
OPA1 links human mitochondrial genome maintenance to mtDNA replication and distribution
- Ghizlane Elachouri1,10,
- Sara Vidoni1,2,10,
- Claudia Zanna2,
- Alexandre Pattyn1,
- Hassan Boukhaddaoui1,
- Karen Gaget1,
- Patrick Yu-Wai-Man3,
- Giuseppe Gasparre4,
- Emmanuelle Sarzi5,
- Cécile Delettre1,
- Aurélien Olichon6,
- Dominique Loiseau7,
- Pascal Reynier7,
- Patrick F. Chinnery3,
- Agnès Rotig5,
- Valerio Carelli8,
- Christian P. Hamel1,9,
- Michela Rugolo2 and
- Guy Lenaers1,11
- 1 INSERM U-583, Institut des Neurosciences de Montpellier, Montpellier, France and Université de Montpellier I et II, Montpellier 34091, France;
- 2 Dipartimento di Biologia Evoluzionistica Sperimentale, Università di Bologna, Bologna 40126, Italy;
- 3 Mitochondrial Research Group, Institute of Ageing and Health, The Medical School, Newcastle University, Newcastle upon Tyne NE1 3BZ, United Kingdom;
- 4 Dipartimento di Biochimica e Biologia Molecolare, E. Quagliariello, Università di Bari, Bari 70126, Italy;
- 5 INSERM U-781, Hôpital Necker-Enfants Malades, Paris 75015, France;
- 6 INSERM U-563, Institut Claudius Regaud, Toulouse 31052, France;
- 7 INSERM U771, CHU d'Angers, Angers 49933, France;
- 8 Dipartimento di Scienze Neurologiche, Università di Bologna, Bologna 40123, Italy;
- 9 CHRU Montpellier, Centre of Reference for Genetic Sensory Diseases, Montpellier 34295, France
-
↵10 These authors contributed equally to this work.
Abstract
Eukaryotic cells harbor a small multiploid mitochondrial genome, organized in nucleoids spread within the mitochondrial network. Maintenance and distribution of mitochondrial DNA (mtDNA) are essential for energy metabolism, mitochondrial lineage in primordial germ cells, and to prevent mtDNA instability, which leads to many debilitating human diseases. Mounting evidence suggests that the actors of the mitochondrial network dynamics, among which is the intramitochondrial dynamin OPA1, might be involved in these processes. Here, using siRNAs specific to OPA1 alternate spliced exons, we evidenced that silencing of the OPA1 variants including exon 4b leads to mtDNA depletion, secondary to inhibition of mtDNA replication, and to marked alteration of mtDNA distribution in nucleoid and nucleoid distribution throughout the mitochondrial network. We demonstrate that a small hydrophobic 10-kDa peptide generated by cleavage of the OPA1-exon4b isoform is responsible for this process and show that this peptide is embedded in the inner membrane and colocalizes and coimmunoprecipitates with nucleoid components. We propose a novel synthetic model in which a peptide, including two trans-membrane domains derived from the N terminus of the OPA1-exon4b isoform in vertebrates or from its ortholog in lower eukaryotes, might contribute to nucleoid attachment to the inner mitochondrial membrane and promotes mtDNA replication and distribution. Thus, this study places OPA1 as a direct actor in the maintenance of mitochondrial genome integrity.
For the last 2 billion yr, eukaryote cells have concomitantly maintained two genomes: one large and diploid located in the nucleus, and one composed of hundreds/thousands copies of a small circular DNA, organized in discrete structures called nucleoids, evenly distributed within the mitochondrial network. Maintenance, organization, and distribution of mitochondrial genomes are crucial to comply with the energetic requirements that have to be fulfilled along cell volumes (Malka et al. 2006; Holt et al. 2007), in particular, in cells with large cytoplasmic extensions like neurons (Chen and Chan 2006). Consequently, many perturbations of mitochondrial genome integrity lead to severe, debilitating human diseases, which in most cases affect highly energy-dependent organs such as skeletal muscles or the peripheral and central nervous systems (Ashley et al. 2007; Sarzi et al. 2007; Copeland 2008; Spinazzola and Zeviani 2009).
The mitochondrial nucleoids include six to 10 copies of the mitochondrial DNA (mtDNA), which are packaged with a growing list of nucleoid related proteins that are organized in layered structures (Holt et al. 2007; Bogenhagen et al. 2008). Nucleoids are assumed to be anchored to the inner mitochondrial membrane (IMM) on the matrix side (Nass 1969; Albring et al. 1977). They form autonomous replication units that segregate independently in the mitochondrial network, allowing for functional complementation (Capaldi et al. 2002; Gilkerson et al. 2008), but without evidence of genetic material exchange (D'Aurelio et al. 2004). Although crucial for understanding cell spatial energetic metabolism and mtDNA inheritance in mitochondrial diseases, the molecular mechanisms responsible for these aspects remain totally unknown.
Beyond the obvious involvement in mtDNA maintenance of the complex machineries required for mtDNA replication and for nucleoid packaging, there is now mounting evidence suggesting that proteins acting on mitochondrial membrane dynamics are also required for this process. First, in yeasts, the MGM1 gene (mitochondrial genome maintenance) in Saccharomyces cerevisiae and the msp1 gene in Schizosaccharomyces pombe, which are the orthologs of the human OPA1 gene (Delettre et al. 2000; Lenaers et al. 2002), encode an intermembrane mitochondrial dynamin involved in both mitochondrial membrane dynamics and genome maintenance (Jones and Fangman 1992; Guan et al. 1993; Pelloquin et al. 1999; Wong et al. 2003; Meeusen et al. 2006; Diot et al. 2009). When mutated or deleted, MGM1 and msp1 cells lose their mtDNA and must rely on anaerobic growth to proliferate, which is only accessible to S. cerevisiae (Jones and Fangman 1992; Guan et al. 1993; Pelloquin et al. 1998). Second, the mammalian DRP1 and MFN large GTPases, acting respectively on the fission and the fusion of the outer mitochondrial membrane (OMM), were also described as indirect contributors to mtDNA integrity and distribution within the mitochondrial network, with particular emphasis in neuronal (Chen et al. 2007; Parone et al. 2008) and muscular cells (Chen et al. 2010). Finally, in a multicentric clinical study, we reported that specific OPA1 mutations, responsible for syndromic Dominant Optic Atrophy, induce the accumulation of mtDNA deletions in skeletal muscle of patients, thus supporting again the possible involvement of mitochondrial dynamics in mtDNA maintenance (Amati-Bonneau et al. 2008; Hudson et al. 2008). Altogether these preliminary observations suggested that altering mitochondrial dynamics can affect the maintenance of mtDNA integrity.
