
Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer
- Andrew E. Teschendorff1,
- Usha Menon2,
- Aleksandra Gentry-Maharaj2,
- Susan J. Ramus2,
- Daniel J. Weisenberger3,
- Hui Shen3,
- Mihaela Campan3,
- Houtan Noushmehr3,
- Christopher G. Bell1,
- A. Peter Maxwell4,
- David A. Savage4,
- Elisabeth Mueller-Holzner5,
- Christian Marth5,
- Gabrijela Kocjan6,
- Simon A. Gayther2,
- Allison Jones2,
- Stephan Beck1,
- Wolfgang Wagner7,
- Peter W. Laird3,
- Ian J. Jacobs2 and
- Martin Widschwendter2,8
- 1 Medical Genomics Group, UCL Cancer Institute, University College London, London WC1E 6BT, United Kingdom;
- 2 Department of Gynecological Oncology, UCL Elizabeth Garrett Anderson Institute for Women's Health, University College London, London W1T 7DN, United Kingdom;
- 3 USC Epigenome Center, University of Southern California, Keck School of Medicine, Los Angeles, California 90089-9601, USA;
- 4 Nephrology Research Group, Centre for Public Health, Queen's University Belfast, Belfast BT9 7AB, Northern Ireland;
- 5 Department of Obstetrics and Gynaecology, Innsbruck Medical University, Innsbruck 6020, Austria;
- 6 Department of Histopathology, University College London, London WC1E 6JJ, United Kingdom;
- 7 Helmholtz Institute for Biomedical Engineering–Cell Biology, Aachen University Medical School, 52074 Aachen, Germany
Abstract
Polycomb group proteins (PCGs) are involved in repression of genes that are required for stem cell differentiation. Recently, it was shown that promoters of PCG target genes (PCGTs) are 12-fold more likely to be methylated in cancer than non-PCGTs. Age is the most important demographic risk factor for cancer, and we hypothesized that its carcinogenic potential may be referred by irreversibly stabilizing stem cell features. To test this, we analyzed the methylation status of over 27,000 CpGs mapping to promoters of ∼14,000 genes in whole blood samples from 261 postmenopausal women. We demonstrate that stem cell PCGTs are far more likely to become methylated with age than non-targets (odds ratio = 5.3 [3.8–7.4], P < 10−10), independently of sex, tissue type, disease state, and methylation platform. We identified a specific subset of 69 PCGT CpGs that undergo hypermethylation with age and validated this methylation signature in seven independent data sets encompassing over 900 samples, including normal and cancer solid tissues and a population of bone marrow mesenchymal stem/stromal cells (P < 10−5). We find that the age-PCGT methylation signature is present in preneoplastic conditions and may drive gene expression changes associated with carcinogenesis. These findings shed substantial novel insights into the epigenetic effects of aging and support the view that age may predispose to malignant transformation by irreversibly stabilizing stem cell features.
Targets of polycomb group proteins (PCGTs) are repressed in human embryonic and adult stem cells (Lee et al. 2006). The repression mechanism involves chromatin modifications and is reversible, allowing stem cells and multipotent progenitors to differentiate into committed cell lineages through expression of specific PCGTs. Recently, we and others have demonstrated that stem cell PCGTs in human embryonic stem cells (hESC) are far more likely to undergo cancer-specific promoter DNA hypermethylation than non-targets, suggesting a stem-cell origin model of cancer. In this model, PCGTs in stem cells would gradually undergo de novo methylation, irreversibly locking cells in an undifferentiated state of self-renewal and thereby predisposing them to subsequent malignant transformation (Ohm et al. 2007; Schlesinger et al. 2007; Widschwendter et al. 2007). However, the mechanisms and factors contributing to this de novo methylation are not yet known.
