestral population. We used a neutral infinite-sites model and assumed that population size is constant (N0) throughout except during bottlenecks that occur immediately after population splits. That is, each population split is accompanied by a bottleneck in which one descendant population experiences a short duration bottleneck, with a subsequent instantaneous recovery of the ancestral population size. Gene flow between populations is assumed not to occur. In this model, the parameters to estimate are θ, the split time t (in units of 2N0 generations), and the bottleneck parameter F. The bottleneck parameter is the fractional reduction in heterozygosity during the bottleneck and can be thought of as the probability that two lineages sampled just after a bottleneck have a most recent common ancestor during the bottleneck.
Under our model, the ancestral population (population 1) has never experienced a bottleneck, and we can estimate θ by π1, the nucleotide diversity observed in population 1. This nucleotide diversity can also be considered an estimate of the nucleotide diversity in the ancestral population at time t, which is denoted πA1. We estimate πA1 by the average of the nucleotide diversity estimates obtained for the Kung and the Pygmy samples (African founder population; π1). Under our model, the expected divergence between population 1 and any of the other populations is

Replacing θ by π1 and E(D1,j) by the average divergence of the African founder for the remaining populations, we obtain the following estimator of t:

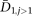 is the average divergence calculated between the African founder and each of the remaining populations. The split time estimate is in 2N0 units; to convert this estimate in years, we assumed a generation time of 25 yr and N0 to be equal to 10,000. Confidence intervals were estimated from the distribution of split times between the African founder and each of the remaining populations. However, because individual split times are not independent from each other, actual confidence intervals may be wider than we estimate. Simulations, which included dropout, showed that our estimates of split times have a small bias (∼10%; see Supplementary Methods).
is the average divergence calculated between the African founder and each of the remaining populations. The split time estimate is in 2N0 units; to convert this estimate in years, we assumed a generation time of 25 yr and N0 to be equal to 10,000. Confidence intervals were estimated from the distribution of split times between the African founder and each of the remaining populations. However, because individual split times are not independent from each other, actual confidence intervals may be wider than we estimate. Simulations, which included dropout, showed that our estimates of split times have a small bias (∼10%; see Supplementary Methods).Under our model, the expected nucleotide diversity for population 2 (π2) is related to θ, F, and t as follows:
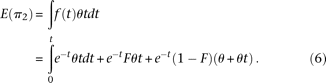
Solving for F, replacing θ by π1, and replacing E(π2) with the observed value of π2 we find the following estimator of F:

Acknowledgments
We thank J. Pritchard, G. Coop, M. Hubisz, J. Shendure, and T. Smith for helpful discussions and preliminary analyses in the beginning of the project. We thank F.G. Sperone for constructing GIS maps and calculating great circle distances. We thank M. Przeworski and J. Wall for valuable comments on an earlier version of this manuscript. We also thank two anonymous reviewers for their helpful comments. This work was supported in part by NIH grants DK56670, GM79558, and GM79558-S1. F.L. was partially supported by a Blanceflor-Foundation Grant for postgraduate studies and by an AHA postdoctoral fellowship (0825792G).
References
- ↵MA Abdulla, I Ahmed, A Assawamakin, J Bhak, SK Brahmachari, GC Calacal, A Chaurasia, CH Chen, J Chen, YT Chen, . 2009. Mapping human genetic diversity in Asia. Science 326: 1541–1545.
- ↵SM Ahn, TH Kim, S Lee, D Kim, H Ghang, DS Kim, BC Kim, SY Kim, WY Kim, C Kim, . 2009. The first Korean genome sequence and analysis: full genome sequencing for a socio-ethnic group. Genome Res 19: 1622–1629.
- ↵JM Akey, MA Eberle, MJ Rieder, CS Carlson, MD Shriver, DA Nickerson, L Kruglyak. 2004. Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol 2: e286. doi: 10.1371/journal.pbio.0020286.
- ↵TJ Albert, MN Molla, DM Muzny, L Nazareth, D Wheeler, X Song, TA Richmond, CM Middle, MJ Rodesch, CJ Packard, . 2007. Direct selection of human genomic loci by microarray hybridization. Nat Methods 4: 903–905.
- ↵D Altshuler, VJ Pollara, CR Cowles, WJ Van Etten, J Baldwin, L Linton, ES Lander. 2000. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature 407: 513–516.
- ↵NA Baird, PD Etter, TS Atwood, MC Currey, AL Shiver, ZA Lewis, EU Selker, WA Cresko, EA Johnson. 2008. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 3: e3376. doi: 10.1371/journal.pone.0003376.
- ↵J Balme, I Davidson, J McDonald, N Stern, P Veth. 2009. Symbolic behaviour and the peopling of the southern arc route to Australia. Quat Int 202: 59–68.
- ↵DR Bentley, S Balasubramanian, HP Swerdlow, GP Smith, J Milton, CG Brown, KP Hall, DJ Evers, CL Barnes, HR Bignell, .. 2008. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456: 53–59.
- ↵LL Cavalli-Sforza, MW Feldman. 2003. The application of molecular genetic approaches to the study of human evolution. Nat Genet 33: 266–275.