In the light of these data and given that OPA1 localization and functions are associated to the inner mitochondrial membrane, the same site where mtDNA-containing nucleoids are believed to be anchored, we investigated the role of the different OPA1 variants in the maintenance of mtDNA integrity and found evidence that a specific OPA1 isoform binds the nucleoids to the inner membrane, a process that we further show to be mandatory for mtDNA replication and distribution.
Results
mtDNA maintenance requires OPA1-exon4b
In previous studies, we evidenced that the OPA1 gene encodes eight isoforms resulting from the alternate splicing of exons 4, 4b, and 5b and that the presence of these exons markedly specifies OPA1 functions either in membrane fusion or in apoptosis (Delettre et al. 2001; Olichon et al. 2007). To assess the possible role of OPA1 isoforms in mtDNA maintenance, we transfected HeLa cells with exons 4, 4b, and 5b specific siRNAs. OPA1 protein levels analyzed by Western blot after 72 h (Fig. 1A) were markedly reduced by using the OPA1-global and exon 4 siRNAs, and only weakly decreased by exon 4b and 5b silencing, in agreement with the respective abundances of each alternate exon in OPA1 transcripts (Olichon et al. 2007). In these cells, mtDNA measurements by quantitative PCR revealed a mild increase in mtDNA copy number compared to control cells, except in those treated with the exon 4b siRNA, which showed a significant 70% decrease of mtDNA copy number, a result confirmed by Southern blot (Fig. 1A). Time-course analysis of mtDNA and OPA1-exon4b abundances in exon 4b–silenced cells showed a rapid and persistent reduction of OPA1-exon4b mRNAs, whereas mtDNA copy number first slightly increased at 24 h, then mildly decreased at 48 h (20%), to finally reach a marked reduction at 72 h (70%) and 96 h (75%) (Fig. 1B), thus revealing a substantial delay between exon 4b silencing and mtDNA loss. Southern blotting confirmed this kinetic and further evidenced the absence of mtDNA degradation or deletion during mtDNA depletion (Fig. 1B). From these results we concluded that mitochondrial genome maintenance in human cells involves OPA1-exon4b isoforms.
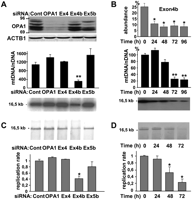
Mitochondrial genome maintenance and replication require OPA1-exon4b. (A) Western blot using OPA1 and ACTB1 antibodies of protein extracts (top), and evaluation by Q-PCR (middle) and Southern blot (bottom) of mtDNA copy number from control HeLa cells (Cont) or cells transfected for 72 h with siRNAs targeting all OPA1 mRNA (OPA1), or those including exon 4 (Ex4), 4b (Ex4b), or 5b (Ex5b), show a specific mtDNA loss in cells silenced for OPA1-exon4b transcripts. Note that because OPA1 isoforms including the exon 4b domain account for <25% of all OPA1 isoforms, their silencing can barely be detected by Western blot. (n = 6, one-way ANOVA; [**] P-value inferior to 0.001). (B) Quantification of the relative abundances of OPA1 transcripts including exon 4b (top) and of mtDNA in HeLa cells transfected by the exon 4b siRNA during a 24-h period time course reveals a delay between OPA1-exon4b silencing and the mtDNA loss, without evidencing mtDNA degradation or deletions on the Southern blot (bottom). (n = 3, one-way ANOVA; P-value inferior to 0.01 [*] and 0.001 [**]). (C) Southern–Western blot using anti-BrdU antibodies on DNA extracts (top) and quantification of the replication rate (bottom) in control HeLa cells (Cont) or cells transfected for 48 h with the siRNAs targeting all OPA1 mRNA (OPA1) or those including exon 4 (Ex4), 4b (Ex4b), or 5b (Ex5b) shows a significant 60% decrease of BrdU incorporation in mtDNA, in cells silenced for OPA1-exon4b (n = 3, one-way ANOVA; [*] P-value inferior to 0.01). (D) Southern–Western blot of DNA extracts (top) and quantification of the replication rate (bottom) from HeLa cells transfected with the exon 4b siRNA for the indicated times illustrates a progressive inhibition of mtDNA replication starting readily after OPA1-exon4b silencing (n = 3, one-way ANOVA; [*] P-value inferior to 0.01).
mtDNA replication requires OPA1-exon4b
The absence of mtDNA degradation or deletions during mtDNA depletion in exon 4b–silenced cells led us to test whether mtDNA replication is affected in these cells. To examine this aspect, HeLa cells transfected with the different OPA1 siRNAs for 48 h were incubated for 2 h with aphidicolin, to inhibit nuclear DNA replication, then for 8 h with BrdU, to label neo-synthesized DNA. After total DNA extraction and digestion with the PvuII restriction enzyme, Southern–Western blots using anti-BrdU antibodies demonstrated in all samples a band at 16.5 kb corresponding to replicated mtDNA (Fig. 1C). Signal quantification revealed almost similar mtDNA replication rates in all conditions, except in exon 4b–silenced cells, in which this parameter was markedly reduced (Fig. 1C). Consequently, we evaluated mtDNA replication during the time course of exon 4b silencing. Southern–Western blots evidenced a progressive reduction of the signal corresponding to neosynthetized mtDNA, down to 80% of inhibition after 72 h (Fig. 1D), and the absence of degraded or intermediate replicative forms of mtDNA (Supplemental Fig. 1). Importantly, the inhibition of mtDNA replication was already apparent at 24 h, well before the initial mtDNA loss shown in Figure 1B, therefore suggesting that the inhibition of mtDNA replication can explain the mtDNA depletion in cells silenced for exon 4b.