Age is by far the strongest demographic risk factor for cancer. Besides time-dependent DNA damage (Hoeijmakers 2009), there is now also substantial evidence that aging affects DNA methylation (DNAm) of specific loci, including cancer-related genes (Issa et al. 1994, 1996; Ahuja et al. 1998; Nakagawa et al. 2001; So et al. 2006; Fraga and Esteller 2007; Fraga et al. 2007; Bjornsson et al. 2008; Christensen et al. 2009). Based on these observations, we hypothesized that age may induce DNAm of PCGTs, and thereby predispose to cancer. Although blood and epithelial cells originate from different germ layers, we speculated that genes that are mandatory for the differentiation of epithelial cells are more likely to become methylated with increasing age in non-epithelial tissue such as blood. Hence, in order to identify age-dependent CpGs that may be important in the biology of epithelial cancers, we first retrieved an age-dependent signature from peripheral blood cells, then validated the age signature in independent blood samples and normal epithelial tissues, and finally tested the biological relevance of this signature in epithelial neoplasias.
Results
Age-dependent hypermethylation of PCGTs is independent of cell type
We first performed DNAm profiling (Illumina Infinium 27k) (Weisenberger et al. 2008) of peripheral blood samples drawn from 261 postmenopausal women spanning a 30-yr age range (Supplemental Fig. 1; Supplemental Tables 1, 2). A stringent quality control and interarray normalization procedure resulted in a normalized data matrix of methylation scores (β-values, 0 < β < 1) across 261 blood samples (148 from healthy women [Set-1], 113 from ovarian cancer cases [Set-2]) and 25,642 CpG sites (Table 1; see Methods, Supplemental material). Unsupervised analysis using singular value decomposition (SVD) revealed significant components of variation associated with age (Supplemental Fig. 2). Next, using linear regressions, we derived a DNAm signature for aging. To see if this signature would be dependent on disease status, this analysis was performed separately for cases and controls. We observed that the age-associated DNAm signature was very similar regardless of disease status (Fig. 1A). We thus combined the samples (n = 261) to derive a core DNA methylation signature for aging (589 CpGs passed a false discovery rate [FDR] threshold of 0.05; Fig. 1A, Supplemental Table 3). While the majority of CpGs were hypomethylated with age, we observed that CpGs mapping to promoters of PCGTs (defined by single occupancy of SUZ12, EED, or H3K27me3 in human embryonic stem cells [hESC] [Lee et al. 2006]) were preferentially hypermethylated (Fig. 1A). Specifically, we identified 69 CpGs mapping to 64 unique PCGT loci (Supplemental Table 4), which was significantly more than the 20 unique gene loci expected by chance (Fisher's exact test, P = 2 × 10−17). We estimated that PCGT loci were approximately fivefold (odds ratio [OR]) more likely (median unbiased mid-p test, P < 10−12, Fig. 1B) to be hypermethylated with age than non-PCGTs, defined by genes that lack occupancy of SUZ12, EED, or H3K27me3 marks in hESC (Lee et al. 2006). Similarly, we observed a fivefold OR enrichment of H3K27me3 marks (Fig. 1C) in hematopoietic stem cells (HSC) (Cui et al. 2009). In contrast, only 11 PCGTs were hypomethylated with age, which was somewhat less than expected by chance (Fig. 1A,B). We verified that PCGT enrichment among hypermethylated CpGs was not due to an overrepresentation of PCGT CpGs within CpG islands, by showing that the enrichment remained when restricting the comparison to those CpGs located within CpG islands (OR = 4.2 [3.0–5.7], P < 10−10). The 69 PCGT CpGs displayed an average methylation profile that increased monotonically over an age range spanning >25 yr (50–80 yr) (Supplemental Fig. 3).

