
Microarray-based multicycle-enrichment of genomic subsets for targeted next-generation sequencing
- Daniel Summerer1,4,
- Haiguo Wu2,
- Bettina Haase1,
- Yang Cheng1,
- Nadine Schracke1,
- Cord F. Stähler1,
- Mark S. Chee3,
- Peer F. Stähler1 and
- Markus Beier1
- 1 febit biomed gmbh, 69120 Heidelberg, Germany;
- 2 febit Inc., Lexington, Massachusetts 02421, USA;
- 3 Prognosys Biosciences Inc., La Jolla, California 92037, USA
Abstract
The lack of efficient high-throughput methods for enrichment of specific sequences from genomic DNA represents a key bottleneck in exploiting the enormous potential of next-generation sequencers. Such methods would allow for a systematic and targeted analysis of relevant genomic regions. Recent studies reported sequence enrichment using a hybridization step to specific DNA capture probes as a possible solution to the problem. However, so far no method has provided sufficient depths of coverage for reliable base calling over the entire target regions. We report a strategy to multiply the enrichment performance and consequently improve depth and breadth of coverage for desired target sequences by applying two iterative cycles of hybridization with microfluidic Geniom biochips. Using this strategy, we enriched and then sequenced the cancer-related genes BRCA1 and TP53 and a set of 1000 individual dbSNP regions of 500 bp using Illumina technology. We achieved overall enrichment factors of up to 1062-fold and average coverage depths of 470-fold. Combined with high coverage uniformity, this resulted in nearly complete consensus coverages with >86% of target region covered at 20-fold or higher. Analysis of SNP calling accuracies after enrichment revealed excellent concordance, with the reference sequence closely mirroring the previously reported performance of Illumina sequencing conducted without sequence enrichment.
Next-generation sequencing (NGS) platforms have transformed genetic variation studies by a massive reduction of cost and sequencing effort (Shendure et al. 2004, 2005; Margulies et al. 2005; Bentley 2006; Johnson et al. 2007; Harris et al. 2008). However, this technology advance has not yet been matched by an equal improvement at the front end: the isolation of target DNA sequences for analysis (Garber 2008). Although untargeted sequencing of even whole human genomes has been shown to be feasible, such large projects exceed the current capacity of NGS instruments and are cost prohibitive for the majority of research laboratories (Bentley et al. 2008; Wang et al. 2008). Many future applications would greatly benefit from focusing on specific genomic subsets. This can be the targeted sequencing of components of a single genome such as the whole exome but also fractions of more complex samples, for example, when applied to microbial communities, host–pathogen mixtures, or somatic variants.
Technologies are thus urgently required to selectively isolate genomic sequences at a scale and specificity that cannot easily be met by traditional enrichment approaches like PCR. An ideal enrichment technology for NGS would allow highly multiplexed access to any desired genomic loci. Enrichment thereby has to be uniform and efficient to enable maximal consensus coverage of the target region with sufficient depth for accurate base calling and with minimal sequencing effort. Furthermore, the method should not interfere with accuracy of base calling by causing allelic bias or dropout.
Several recent studies have started to address this bottleneck by using solution- or microarray-based sequence capture relying on hybridization. Two studies using solution-phase sequence capture with padlock or molecular inversion probes have been published that targeted large numbers of small genomic regions in a single reaction. Although the multiplexing level of one of these methods was high, low uniformity of coverage was reported as a serious drawback of both of these approaches (Dahl et al. 2007; Porreca et al. 2007). Still another approach made use of long, biotinylated RNA probes for solution-phase hybridization. However, the overall workflow depended on multistep enzymatic processing of DNA capture probes including PCR and in vitro transcription, possibly introducing bias and errors into the probe library. Moreover, very long hybridization times of several days were applied (Gnirke et al. 2009), which is rather time-consuming even compared with approaches relying on solid-phase hybridization.
Recently, sequence enrichment using solid-phase hybridization to DNA microarrays with flexible content has been described (Albert et al. 2007; Hodges et al. 2007; Okou et al. 2007; Bau et al. 2009). For several projects targeting different regions, enrichment factors of several hundred- to a 1000-fold have been reported, resulting in good depth of coverage for at least a fraction of the target region. However, covering the full target region with the depth sufficient for reliable base calling has emerged as a key challenge (Garber 2008).
In fact, no method has so far been able to reach an enrichment performance that allows for full consensus coverage of a target with satisfactory depth, and before now, it was not clear whether optimization of the most obvious experimental variables such as hybridization stringency, probe design, or blocking conditions would overcome this problem. Given that reported target sizes are typically in the range of kilobases to megabases, the fraction of target sequence in a human DNA sample relative to background is only 3.1 × 10−5% to 3.1 × 10−2% for 1 kb and 1 Mb, respectively. This range of concentration presents a serious purification challenge, e.g., similar to the most demanding protein purifications. Although the specificity of protein–protein interactions employed in protein purifications (e.g., antibody–antigen interactions or affinity tag binding) can be much higher than the specificity of Watson-Crick base pairing, the application of multiple rounds of chromatography is a standard procedure to obtain target protein of sufficient purity (Coligan et al. 2008).