OPA1-exon4b silencing disrupts mtDNA distribution in the mitochondrial network
We then evaluated how the residual mtDNA is distributed in the mitochondrial network using anti-DNA antibodies (Fig. 2) and the PicoGreen dye (Supplemental Fig. 2). In control HeLa cells, about 240 green cytoplasmic dots with a homogeneous fluorescence appear evenly distributed within the mitochondrial network (Fig. 2A). Conversely, 72 h after exon 4b silencing, although the structure of the mitochondrial network remained tubular as in control cells, we observed a striking variation in the amount and distribution of cytoplasmic anti-DNA immunofluorescence, revealing many regions devoid of fluorescence and some with intense dots (Fig. 2A; Supplemental Fig. 2). To infer statistical analyses, we evaluated the total cytoplasmic green fluorescence, the number of nucleoids per cell, and the fluorescence intensity of each nucleoid. Results showed a global 50% reduction of anti-DNA immunofluorescence, confirming the tendency observed by Q-PCR in previous experiments, and a marked decrease in the number of nucleoids per cell, from 243 ± 38 (n = 8) in control to 97 ± 19 (n = 8) in exon 4b–silenced cells (Fig. 2B). Furthermore, the distribution of nucleoid fluorescence, instead of forming a normal profile as in control cells, demonstrated two populations: one displaying fluorescence levels with a similar distribution as in control cells, and one with higher fluorescence levels, corresponding to clumpy nucleoids that are not present in control cells (Fig. 2C). Similar observations and counting were obtained using the PicoGreen dye (Supplemental Fig. 2), although the number of nucleoids was underestimated by this labeling procedure. Together, these data indicate that OPA1-exon4b is required to achieve the even distribution of mtDNA molecules within nucleoids and to distribute nucleoids within the mitochondrial network.
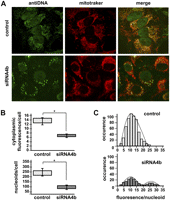
mtDNA and nucleoid distributions are dismantled by OPA1-exon4b silencing. (A) Fluorescence images using anti-DNA and Mitotracker of HeLa cells (control, top) and cells transfected with the exon 4b siRNA (siRNA4b, bottom) reveal a dramatic change in mtDNA distribution in cells silenced for exon 4b, whereas the mitochondrial network remains tubular as in control cells. (B) Quantifications of cytoplasmic anti-DNA fluorescence (top, relative units) and of the number of nucleoids per cell (bottom) in control (Cont) and exon 4b siRNA-transfected cells (si4b) disclose a decrease in mtDNA-associated fluorescence (50%) and in the number of nucleoids (60%), in exon 4b–silenced cells (n = 8; Student's t-test; [*] P-value inferior to 0.01). (C) Distribution of the anti-DNA fluorescence per nucleoid (relative units) in control (Control, top) and exon 4b siRNA-transfected (si4b, bottom) cells (n = 8) reveals a population with higher than normal fluorescence levels in exon 4b–silenced cells.
OPA1-exon4b N terminus peptide colocalizes with the mtDNA and rescues the silencing of exon 4b
In all cells so far tested, OPA1-exon4b isoforms are systematically cleaved in the peptide sequence following the exon 4b domain by YME1L, an inner membrane peptidase (Duvezin-Caubet et al. 2007; Griparic et al. 2007; Olichon et al. 2007; Song et al. 2007), thus generating two peptides: the large 84-kDa OPA1 C terminus including the dynamin domains (GTPase, central, and GTPase effector domain), which is common to all isoforms, and a 10-kDa N terminus sequence situated between the import cleavage site in the mitochondrial targeting sequence (amino acid 88) and a second cleavage site recognized by YME1L situated in the exon 5 domain (amino acid 175). Because this peptide, which we will hereafter call NT-OPA1-exon4b, includes the domain corresponding to exon 4b (see Fig. 6, below), we hypothesized that it represents the key element for the process described here. To address this question, we analyzed its colocalization with mtDNA, comparatively to that of the peptide including exon 4 instead of exon 4b (NT-OPA1-exon4), both being fused to the fluorescent Cherry protein. In all transfected cells, the red fluorescence appeared as regular dots closely localized with the mtDNA labeled with anti-DNA antibodies (Fig. 3A). Nevertheless, calculation of fluorescence superposition revealed a significant preferential colocalization of mitochondrial nucleoids with NT-OPA1-exon4b, compared with that of NT-OPA1-exon4 (Fig. 3A). Similar results were obtained using PicoGreen staining instead of anti-DNA antibodies (data not shown).
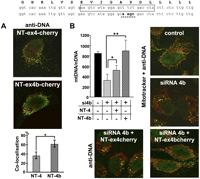
OPA1-exon4b N terminus colocalizes with mtDNA and rescues exon 4b silencing. The human exon 4b peptide sequence (top) stands above the corresponding wild-type and mutagenized exon 4b nucleotide sequences. The sequence targeted by exon 4b siRNA is boxed on the wild-type sequence, and the mutagenized sequence used in expression vectors to prevent cross-reaction with the siRNA 4b is underlined on the bottom sequence. (A) Merged Cherry and anti-DNA fluorescences from HeLa cells transfected by the plasmids expressing NT-OPA1-exon4-cherry (top) and NT-OPA1-exon4b-cherry (bottom) show the preferential colocalization of the mitochondrial nucleoid with the NT-OPA1-exon4b peptide. (Bottom) Quantification of the colocalization in both conditions (n = 12 cells, t-test; [*] P-value inferior to 0.01). (B, left) Quantification of mtDNA copy number from wild-type cells, cells transfected by the exon 4b siRNA (si4b), or cells transfected with exon 4b siRNA plus a plasmid expressing NT-OPA1-exon4 (si4b + NT-4) or NT-OPA1-exon4b (si4b + NT-4b) illustrates the full recovery of mtDNA abundance in cells transfected by exon 4b siRNA and expressing the NT-OPA1-exon4b peptide and partial recovery in those expressing the NT-OPA1-exon4 peptide. (n = 3, one-way ANOVA; P-value inferior to 0.01 [*] and 0.001 [**]). (Right) Merged fluorescence pictures using anti-DNA and Mitotracker, showing the mtDNA distribution in control and exon 4b–silenced HeLa cells. (Bottom) Merged fluorescences of antiDNA and Cherry protein in cells cotransfected by the exon 4b siRNA and the NT-OPA1-exon4 (left) and NT-OPA1-exon4b encoding plasmids, showing the rescue of mtDNA distribution in cells expressing NT-OPA1-exon4b.