DNAm signatures for aging and enrichment of PCGTs. (A) Flowchart depicting the derivation of the “core” DNA methylation signature for aging. First, the supervised analysis was performed separately for the blood samples from 148 healthy and 113 ovarian cancer cases. This yielded 293 CpGs and 420 CpGs passing a FDR (q) cut-off of 0.3. There was a strong overlap between these two signatures (Fisher's exact test, P = 10−30) with >80% concordance. Healthy and pretreatment samples were thus combined and supervised analysis was performed on this larger set to identify with more confidence a DNA methylation signature for aging. This gave 589 age-associated CpGs (q < 0.05), termed the “core” aging signature. Distribution of these 589 CpGs in terms of hyper- and hypomethylation patterns demonstrated a skew toward hypomethylation (binomial test, P = 6 × 10−9). Among the 226 hypermethylated CpGs, 69 mapped to polycomb group targets (PCGTs) (64 unique gene loci), while among the 363 hypomethylated CpGs this number was only 20 (11 unique gene loci). Thus, relative to the “core” aging signature, PCGTs were preferentially hypermethylated (69 vs. 20 compared with 226 vs. 363, Fisher's exact test, P < 4 × 10−12). Here, PCGTs were defined by promoter occupancy of any one of SUZ12, EED, or H3K27me3 in human embryonic stem cells (Lee et al. 2006). (B,C) Enrichment odds ratios with 95% confidence intervals for PCGTs (B) and for H3K27me3 marks (C), among the 226 age-hypermethylated and 363 age-hypomethylated CpGs. H3K27me3 marks were defined by trimethylation of H3K27 within gene body, promoter, and gene body + promoter regions in CD133+ hematopoietic stem cells (HSC) (Cui et al. 2009). (D,E) Independent validation: enrichment odds ratios with 95% confidence intervals for PCGTs among CpGs undergoing significant hyper- and hypomethylation with age in 188 blood samples from patients with type-1 diabetes (D) and 177 ovarian cancer samples (E). (Dashed line) Line of unit odds ratio. Two-tailed P-values of enrichment (i.e., deviation from this line) are given.
Main methylation data sets used in this study
To investigate the generality of this epigenetic phenomenon, we next applied the same linear regression approach to derive DNAm signatures for aging in two independent data sets (Table 1, Set-5 and Set-6): whole blood (WB) samples from 188 patients (95 women and 93 men) with type 1 diabetes (T1D), and tumor tissue samples from 177 women with ovarian cancer (OvC). Using the same FDR cutoff of 0.05, we observed many age-associated CpGs in WB and OvC tissue (Supplemental Tables 5, 6), with a highly significant OR enrichment of PCGTs among CpGs undergoing hypermethylation with age, but not so among CpGs undergoing hypomethylation (Fig. 1D,E). For the WB T1D samples, we verified that PCGT enrichment was independent of sex (Supplemental Fig. 4).
In addition, using data generated on a different platform, with a different set of CpGs (Goldengate assay; Christensen et al. 2009), we confirmed that PCGTs undergo preferential hypermethylation with age in normal tissues other than blood, including normal pleura and lung samples (Supplemental Fig. 5).
Given the common enrichment of PCGTs across multiple tissue types, we next asked if this result could be due to a specific “core” subset of PCGTs, or if instead the age-PCGT signature is largely tissue-specific. To address this question, we took the specific subset of 69 PCGT CpGs, as identified in the training set of 261 WB samples, and asked if they showed a consistent pattern of increased methylation with age in the validation data sets (Table 1). We found that the average methylation profile of the 69 CpGs correlated significantly with age in blood samples from 108 healthy individuals (Fig. 2A, Set-3), 122 ovarian cancer cases (Supplemental Fig. 3, Set-4), 188 patients with T1D (Fig. 2B, Set-5), and in ovarian cancer tissue from 177 women (Fig. 2C, Set-6). Moreover, we observed that the 69 PCGT CpGs exhibited a significant skew toward hypermethylation with age in all validation sets examined (Fig. 2E–G; Supplemental Fig. 3; Supplemental Table 4). The skew toward hypermethylation remained significant relative to random choices of 69 CpGs. Furthermore, we observed that the 69 PCGT CpGs exhibited higher levels of methylation than non-age-associated PCGT CpGs and that the difference in methylation between these two groups increased with age in all validation sets examined (Supplemental Fig. 6).