- ↵B Daines, H Wang, Y Li, Y Han, R Gibbs, R Chen. 2009. High-throughput multiplex sequencing to discover copy number variants in Drosophila. Genetics 182: 935–941.
- ↵JF Degner, JC Marioni, AA Pai, JK Pickrell, E Nkadori, Y Gilad, JK Pritchard. 2009. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics 25: 3207–3212.
- ↵SW Doniger, HS Kim, D Swain, D Corcuera, M Williams, SP Yang, JC Fay. 2008. A catalog of neutral and deleterious polymorphism in yeast. PLoS Genet 4: e1000183. doi: 10.1371/journal.pgen.1000183.
- ↵NJ Fagundes, N Ray, M Beaumont, S Neuenschwander, FM Salzano, SL Bonatto, L Excoffier. 2007. Statistical evaluation of alternative models of human evolution. Proc Natl Acad Sci 104: 17614–17619.
- ↵J Felsenstein. 2006. Accuracy of coalescent likelihood estimates: do we need more sites, more sequences, or more loci? Mol Biol Evol 23: 691–700.
- ↵RE Green, J Krause, AW Briggs, T Maricic, U Stenzel, M Kircher, N Patterson, H Li, W Zhai, MH Fritz, . 2010. A draft sequence of the Neandertal genome. Science 328: 710–722.
- ↵MF Hammer, AE Woerner, FL Mendez, JC Watkins, MP Cox, JD Wall. 2010. The ratio of human X chromosome to autosome diversity is positively correlated with genetic distance from genes. Nat Genet 42: 830–831.
- ↵I Hellmann, Y Mang, Z Gu, P Li, FM de la Vega, AG Clark, R Nielsen. 2008. Population genetic analysis of shotgun assemblies of genomic sequences from multiple individuals. Genome Res 18: 1020–1029.
- ↵BM Henn, CR Gignoux, M Jobin, JM Granka, JM Macpherson, JM Kidd, L Rodriguez-Botigue, S Ramachandran, L Hon, A Brisbin, . 2011. Feature article: Hunter-gatherer genomic diversity suggests a southern African origin for modern humans. Proc Natl Acad Sci 108: 5154–5162.
- ↵LW Hillier, V Reinke, P Green, M Hirst, MA Marra, RH Waterston. 2009. Massively parallel sequencing of the polyadenylated transcriptome of C. elegans. Genome Res 19: 657–666.
- ↵PL Johnson, M Slatkin. 2006. Inference of population genetic parameters in metagenomics: a clean look at messy data. Genome Res 16: 1320–1327.
- ↵PL Johnson, M Slatkin. 2008. Accounting for bias from sequencing error in population genetic estimates. Mol Biol Evol 25: 199–206.
- ↵A Keinan, JC Mullikin, N Patterson, D Reich. 2007. Measurement of the human allele frequency spectrum demonstrates greater genetic drift in East Asians than in Europeans. Nat Genet 39: 1251–1255.
- ↵ES Lander, LM Linton, B Birren, C Nusbaum, MC Zody, J Baldwin, K Devon, K Dewar, M Doyle, W FitzHugh, . 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921.
- ↵G Laval, E Patin, LB Barreiro, L Quintana-Murci. 2010. Formulating a historical and demographic model of recent human evolution based on resequencing data from noncoding regions. PLoS ONE 5: e10284. doi: 10.1371/journal.pone.0010284.
- ↵JZ Li, DM Absher, H Tang, AM Southwick, AM Casto, S Ramachandran, HM Cann, GS Barsh, M Feldman, LL Cavalli-Sforza, . 2008. Worldwide human relationships inferred from genome-wide patterns of variation. Science 319: 1100–1104.
- ↵M Lynch. 2008. Estimation of nucleotide diversity, disequilibrium coefficients, and mutation rates from high-coverage genome-sequencing projects. Mol Biol Evol 25: 2409–2419.
- ↵V Macaulay, C Hill, A Achilli, C Rengo, D Clarke, W Meehan, J Blackburn, O Semino, R Scozzari, F Cruciani, . 2005. Single, rapid coastal settlement of Asia revealed by analysis of complete mitochondrial genomes. Science 308: 1034–1036.
- ↵GT Marth, E Czabarka, J Murvai, ST Sherry. 2004. The allele frequency spectrum in genome-wide human variation data reveals signals of differential demographic history in three large world populations. Genetics 166: 351–372.
- ↵KJ McKernan, HE Peckham, GL Costa, SF McLaughlin, Y Fu, EF Tsung, CR Clouser, C Duncan, JK Ichikawa, CC Lee, . 2009. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res 19: 1527–1541.
- ↵G McVicker, D Gordon, C Davis, P Green. 2009. Widespread genomic signatures of natural selection in hominid evolution. PLoS Genet 5: e1000471. doi: 10.1371/journal.pgen.1000471.
- ↵P Mellars. 2006a. Going east: new genetic and archaeological perspectives on the modern human colonization of Eurasia. Science 313: 796–800.
- ↵P Mellars. 2006b. Why did modern human populations disperse from Africa ca. 60,000 years ago? A new model. Proc Natl Acad Sci 103: 9381–9386.