We then checked if the expression of NT-OPA1-exon4b could rescue the effects of exon 4b silencing. To prevent a direct interaction between the exon 4b siRNA and the transcripts generated by the vector coding NT-OPA1-exon4b, we mutagenized four nucleotides in the exon 4b sequence from the vector (Fig. 3, top) and verified that the expression levels of NT-OPA1-exon4b and NT-OPA1-exon4 were equivalent in the presence of the exon 4b siRNA (data not shown). In cells silenced for exon 4b, we found a significant reduction of mtDNA abundance, whereas cotransfection with the plasmid encoding NT-OPA1-exon4b maintained a level of mtDNA abundance similar to that of untransfected cells, and cotransfection with the plasmid expressing NT-OPA1-exon4 peptide led to a partial rescue (Fig. 3B). We then evaluated the mtDNA distribution in these experimental conditions. Anti-DNA fluorescence observations disclosed only a slight alteration of mtDNA distribution with rare aggregated nucleoids in cells expressing NT-OPA1-exon4b, whereas dramatic changes with clumpy nucleoids were apparent in all other silenced conditions (Fig. 3B), as also observed with PicoGreen (data not shown). Together, these results indicate that the 10-kDa NT-OPA1-exon4b peptide is a key actor in the maintenance and distribution of mtDNA.
NT-OPA1-exon4b binds to the inner mitochondrial membrane and to the nucleoids
NT-OPA1-exon4b structure is composed of two transmembrane (TM) domains, one common to all OPA1 isoforms, and one encoded by exon 4b, plus a 35-amino-acid intermediate domain without signature motif. This peculiar structure together with the previous results suggest a scenario in which NT-OPA1-exon4b could be associated with the IMM and the nucleoids. To assess the mitochondrial localization of NT-OPA1-exon4b, we transfected HeLa cells with a plasmid expressing a C-terminal Flag-tagged version of NT-OPA1-exon4b and purified the mitochondrial fraction. After breaking down mitochondria by sonication, we observed that NT-OPA1-exon4b segregated in the membrane fraction, as did OPA1 isoforms. Incubation with 1 M NaCl did not solubilize NT-OPA1-exon4b or OPA1 isoforms from the pellet fraction, but incubation with 3% SDS did extract NT-OPA1-exon4b and OPA1 from the membranes (Fig. 4A, left). Thus, these data suggest that NT-OPA1-exon4b is tightly bound to the mitochondrial membrane fraction. We then examined the sub-mitochondrial localization of NT-OPA1-exon4b by a protease protection assay on permeated mitochondria. In intact mitochondria, the Flag-tagged peptide was undigested by trypsin treatment (Fig. 4A, right) as OPA1 and HSPD1 (HSP60), two inner mitochondrial proteins localized, respectively, in the intermembrane space and the matrix (Olichon et al. 2002). Only when the OMM was permeabilized by digitonin treatment was NT-OPA1-exon4b digested by trypsin along with OPA1 isoforms, whereas HSPD1 remained protected. Further addition of Triton X-100, which solubilizes the IMM, led to the complete digestion of HSPD1 (Fig. 4A, right). Thus, these results suggest that NT-OPA1-exon4b is embedded in the IMM, possibly with the Flag-tag C-terminal end localized in the intermembrane space.
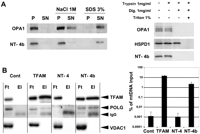
NT-OPA1-exon4b is embedded in the inner mitochondrial membrane and interacts with the nucleoid. (A, left) Western blot showing the presence of OPA1 and NT-OPA1-exon4b in the pellet (P) or supernatant (SN) fractions of mitochondria, either untreated, or treated with 1 M NaCl or 3% SDS. (Right) Western blot showing the presence or absence of OPA1, HSPD1, and NT-OPA1-exon4b in mitochondrial fractions treated successively with trypsin, digitonin, and triton. (B, left) Western blots using TFAM, POLG, and VDAC1 antibodies, performed on the flow-through (Ft) and eluted (El) fractions from immunoprecipitated mitochondrial proteins purified from mocked cells or cells transfected with plasmids expressing Flag-tagged TFAM, NT-OPA1-exon4, or NT-OPA1-exon4b, evidence the specific presence of TFAM and POLG but not VDAC1, in the eluted fractions of cells specifically expressing the TFAM and NT-OPA1-exon4b proteins. (Right) Measurements of mtDNA recovery, as the ratio between the amounts of mtDNA in the immunoprecipitates from that in the input.