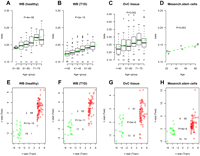
External validation of specific age-associated PCGT DNAm signature. (A–D) Average beta-methylation values over the 69 age-hypermethylated PCGTs (y-axis) as a function of age (x-axis) in validation data sets. Number of samples in each age group are given above the x-axis. t-test P-values for linear trend derived from a robust linear regression are given; (green dashed line) best linear fit. (E–H) Validation of age-associated (69 hypermethylated and 20 hypomethylated) PCGT CpGs in test sets. (X-axis) t-statistic of the linear regression test of age vs. methylation in the training set (blood samples from 148 healthy + 113 pretreatment ovarian cancer cases). Colors reflect directionality: (red) hypermethylated, (green) hypomethylated. (Y-axis) t-statistic of the linear regression test of age vs. methylation in the test set. We provide the number of CpGs displaying significant hyper/hypomethylation in the training set and hyper/hypomethylation in the test set, as well as the corresponding Fisher's exact test P-value. (A,E) Test set of blood samples from an independent set of 108 healthy individuals spanning an age range of 50–80 yr. In A, age was categorized into six age groups (50–55, 56–60, 61–65, 66–70, 71–75, >75). (B,F) Test set of blood samples from 188 T1D patients spanning an age range of 24–74 yr. In B, age was categorized into six age groups (≤35, 36–40, 41–45, 46–50, 51–60, >60). (C,G) A test set of ovarian cancer samples from 177 ovarian cancer patients spanning an age range 24–88 yr. In C, age was categorized into six age groups (≤40, 41–50, 51–60, 61–70, 71–75, >75). (D,H) A test set of eight bone marrow mesenchymal stromal cell samples from healthy donors of the following ages: 21, 24, 25, 50, 53, 79, 85, 85 (Bork et al. 2010).
We next asked if the 69 PCGT CpG DNAm signatures could be reflected in multipotent progenitor and stem cell pools. To this end, we investigated the DNA methylation profiles of cultured mesenchymal stromal/stem cells (MSC) derived from the bone marrow of eight healthy individuals spanning a wide age range (21–85 yr; Set-7) (Bork et al. 2010). Despite the small sample size, the average methylation profile exhibited a significant linear increase with age (t-test for linear trend, P = 0.003, Fig. 2D), with 59 of the 69 age-hypermethylated CpGs demonstrating corresponding increases in methylation, while 14 of the 20 age-hypomethylated CpGs demonstrated coordinate decreases (Fisher's exact test, P = 4 × 10−6; Fig. 2H; Supplemental Table 4).
All these results demonstrate that although the magnitude of methylation changes differed between studies and tissues (Supplemental Table 4), the 69 PCGT CpGs (henceforth “age-PCGT” CpGs) defined a robust age-related DNAm signature, exhibiting the same directional DNAm changes independently of disease state, sex, tissue, and cell type.
The age-PCGT signature discriminates normal from preinvasive and invasive cancer
We observed that in ovarian cancer tissue, methylation levels of age-PCGT CpGs were higher than those of PCGT CpGs not associated with age (Supplemental Fig. 7). This suggested to us that the implicated genes could be contributing to carcinogenesis. We therefore hypothesized that this age-PCGT signature could be present in preinvasive lesions. As there is still debate over the cell of origin, and there is no well-defined preneoplastic lesion for ovarian cancer, we used the uterine cervix as a model to test this hypothesis. We performed DNAm profiling of 48 age-matched cervical smear samples from premenopausal women (Table 1, Set-10) with normal smears (HPV-positive and -negative) and smears exhibiting dysplasia (all HPV-positive; Supplemental material). We verified that the age of samples with dysplasia did not differ from the normal smears (Wilcoxon test, P = 0.86). Despite the relatively small sample size and narrow age range of this premenopausal sample set, we found that PCGTs and our 69 age-PCGT CpG subset were preferentially hypermethylated with age (Supplemental Fig. 8). In addition, we observed that the 69 age-PCGT CpGs were more highly methylated in the HPV-positive samples exhibiting dysplasia compared with HPV-positive and -negative normal samples (Fig. 3A). In contrast, DNAm of PCGT CpGs that underwent hypomethylation in whole blood did not correlate with progression (Fig. 3B), and non-age-associated PCGT CpGs also did not exhibit methylation differences between dysplasia and normal conditions (P = 0.47, Fig. 3C). In only 0.5% of 10,000 random choices of other 69 PCGTs CpGs did we observe an association as strong as the one provided by the age-PCGTs (P < 0.01, Fig. 3C). Clustering the 48 samples over the 69 CpG methylation profiles also demonstrated that inferred clusters correlated significantly with dysplasia (Fisher's exact test, P < 0.001, Fig. 3D).