- ↵JF O'Connell, J Allen. 2004. Dating the colonization of Sahul (Pleistocene Australia-New Guinea): A review of recent research. J Archaeol Sci 31: 835–853.
- ↵DT Okou, KM Steinberg, C Middle, DJ Cutler, TJ Albert, ME Zwick. 2007. Microarray-based genomic selection for high-throughput resequencing. Nat Methods 4: 907–909.
- ↵S Oppenheimer. 2009. The great arc of dispersal of modern humans: Africa to Australia. Quat Int 202: 2–13.
- ↵S Ossowski, K Schneeberger, RM Clark, C Lanz, N Warthmann, D Weigel. 2008. Sequencing of natural strains of Arabidopsis thaliana with short reads. Genome Res 18: 2024–2033.
- ↵A Pluzhnikov, P Donnelly. 1996. Optimal sequencing strategies for surveying molecular genetic diversity. Genetics 144: 1247–1262.
- ↵JE Pool, I Hellmann, JD Jensen, R Nielsen. 2010. Population genetic inference from genomic sequence variation. Genome Res 20: 291–300.
- ↵D Pushkarev, NF Neff, SR Quake. 2009. Single-molecule sequencing of an individual human genome. Nat Biotechnol 27: 847–852.
- ↵L Quintana-Murci, O Semino, HJ Bandelt, G Passarino, K McElreavey, AS Santachiara-Benerecetti. 1999. Genetic evidence of an early exit of Homo sapiens sapiens from Africa through eastern Africa. Nat Genet 23: 437–441.
- ↵S Ramachandran, O Deshpande, CC Roseman, NA Rosenberg, MW Feldman, LL Cavalli-Sforza. 2005. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc Natl Acad Sci 102: 15942–15947.
- ↵RG Roberts, M Jones, MA Smith. 1990. Thermoluminescence dating of a 50,000-year-old human occupation site in northern Australia. Nature 345: 153–156.
- ↵A Rokas, P Abbot. 2009. Harnessing genomics for evolutionary insights. Trends Ecol Evol 24: 192–200.
- ↵SC Schuster, W Miller, A Ratan, LP Tomsho, B Giardine, LR Kasson, RS Harris, DC Petersen, F Zhao, J Qi, . 2010. Complete Khoisan and Bantu genomes from southern Africa. Nature 463: 943–947.
- ↵CB Stringer, P Andrews. 1988. Genetic and fossil evidence for the origin of modern humans. Science 239: 1263–1268.
- ↵JX Sun, JC Mullikin, N Patterson, DE Reich. 2009. Microsatellites are molecular clocks that support accurate inferences about history. Mol Biol Evol 26: 1017–1027.
- ↵F Tajima. 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595.
- ↵K Tamura, J Dudley, M Nei, S Kumar. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24: 1596–1599.
- ↵SA Tishkoff, FA Reed, FR Friedlaender, C Ehret, A Ranciaro, A Froment, JB Hirbo, AA Awomoyi, JM Bodo, O Doumbo, . 2009. The genetic structure and history of Africans and African Americans. Science 324: 1035–1044.
- ↵PA Underhill, P Shen, AA Lin, L Jin, G Passarino, WH Yang, E Kauffman, B Bonne-Tamir, J Bertranpetit, P Francalacci, . 2000. Y chromosome sequence variation and the history of human populations. Nat Genet 26: 358–361.
- ↵CP Van Tassell, TP Smith, LK Matukumalli, JF Taylor, RD Schnabel, CT Lawley, CD Haudenschild, SS Moore, WC Warren, TS Sonstegard. 2008. SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat Methods 5: 247–252.
- ↵BF Voight, AM Adams, LA Frisse, Y Qian, RR Hudson, A Di Rienzo. 2005. Interrogating multiple aspects of variation in a full resequencing data set to infer human population size changes. Proc Natl Acad Sci 102: 18508–18513.
- ↵NV Volodko, EB Starikovskaya, IO Mazunin, NP Eltsov, PV Naidenko, DC Wallace, RI Sukernik. 2008. Mitochondrial genome diversity in arctic Siberians, with particular reference to the evolutionary history of Beringia and Pleistocenic peopling of the Americas. Am J Hum Genet 82: 1084–1100.
- ↵JD Wall, MP Cox, FL Mendez, A Woerner, T Severson, MF Hammer. 2008. A novel DNA sequence database for analyzing human demographic history. Genome Res 18: 1354–1361.
- ↵DA Wheeler, M Srinivasan, M Egholm, Y Shen, L Chen, A McGuire, W He, YJ Chen, V Makhijani, GT Roth, . 2008. The complete genome of an individual by massively parallel DNA sequencing. Nature 452: 872–876.
- ↵A Yu, C Zhao, Y Fan, W Jang, AJ Mungall, P Deloukas, A Olsen, NA Doggett, N Ghebranious, KW Broman, . 2001. Comparison of human genetic and sequence-based physical maps. Nature 409: 951–953.
Notes
[1] [Supplemental material is available for this article. The sequencing data from this study have been submitted to the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi) under accession no. SRA030837.]
[2] Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.119792.110.