We transferred this purification strategy to DNA sequence isolation by performing two instead of one cycles of enrichment using microfluidic Geniom biochips before Illumina NGS. We show that for different target sequences enrichment performance dramatically increases from the first to the second cycle, indicating a multiplicative effect. This effect on enrichment performance is accompanied by a significant increase of the percentage of target region being covered. This results in higher enrichment factors than previously reported for sequence capture methods prior to Illumina NGS (Hodges et al. 2007; Gnirke et al. 2009). A comprehensive analysis of SNP calling performance after enrichment shows that the method does not interfere with base-calling accuracy.
Using a microfluidic array platform with integrated hardware thereby results in several advantages. The hybridization steps employed are four times shorter than in other methods, which results in shorter overall process times. Furthermore, the process can be highly automated, which supports improved handling effort, reduces contamination risk, and increases reproducibility.
Results and Discussion
The sequence enrichment technology reported here, called HybSelect, is conducted in three main steps: hybridization, washing, and elution. First, a genomic DNA library is hybridized to a Geniom biochip containing target-specific DNA capture probes. After washing and elution, the sample is subjected to a second cycle of enrichment and analyzed by an NGS platform. Though the process should be applicable to any NGS platform, experiments for this study were analyzed using the Illumina Genome Analyzer II (GAII).
Capture of cancer-related genes
We chose the human genes BRCA1 and TP53 as our first targets for enrichment, because of their well-known role in the development of certain cancers.
We designed an array of 50mer DNA oligonucleotide probes with a tiling density of 8 bp. A Geniom biochip is composed of eight individual microfluidic channels, each having a capacity for >15,000 capture probes; we used part of one channel for synthesis of the tiling array. To prevent the enrichment of repetitive elements, we excluded low-complexity probes from the array design, which reduces the region of interest (ROI) of 100 kb to a core region of 54 kb actually covered by capture probes (hybselected region [HR]). This corresponds to a capacity of >1.8 Mb ROI or >1 Mb HR per biochip. Next, we subjected a human Illumina paired-end library to a first round of hybridization on the biochip for 16 h with active mixing of the sample.
Two independent experiments, A and B, were conducted in parallel to test the reproducibility of the process. After four consecutive washing steps, we eluted the samples and amplified them using the Illumina paired-end primers, which afforded sufficient amounts for a second hybridization step. Processing of the enriched samples on an Illumina GAII instrument yielded 8,217,673 and 7,624,181 paired-end reads of 2 × 36 bp for the individual samples. The reads were used for further analysis after homopolymeric and ambiguous sequences were filtered out.
After this first cycle of enrichment, mapping of the reads to the ROI revealed that 61.8% to 88.8% of the HR was covered at least once, exhibiting a similar range to what was previously reported for one cycle of microarray-based sequence enrichment and Illumina sequencing (Table 1). In this study, between 12% and 91% of target sequence were reported to be covered at least once, depending on sequence context and library fragment size (Hodges et al. 2007). The average depth of coverage was between 2.9- and 5.0-fold for all target regions for both experiments (Table 1). Overall, the data suggest similar or better reproducibility than previously reported for microarray-based sequence capture (Albert et al. 2007; Hodges et al. 2007; Okou et al. 2007; Bau et al. 2009). Importantly, analysis of the uniqueness of obtained read pairs revealed that more than 98% for both runs, were unique, which is higher than previously reported for standard Illumina GAII sequencing without any enrichment method (Quail et al. 2008). This clearly shows that no detectable library representation bias has been introduced during the HybSelect process that would compromise the information value of obtained reads.
Mapping data of reads obtained from one or two cycles of array-based sequence enrichment of human genomic DNA samples for different target regions and Illumina GAII paired-end sequencing
Impact of a second enrichment cycle on capture performance
We next subjected the enriched sample from experiment A to a hybridization process under the same conditions applied in the first enrichment cycle. Sequencing yielded 7,433,555 paired end reads of 2 × 36 bp that were filtered as described above.
Figure 1 shows a graphic view of the ROI with HR regions and coverage depth distribution of mapped reads from the first and second cycle for a representative region of TP53. Reads were obtained almost exclusively in the HR that is covered by capture probes with some overlap to adjacent regions. Moreover, the second cycle experiment strongly increased depth of coverage and apparently also uniformity over the whole region compared with the first cycle of enrichment. Overall, 96.5% and 98.5% of BRCA1 and TP53 were covered at least once after this second enrichment cycle (Table 1). The individual enrichment factors (representation of HR sequence in the obtained sequence reads divided by their representation in the human genome) for the two genes obtained from the second cycle were 15.6- and 22.6-fold, respectively, similar to the enrichment factors for the first cycle (22.9- and 27.3-fold), which indicates a multiplicative enrichment effect. This resulted in final enrichment factors for the overall process of 356.4- and 616.9-fold. Interestingly, quantitative analyses suggest that biochips that are reused for the second enrichment cycle result in comparable enrichment factors as observed for the standard process (Supplemental Fig. 1).