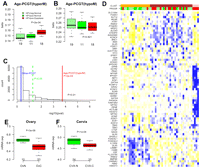
Biological and clinical significance of age-PCGT DNAm signature. (A,B) Average methylation values of the 69 age-hypermethylated and 20 age-hypomethylated PCGT CpGs as a function of disease status in 48 cervical cytology samples. (HPVneg) Normal cervical sample not infected with HPV, (HPVpos) normal cervical sample infected with HPV, (HPVpos-Dysplasia) samples infected with HPV and displaying dysplasia. Wilcoxon test P-value between normal and dysplastic condition is given. Number of samples in each group given below boxplots. (C) Histogram distribution of −log10(P-values) from 1000 randomly selected 69 non-age-associated PCGT CpGs. P-values were derived from the Wilcoxon test. (Red line) −log10(P-value) for the 69 age-hypermethylated PCGT CpGs, (blue line) −log10(P-value) for PCGT CpGs not mapping to age-PCGTs. In less than 0.5% of runs (P < 0.01) were P-values as extreme as the observed one, indicating that the age-PCGTs discriminate the dysplastic condition better than a random set of PCGTs. (D) Heatmap of the 48 cervival samples over the 69 age-hypermethylated PCGT CpGs. Samples were clustered using a Gaussian mixture model and three optimal clusters were inferred using the Bayesian Information Criterion (see Supplemental material). (Orange, brown, pink) Distinct clusters. The disease status of samples is labeled as a color bar (PROGR): (light green) HPVneg, (green) HPVpos+normal, (red) HPVpos+dysplasia. CpGs were clustered according to hierarchical clustering with a Pearson correlation metric. Prior to sample and CpG clustering, methylation profiles of invividual CpGs were renormalized to mean zero and unit standard deviation. Heatmap reflects, for each CpG, relative methylation levels across samples as determined by the renormalized methylation profile. (Blue) Relative high methylation, (yellow) relative low methylation. (E,F) Average gene expression intensity (Affymetrix) values for the 64 age-hypermethylated PCGTs in normal ovarian (OvN) and ovarian cancer tissue (OvC) and in normal cervix (CVX-N) and cervical cancer (CVX-T). Number of samples of each type and Wilcoxon test P-values are given.
Of the 64 age-PCGT genes, many have been reported to undergo hypermethylation in cancer (Ongenaert et al. 2008). Notably, TP73 and SFRP1 have been reported to undergo hypermethylation in not less than 10 different cancers (Supplemental Table 7). In line with this, we also observed that methylation levels of age-PCGTs discriminated other cancers from their normal counterparts (Supplemental Fig. 9; Bibikova et al. 2006).
In addition to frequent hypermethylation, PCGTs also exhibit frequent underexpression in cancer (Ben-Porath et al. 2008). We therefore compared gene expression profiles in ovarian and cervical cancer samples with their respective normal tissues (Scotto et al. 2008; Mok et al. 2009). In both cases, we observed that age-PCGTs exhibited average expression profiles that were significantly lower in cancer compared with normal tissue (Fig. 3E,F). We also observed that age-PCGTs were generally better discriminators of ovarian cancer than 1000 random choices of other 64 PCGTs (P = 0.06). Clustering over age-PCGTs further confirmed their power to discriminate ovarian and cervical cancer from their respective normal tissues (Supplemental Fig. 10). Interestingly, age-PCGT mRNA expression also showed a gradual decrease with cancer progression in a data set including preneoplastic lesions (Supplemental Fig. 11; Wurmbach et al. 2007).