We further assessed the physical interaction between NT-OPA1-exon4b and nucleoids. Flag-tagged NT-OPA1-exon4, NT-OPA1-exon4b, or TFAM, an abundant mitochondrial transcription factor used as a positive control, were expressed in HeLa cells, then immunoprecipitated from purified mitochondria using anti-Flag antibodies. Western blot analysis revealed the presence of TFAM and POLG, two mtDNA-associated proteins, but not VDAC1, a mitochondrial protein absent from nucleoids, nor any OPA1 isoform (data not shown), in NT-OPA1-exon4b and TFAM immunoprecipitates, but not in those from control cells or from cells expressing NT-OPA1-exon4 (Fig. 4B, left). In addition, under similar experimental conditions, dual-step chromatin immunoprecipitation experiments recovered a significant amount of mtDNA from cells expressing NT-OPA1-exon4b and TFAM, but not from control cells or cells expressing NT-OPA1-exon4 (Fig. 4B, right). These results demonstrate that NT-OPA1-exon4b directly interacts with mitochondrial nucleoids. Altogether, they further suggest that the intermediate loop localized between the two TM domains should be involved in nucleoid binding.
mtDNA copy number and exon 4b abundance in OPA1 transcripts are coregulated
Former results suggest that NT-OPA1-exon4b could form a bridge in between the mitochondrial inner membrane and the mitochondrial nucleoids. Consequently, the abundance of mtDNA copy number should parallel the amount of NT-OPA1-exon4b and that of exon 4b in OPA1 transcripts. We tested this hypothesis in different cell types and organs by monitoring mtDNA and OPA1 variant abundances by quantitative PCR and RT-PCR, respectively, on total DNA and RNA. In mouse organs, the results showed a tight correlation between exon 4b abundance and mtDNA copy number (r = 0.96), whereas this correlation was lower for exon 5b (r = 0.82) and negative for exon 4 (r = −0.52) (Fig. 5A). In fibroblasts from two patients with a mitochondrial disease (Cornille et al. 2008), we found that the increase in mtDNA copy number paralleled an increase in OPA1-exon4b abundance, whereas exon 4 and 5b abundances were reduced (Fig. 5B). These results suggest that in physiological and in pathological conditions the abundances of mtDNA and exon 4b in OPA1 transcripts are coordinately regulated.

mtDNA copy number and Ex4b abundance in OPA1 transcripts are coordinately regulated. (A) Mean mitochondrial genome copy number and relative abundance of the OPA1 alternate spliced exons 4, 4b, and 5b among total OPA1 transcripts in mouse brain (Br.), heart (He.), liver (Li.), and muscle (Mu.) from three animals. (B) Mean mitochondrial genome copy number and relative abundance of the alternate OPA1 exons in fibroblasts from two controls and two patients with a mitochondrial disease (n = 3, t-test; [*] P-value inferior to 0.01). (C) Mitochondrial genome copy number and relative abundance of the alternate OPA1 exons in HepG F2 rho(+) and rho(0) cells (n = 3, t-test; [*] P-value inferior to 0.01). (D) Western blot showing the expression of OPA1 in control cells and in cells overexpressing OPA1-exon4b and the corresponding quantification of mtDNA abundance (n = 3, t-test, not significant).
Conversely, in HepG2 rho(0) cells (Loiseau et al. 2009) in which mtDNA was eliminated, the expression level of exon 4b is barely decreased comparatively to the HepG2 rho(+) cells (Fig. 5C). Similarly, the overexpression of OPA1-exon4b in HeLa cells remained without effect on mtDNA copy number (Fig. 5D). Thus these data suggest that NT-OPA1-exon4b is not a regulator of mtDNA abundance and that the mtDNA is not a regulator of NT-OPA1-exon4b abundance, but rather that mtDNA abundance and alternate splicing in OPA1 mRNAs might be controlled by a common regulatory pathway leading to stoichiometric abundances of mtDNA and NT-OPA1-exon4b.
OPA1 gene screening in mitochondrial depletion syndromes
Our data suggest that alterations of NT-OPA1-exon4b sequence or abundance may lead to mtDNA loss and be relevant to mitochondrial depletion syndromes (MDS) in humans. Of note, this idea is reinforced by the fact that no mutation in exon 4b and the surrounding exons has been reported in dominant optic atrophy patients (Ferre et al. 2009). Consequently, we screened the OPA1 gene region involved in the formation of the NT-OPA1-exon4b peptide, by sequencing exons 2, 3, 4, and 4b from 46 French and German MDS patients with unknown etiology. None of them presented pathogenic mutation, suggesting that OPA1-associated MDS might be an extremely rare event or that mutation in both alleles of these exons might be embryonic-lethal. Nevertheless, we found a single nucleotide polymorphism in exon 4 that preferentially segregates with the MDS disease condition (Supplemental Fig. 3). Although in silico analysis of this mutation suggests that it might be involved in regulating OPA1 splicing, its (patho-)physiological significance remains to be elucidated.
Discussion
This study provides extensive experimental evidence that the mitochondrial fusion protein OPA1 generates a peptide including the exon 4b domain associated with the inner membrane, which is crucial for the maintenance of mtDNA. This peptide directly interacts with the nucleoids, allowing their distribution within the mitochondrial network and promoting mitochondrial genome replication. Based on these results, we propose the model summarized in Figure 6 to explain the molecular bases of OPA1 function in mtDNA physiology. This model integrates two main processes: the generation of the NT-OPA1-exon4b peptide from the OPA1 gene, which is vertebrate-specific, and the involvement of this peptide in mtDNA maintenance, which we propose to be evolutionarily conserved among all eukaryotes.
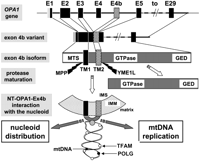
Summarizing model. Schematic representation showing the different steps from the OPA1 gene to the generation of NT-OPA1-exon4b peptide and its interaction with the mitochondrial nucleoid. (E1 to E29) OPA1 exon 1 to exon 29; (MTS) mitochondrial targeting sequence; (GED) GTPase effector domain; (TM) transmembrane domain; (MPP) mitochondrial processing peptidase; (YME1L) inner membrane protease cleaving OPA1-exon4b; (IMS) inner membrane space; (IMM) inner mitochondrial membrane. (White spot) TFAM protein; (black spot) POLG; (gray spot) an unknown protein linking NT-OPA1-exon4b peptide to the mtDNA.