Discussion
In this paper we have described a consistent directional change of DNAm with age, characterized by hypermethylation of PCGTs (Fig. 1; Supplemental Figs. 4, 8). While effect and sample sizes were not large enough for us to ascertain which genes undergo age-associated hypermethylation in a tissue-specific manner, the fact that we were able to identify a subset of 64 PCGTs exhibiting a clear trend toward hypermethylation with age across multiple cell types (blood, ovarian cancer, cervix, mesenchymal stem cells) indicates that a component of the identified signature is largely nonspecific (Fig. 2; Supplemental Figs. 6–8). It is also very unlikely that the identified age-PCGT signature is caused by age-related variation in cell-type composition. Indeed, as demonstrated in our recent work (Teschendorff et al. 2009), we were able to correlate the age-associated hypomethylation signature in blood with changes in blood cell-type composition, but not so for the age-hypermethylated signature. Consistent with this, we were also not able to validate the age hypomethylation signature in tissues other than blood (Supplemental Fig. 12), or to implicate it in carcinogenesis (Fig. 3B; Supplemental Fig. 13). In contrast, the age-hypermethylated PCGT signature was able to discriminate preneoplastic from normal cells and was found to be aggravated in invasive cancer leading to reduced expression of affected genes (Fig. 3; Supplemental Figs. 9–11). Besides the 64 age-PCGTs identified here, it is likely that other non-PCGT genes that also undergo hypermethylation with age in blood may also be broadly implicated in aging and carcinogenesis (Supplemental Figs. 12, 13).
To obtain direct functional proof that simultaneous silencing of age-PCGT genes predisposes a cell to become malignant is not possible with currently available technology. In the absence of a functional test, there are, however, other lines of evidence supporting the role of age-PCGTs in carcinogenesis: (1) 36% (24/64) of the 64 age-PCGT genes have already been published to be aberrantly methylated and deregulated in cancer; (2) 34% (22/64) of these genes are transcription factors known to be involved in normal differentiation. For instance, FOXC1 has been shown to play an essential role in development (Myatt and Lam 2007) and is also implicated in cancer (Bloushtain-Qimron et al. 2008). GATA4 belongs to the family of zinc finger–containing GATA transcription factors, which play critical roles in cell lineage specification during early embryonic development and organ formation. GATA4 is expressed in human ovarian surface epithelial cells and is important for the formation and maintenance of the differentiated state of these cells (Capo-chichi et al. 2003; Caslini et al. 2006). Loss of GATA4 expression precedes neoplastic transformation of ovarian surface epithelia (Cai et al. 2009), and GATA4 is also heavily methylated in ovarian cancer (Wakana et al. 2006). Another age-PCGT transcription factor is DLX5, which is increasingly methylated and silenced in MSC by both replicative senescence in vitro and aging in vivo (Bork et al. 2010). As a final example, TP73 (also known as p73) shares many functional properties with the TP53 (also known as p53) tumor suppressor and is also involved in mediating DNA damage-induced apoptosis as well as suppressing polyploidy and aneuploidy when p53 is inactivated, which suggests that age-dependent methylation and suppression of TP73 may potentially lead to genetic alterations and increased predisposition to cancer (Irwin et al. 2000; Moll and Slade 2004; Talos et al. 2007). (3) Finally, alongside transcription factors, there are numerous other genes in the age-PCGT panel that have been demonstrated to be involved in carcinogenesis, including ALOX5 (Catalano et al. 2005), SFRP1 (Wnt pathway) (Baylin and Ohm 2006), and KLF14 (TGF-beta signaling) (Truty et al. 2009).
In summary, we have found that age may contribute to carcinogenesis by irreversibly silencing genes that are suppressed in stem cells. To our knowledge, this constitutes the first report of a molecular (epigenetic) signature common to the processes of aging and carcinogenesis. Our findings may have broad implications for cancer prevention, risk prediction, detection, prognosis, and therapy.