In humans, the generation of the NT-OPA1-exon4b peptide requires two specific steps: the insertion of the exon 4b sequence in OPA1 transcripts, and the cleavage of the corresponding isoforms by the YME1L inner mitochondrial membrane protease (Griparic et al. 2007; Song et al. 2007), two mechanisms with marked evolutionary divergences from nonvertebrate models. Our previous work and the present study indicate that the different OPA1 isoforms have clear-cut different functions in mitochondria that are specified by the presence of exons 4, 4b, and 5b (Olichon et al. 2007), in contrast to lower eukaryotes in which the OPA1 ortholog gene is transcribed in a single mRNA. Indeed, we showed that the abundance of OPA1 variants greatly varies from one organ or cell line to another, possibly to comply with the different demands in energy metabolism, mitochondrial dynamics, and regulation of apoptosis. Consistent with the tissue-specific fingerprint of OPA1 variant expression levels, we provide now evidence that the abundance of exon 4b in OPA1 transcripts correlates with mtDNA copy number, suggesting a common regulatory mechanism for both processes. These observations emphasize the critical involvement of alternate splicing in regulating the expression of OPA1 variants, allowing a fine and independent tuning of the corresponding isoforms and their functions in mitochondria. Consequently, the identification of the splicing factor specific to exon 4b represents an important challenge to elucidate the mechanisms underlying mitochondrial genome maintenance and possibly providing a candidate gene for mitochondrial depletion syndromes.
In order to understand the role of NT-OPA1-exon4b in the maintenance of the mitochondrial genome, one should consider the structure of this peptide and its counterpart in other models. Although no sequence homology can be detected between their sequences, they present a highly conserved structure including two TM domains plus an intermediate loop. In vertebrates, the second TM domain consisting in a 15-amino-acid-long hydrophobic sequence is encoded by exon 4b, whereas in yeasts and Drosophila, the second TM domain is systematically present in the unique isoform. Our results highlight the critical role played by exon 4b, as its presence specifies the colocalization and coimmunoprecipitation of NT-OPA1-exon4b with the nucleoids. Converging results were recently obtained in yeast, proving that the integrity of the second TM domain is mandatory for mtDNA maintenance, but not for the fusion activity (Herlan et al. 2003; Diot et al. 2009). In addition, results from the trypsin protection experiments suggest that the intermediate loop is oriented toward the matrix, a hypothesis that fits also with the way OPA1 is imported through the IMM. This concept is also supported by the fact that NT-OPA1-exon4b coimmunoprecipitates with nucleoids, suggesting that the intermediate loop interacts with the nucleoids in the matrix. Nevertheless, in silico analysis of NT-OPA1-exon4b sequence does not reveal the presence of a DNA binding domain, thus excluding a direct interaction with mtDNA. Consequently, the identification of the protein that stands in between NT-OPA1-exon4b and mtDNA remains another future important challenge. All considered, our study provides the first evidence for an evolutionarily conserved mechanism that could anchor mtDNA to the inner mitochondrial membrane.
It is further tempting to speculate that NT-OPA1-exon4b may form a specific structure embedded in the membrane to organize the multiple mtDNA copies inside a single nucleoid unit. Indeed, we observed that NT-OPA1-exon4b forms regular dots in mitochondria, instead of decorating the network homogeneously. In addition, results from exon 4b silencing experiments suggest that inside nucleoids, the amount of mtDNA could vary according to changes in the abundance of NT-OPA1-exon4b. Thus, the assembly of NT-OPA1-exon4b in a membrane structure might form a basement defining the number of mtDNA copies per nucleoid.
Furthermore, our results indicate that nucleoid anchoring is required for mtDNA replication and nucleoid distribution within the network, supporting an interaction between NT-OPA1-exon4b and the replication machinery, and with proteins involved in membrane dynamics. These observations, which have never been reported to date in other eukaryotic models, are highly reminiscent of the coupling between chromosome replication and partitioning in bacteria (Thanbichler and Shapiro 2006). The role played by NT-OPA1-exon4b interfacing the mitochondrial genome organization and its replication machinery with the membrane dynamics may also provide an explanatory basis for our recent observation showing that specific mis-sense mutations in the OPA1 GTPase domain induce accumulation of mtDNA multiple deletions in post-mitotic tissues of syndromic DOA patients (Amati-Bonneau et al. 2008; Hudson et al. 2008). In conclusion, this study defines a novel fundamental function for OPA1 in mtDNA maintenance and highlights OPA1 as an essential gatekeeper of the mitochondrial genome. This new concept opens novel avenues in understanding mtDNA copy number adaptations to the cell's energetic requirements, with likely relevance to human mitochondrial diseases.
Methods
Antibodies
OPA1 (1:300) (Olichon et al. 2002), ACTB1 (beta-actin, 1:10,000; Chemicon), anti-BrdU (1:600; BD Pharmingen), TFAM (1:1000; Santa Cruz), POLG (DNA polymerase gamma, 1:500; Abcam), VDAC1 (Porin, 1:10,000; Invitrogen), HSPD1 (HSP60, 1:1000; ABCAM); monoclonal anti-Flag (1:1000; Sigma), anti-DNA (1:200; Progen), secondary anti-rabbit IgG-alkaline phosphatase (1:10,000; Invitrogen), and secondary anti-mouse and anti-rabbit IgG-Hrp conjugate (Jackson Immunoresearch; 1:2000).
Cell culture, plasmid transfection, and Western blots
HeLa cells were cultured under standard conditions. HepG2 Rho(+) and Rho(0) cells were grown in RPMI 1640 + 10% FCS + 50 μg/mL uridine and 100 μg/mL pyruvate (Loiseau et al. 2009). Plasmid (2 μg/experiment) and siRNA (100 nM final) transfections of HeLa cells were performed with Lipofectamine 2000 reagent (Invitrogen). To detect OPA1 and ACTB1 by Western blots, transfected cells were harvested and washed once in PBS, and equal amounts of proteins were solubilized in 50 μL of Laemmli sample buffer and boiled for 10 min. Samples were run on 7% polyacrylamide gels and transferred to Hybond nitrocellulose (Amersham). After incubation with the primary OPA1 and ACTB1 antibodies, immunodetection was performed with the anti-rabbit IgG-alkaline phosphatase and revealed by BCIP/NBT.