Methods
Clinical samples
All DNAm data sets used in the study are summarized in Table 1. The primary sample set consisted of 491 whole blood samples drawn from the United Kingdom Ovarian Cancer Population Study (UKOPS) (Table 1; Supplemental Table 1, Data Sets 1–4; Song et al. 2009). Blood samples were taken at ages spanning a wide age range (50–85 yr) (Table 1). A total of 256 samples were from healthy postmenopausal women (Set-1 and Set-3). The remaining samples (n = 235) consisted of postmenopausal women diagnosed with primary epithelial ovarian cancer. About half of these (pre-treatment [preT] cases; n = 113; Set-2) gave their blood at the time of their diagnosis prior to treatment, and the other half (post-treatment [posT] cases; n = 122; Set-4) gave their blood at some stage during their follow-up visits after primary treatment (mean 2.4 ± 2.7 yr between diagnosis and blood sample taken). The distribution of all these samples across batches is given in Supplemental Table 2. Set-5 consisted of 188 whole blood samples from patients with type 1 diabetes mellitus (CG Bell, AE Teschendorff, V Rakyan, AP Maxwell, S Beck, and DA Savage, in prep.). Set-6 consisted of 177 ovarian cancer tissue specimens from pre-and postmenopausal women. Clinical characteristics of this cohort are provided in Supplemental Table 8. Details of the age distribution of samples per study is shown in Supplemental Figure 1. Full experimental methods and descriptions of other sample sets used in this study and any associated references are available in the Supplemental material. Ethical approval has been obtained for all sample sets.
DNA methylation profiling and quality control
Methylation analysis was performed using the validated Illumina Infinium Human Methylation27 BeadChip (Weisenberger et al. 2008). The methylation status of a specifc CpG site was calculated from the intensity of the methylated (M) and unmethylated (U) alleles, as the ratio of fluorescent signals β = Max(M,0)/[Max(M,0) + Max(U,0) + 100]. On this scale, 0 < β < 1, with β-values close to 1 (0) indicating methylation (no methylation). Quality control procedures are described in the Supplemental material. After quality control, singular value decompositions (SVD) were used to assess unwanted variation caused by experimental factors (variable bisulfite conversion efficiency, plate and chip effects) and to test the efficiency of interarray normalization procedures (full details are in the Supplemental material).
All primary data used in this study are available at the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE19711, GSE20067, and GSE20080.
Statistical analysis
Unsupervised analysis was performed using singular value decomposition (SVD), adapted to the methylation data, to determine the number of significant components of variation and their association with phenotypes (here, age). Supervised analyses were performed for each CpG site separately, using a robust linear regression model with age as the response and DNAm as the predictor, including covariates to model the batch, DNA input, and bisulfite conversion efficiency effects. FDRs were evaluated analytically (q-values) (Storey and Tibshirani 2003) as well as using random permutation of sample labels to take potential correlations between CpG sites into account. When data from potentially confounding experimental factors were not available, we used the surrogate variable analysis (SVA) framework (Leek and Storey 2007, 2008) to perform the supervised analysis and FDR estimation. Further details of methodology and software used are available in the Supplemental material.
Acknowledgments
We thank all the individuals who took part in this study and all the researchers, clinicians, and administrative staff who have enabled the many studies contributing to this work. In particular, we thank Andy Ryan, Jeremy Ford, Eva Wozniak, and Nyaladzi Balogun. This work was supported by the Eve Appeal and undertaken at UCLH/UCL, which received a proportion of its funding from the Department of Health NIHR Biomedical Research Centres funding scheme. A.E.T. was supported by a Heller Research Fellowship. S.B. was supported by the Wellcome Trust. D.S and P.M acknowledge the support of the Renal Unit Fund, Belfast Health and Social Care Trust. This work was supported in part by the Ovarian Cancer Research Fund (OCRF) and NIH/NCI grant R01-CA096958 (P.W.L.). We also thank Keji Zhao and Chongzhi Zang for sending us processed ChIP-seq data.
Footnotes
-
↵8 Corresponding author.
E-mail m.widschwendter{at}ucl.ac.uk.
-
[Supplemental material is available online at http://www.genome.org. The microarray data from this study have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession nos. GSE19711, GSE20067, and GSE20080.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.103606.109.
-
- Received November 26, 2009.
- Accepted February 11, 2010.
- Copyright © 2010 by Cold Spring Harbor Laboratory Press