Microscopy
Cells were seeded in 36-mm-diameter dishes with glass bottoms. Mitochondria were labeled by incubating cells with 100 nM CMXros Mitotracker Red (Molecular Probes) for 30 min. mtDNA and nuclear DNA were labeled by incubating fixed cells with anti-DNA antibodies (He et al. 2007) or live cells with 3 μL/mL PicoGreen (Invitrogen) (Ashley et al. 2005) for 1 h, followed by two PBS washes and direct observation with a confocal microscope.
For immunostaining cells were washed with DMEM and fixed in 3.7% paraformaldehyde in PBS for 20 min at 37°C, then washed three times with PBS, permeabilized in 0.5% Triton X-100 in PBSF (PBS containing 10% FBS) for 5 min at room temperature, followed by washing three times with PBS. Primary anti-DNA mouse IgM antibodies (PROGEN) were diluted 1 μg/mL in PBSF and incubated with cells for 2 h at 37°C. After washing three times with PBSF, the cells were incubated for 1 h at 37°C with the fluorescent secondary goat anti-mouse IgG-Alexa-Fluor 488 antibody (Molecular Probes), diluted 2 μg/mL in PBSF. Cells were washed with PBSF three times and once with PBS, Optimem was added, and dishes were immediately observed. The images were acquired using an Inverse1 Zeiss Axioobserver/LSM 5 LIVE DUO Confocal microscope (Carl Zeiss Microscopy), equipped with a 63× oil objective.
For all imaging, Z-stack images (1024 × 1024 pixels) were acquired and processed with the Bitplane Imaris 4 software. At least eight cells from three independent experiments were analyzed for each condition. To measure colocalization in the whole cell, images were processed using Imaris 4.0 (Bitplane) software using the automatic thresholding feature for colocalization. Nucleoid fluorescence intensities were obtained after maximal projection of the images using MetaMorph 5.0 software (Molecular Devices).
Molecular cloning
Plasmids expressing the full-length OPA1 variants are from Olichon et al. (2007). OPA1 open reading frames corresponding to NT-OPA1-Exon4 or NT-OPA1-Exon4b peptides were cloned by PCR using, respectively, the OPA1 variants 1 and 3, with primers introducing a XhoI site at the ATG initiation codon and a BamHI site at the end of exon 4 and exon 4b sequences (see list of primer sequences: HsOPA1-ATG Xho S, HsOPA1-Ex4 Bam AS, HsOPA1-Ex4b Bam AS). Exon 4b sequence was further mutagenized according to the protocol (QuikChange II Site-Directed Mutagenesis kit; Stratagene) with the Mut4b S and AS primers (see list of primer sequences). All amplified fragments were cloned in the Topo-pCRII vector (Invitrogen), sequenced, and recloned in the pCC-Cherry and pFlagTag vectors. Similarly, the human TFAM open reading frame was cloned in the pFlagTag vector.
Mitochondrial DNA measurement
Total DNA purifications were performed using the DNeasy kit (QIAGEN) and precisely quantified by spectrophotometry (Nano-Drop 1000). Nuclear and mitochondrial DNA respective abundances were quantified in duplicate by PCR in standard conditions using the LightCycler FastStart DNA Master SYBR Green I kit (Roche), with the human HunuclS, HunuclAS, HumitS, and HumitAS primers, or the corresponding ones in mouse: MmnuclS, MmnuclAS, MmmitS, and MmmitAS (see Supplemental Table for primer sequences). The ratios between mtDNA and nuclear DNA concentrations are reported on graphs.
Determination of OPA1 variant expression abundance
Total RNA was purified from organs or cells using the RNeasy kit (QIAGEN). After boiling and fast chilling the RNA (1 μg) mixed with the oligo-dT (0.5 μg), cDNAs were synthesized with 200 units of Superscript II reverse-transcriptase (Invitrogen) for 50 min at 42°C. OPA1 mRNA different variant abundances were determined in duplicate by quantitative PCR in human samples as described in Olichon et al. (2007) and in mouse samples, using the same protocol and the OPA1Mm S1, S2, S4, and S4 forward primers and OPA1Mm AS5 and AS6 reverse primers (see Supplemental Table for primer sequences). Relative abundances of each alternate spliced exon were calculated as the sum of the abundance of the four variants including this exon, divided by the sum of the abundance of the eight different OPA1 variants. Each experiment was performed three times on different biological samples.
Southern blots
Qualitative mtDNA analyses by Southern blots were performed using standard protocols (Sambrook and Russell 2001). Briefly, after overnight digestion of 5 μg of total DNA by PvuII, DNAs were separated on a 0.8% agarose gel then transferred to Hybond nitrocellulose (Amersham) and hybridized to a random primed (Stratagene) labeled 32P probe, corresponding to four 1-kb DNA fragments originating from different regions of the mitochondrial DNA. After the washes, membranes were exposed on BioMax films (Kodak). Southern blots were repeated on DNA samples from three different experiments.
Southern–Western blots
To evaluate mtDNA replication, cells were treated with 2 μM aphidicolin for 2 h, then incubated with 10 μM BrdU for an additional 8 h (Garrido et al. 2003). Total DNA was extracted from cell pellets and processed as for Southern blots, except that after the transfer onto nitrocellulose, replicated DNA was detected by a 2-h anti-BrdU incubation using a classical Western blot protocol. Quantification of band intensity from three separate experiments was performed using the Definiens Developer 7.0 program (Statitistics).
Isolation of mitochondria from cultured cells
Cells were washed once with PBS, scraped in PBS, and centrifuged for 10 min at 500g. The cell pellet was suspended in mitochondria isolation buffer (200 mM mannitol, 70 mM sucrose, 1 mM EGTA, 10 mM HEPES at pH 7.6), with protease inhibitor cocktail (Roche) and homogenized by 30 strokes with a Dounce homogenizer at 4°C. The homogenate was centrifuged twice for 10 min at 400g, the pellet was discarded, and the supernatant was re-centrifuged for 20 min at 10.000g to isolate the mitochondrial fraction.
Mitochondrial membrane association and sub-compartmentalization experiments
To assess membrane association, mitochondria suspended in mitochondria isolation buffer were sonicated three times for 30 sec on ice. Residual mitochondria were removed by centrifugation (10.000g, 4°C, 10 min), and the supernatant was treated either with 1 M NaCl or 3% SDS and incubated for 20 min on ice with frequent vortexing. The suspension was centrifuged (100.000g, 4°C, 45 min) to separate the supernatant from the membrane pellet. The different fractions (50 μg of mitochondrial proteins) obtained were analyzed by Western blotting using OPA1 and anti-Flag antibodies.
To determine protein sub-compartmentalization, the mitochondrial pellet was suspended in mitochondria isolation buffer and treated with trypsin (1 μg/μL), then digitonin (1 μg/μL), and 1% Triton X-100 for 20 min at 4°C. Proteolysis was terminated by the addition of 2× protease inhibitor cocktail (Roche) and 2× Laemli's sample buffer. The samples (30 μg of mitochondrial proteins) were separated on a 12% SDS-PAGE and analyzed by Western blotting using OPA1, HSPD1, and anti-Flag antibodies.
Coimmunoprecipitation and Western blotting
HeLa cells (2.5 × 106) were transfected with pCMV empty vector (control cells), pCC-TFAM-flag, pCC-NT-OPA1-Ex4-flag, or Ex4b-flag, and after 24 h mitochondria were isolated by standard differential centrifugation. Mitochondrial pellets were suspended in 100 μL of RIPA buffer (50 mM Tris-HCl at pH 7,6, 150 mM NaCl, 1% NaDOC, 1% SDS, 5 mM EDTA), and protein content was determined. IP was performed following the protocol previously described (Zanna et al. 2008) with minor modifications. Two-hundred-and-fifty micrograms of mitochondrial proteins was incubated with 5 μg of anti-Flag antibody cross-linked to protein A-Sepharose (Sigma) for 3 h at 4°C. Immunoprecipitated complexes were loaded on Bio-Rad columns and centrifuged for 1 min at 500g, and 20 μL of the flow-through fractions was collected and solubilized with 5 μL of Laemmli buffer. Columns were washed five times with PBS then eluted in 400 μL of 7 M Urea, 2 M Thio-urea, and 2% Chaps. Eluates were precipitated overnight with acetone at −20°C and resuspended in 25 μL of Laemmli buffer. Samples were separated on 8% SDS-PAGE and analyzed by Western blot using the Flag-tag, POLG, TFAM, and VDAC1 primary antibodies.
Dual-step cross-linking chromatin immunoprecipitation
HeLa cells (2.5 × 106) were transfected as for the IP experiment. The dual-step chromatin immunoprecipitation was performed according to published protocol (Nowak et al. 2005) with some modifications. Cells were collected in 2 mL of PBS/1 mM PMSF, then incubated with 2 mM disuccinimidyl glutarate for 45 min at room temperature, rinsed twice, and further incubated in 2 mL of PBS/1 mM PMSF with 1% formaldehyde for 15 min. Cross-linking was quenched by addition of 0.125 M glycine. After three washes with PBS/PMSF, cells were lysed in 100 μL of RIPA buffer in the presence of the protease inhibitor cocktail (Roche) and sonicated for 45 min with a Branson Sonifier 1211. Samples were centrifuged (5000g for 5 min) at 4°C, and the supernatants were incubated with 5 μg of anti-Flag antibody (Sigma) overnight on a rotating wheel at 4°C. Aliquots of supernatants were collected and used as “input DNA.” The immunoprecipitates were incubated with protein A-Sepharose. Beads were washed three times with RIPA buffer, three times with LiCl buffer (100 mM Tris-HCl at pH 8, 500 mM LiCl, 1% NaDoc, 1% NP-40), and three times with TE buffer (10 mM Tris-HCl at pH 8.0, 1 mM EDTA). Beads were then suspended in 200 μL of TE buffer and incubated with 10 μg of RNase A for 30 min at 37°C, then with Proteinase K for 6 h at 65°C. Samples were centrifuged at 16,000g for 10 min at 4°C and the supernatants were processed for DNA extraction. Quantification of mtDNA was performed by Q-PCR as described above.
Statistics
All experiments were repeated at least three times. Statistics were inferred using the Student's t-test or ANOVA test from the GraphPad Prism software (GraphPad) as indicated in the legends of the figures.
Acknowledgments
We thank the INSERM, CNRS, and Université Montpellier I and II for providing institutional support and the patient associations Retina France and Ouvrir Les Yeux for their financial support. G.E. was supported by the Association Française contre les Myopathies. S.V. was supported by a Progetto Marco Polo fellowship from the University of Bologna, and V.C. received a Telethon-Italy grant (GGP06233). P.Y.W.M. is an MRC Clinical Research Fellow, and P.F.C. is a Wellcome Trust Senior Fellow in Clinical Science. P.F.C. also receives funding from the Parkinson's Disease Society (UK), the Medical Research Council Translational Muscle Centre, and the UK NIHR Biomedical Research Centre in Ageing and Age Related Disease. We thank Dr. Agnel (Montpellier, France) and Dr. Bonzarone (Bologna, Italy) for generating helpful discussions.
Footnotes
-
↵11 Corresponding author.
E-mail guy.lenaers{at}inserm.fr; fax (33) 499-63-60-20.
-
[Supplemental material is available online at http://www.genome.org.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.108696.110.
- Received April 3, 2010.
- Accepted October 12, 2010.
- Copyright © 2011 by Cold Spring Harbor Laboratory Press