Graphic overview of mapping analysis of an Illumina paired-end sequencing run with a human genomic DNA sample enriched for the genes BRCA1 and TP53. Shown is the capture probe region used for array-based enrichment (black line at top), coverage depth distribution obtained from the first enrichment cycle (middle), and coverage depth distribution from the second enrichment cycle (bottom) to a representative part of the TP53 gene (nucleotides ∼9500–14,000). The obtained consensus sequences are shown as black lines. X-axis, the nucleotide position of the gene; y-axis, the fold coverage depth. Note that the scale of the y-axis varies between the two mappings.
Further analysis revealed that the average depth of coverage was also higher for both regions after the second enrichment cycle, being 58.1- and 101.3-fold for BRCA1 and TP53, respectively.
However, the most striking effect was observed for consensus coverages of the HR (percent of HR covered with reads) at increased minimum coverage depths. These numbers are especially important, since a certain minimal depth of coverage is generally required for base calling. This makes a consensus coverage with the minimal depth for reliable base calling the most relevant parameter of an experiment in terms of analytical value for the targeted region. Recent whole human genome sequencing projects using Illumina technology revealed that >95% of both homo- and heterozygous single nucleotide polymorphisms (SNPs) can be accurately called at a coverage depth of 20-fold or higher when paired-end reads are used (Bentley et al. 2008; Wang et al. 2008). The consensus coverage of the HR (i.e., target region) at more than 20-fold depth of coverage can therefore be considered a key parameter for targeted NGS using Illumina instruments.
Strikingly, the consensus coverage with at least 20-fold coverage depth increased between 46- and 96-fold for the two genes from the first to the second cycle of enrichment (Table 1). In total, 68.8%–86.2% of the target regions were covered at ≥20-fold, exceeding previously reported data for targeted sequencing using microarray-based enrichment and Illumina NGS (Hodges et al. 2007).
Capture of 1000 SNP loci
A crucial performance criterion of an enrichment method is its accuracy of base calling. In principle, several steps of the overall process could lead to allelic bias or dropout, which would prevent the practical use of the method for resequencing studies.
To evaluate our method in this direction, we aimed at the enrichment of 1000 nonoverlapping loci of 500-bp size throughout the human genome, each harboring a central dbSNP position. Capture probes with a tiling density of 8 bp were synthesized on four channels of a Geniom biochip, and genomic DNA of two CHB individuals (Chinese individuals from Beijing, HapMap IDs NA18558 and NA18561) was subjected to the two-cycle HybSelect process as described above.
A total of 19,762,440 and 19,405,469 paired end reads of 2 × 36 bp were obtained that were mapped to the ROI after filtering. For the two samples, enrichment factors of 713.3- and 1061.9-fold were obtained. This resulted in average depths of coverage of 315.6- and 469.1-fold over the whole HR (Table 1). Importantly, 80.4% or 85.5% of the HR for all 1000 regions was covered with a depth of at least 20-fold, corresponding well to the obtained consensus coverages for BRCA1 and TP53. This should allow for reliable analysis of most nucleotide positions within the targeted sequence regions.
We performed detailed analysis of consensus coverages and read distributions on the level of the individual loci (a list containing the locus-wise analysis of obtained reads, consensus coverages at one-, five-, 10-, and 20-fold depth of coverage, enrichment factors, and average coverage depths can be found in Supplemental Table 1). Figure 2 shows a histogram of the average depths of coverage for all loci. Remarkably, most regions were covered at a depth of between 250- to 500-fold, with decreasing numbers for higher and lower coverage depths. On average, 90% and 94% of the regions were covered at ≥20-fold, respectively.
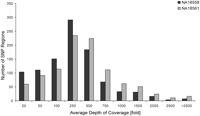
Statistical analysis of average coverage depths and consensus coverages of 1000 human 500-bp loci obtained from mapping analysis after sequence enrichment from the two HapMap reference samples NA18558 and NA18561 and Illumina GAII sequencing. Shown is a histogram of average coverage depths for the HR of individual 500-bp loci for both samples as depicted in the figure.
Next, we analyzed the uniformity of coverage depth for the whole set of loci. For the most cost-effective sequence capture, uniformity should be maximal since this avoids redundant reads in overcaptured regions. We found that across all regions a fraction of 27%–30% exhibited the average depth of coverage or more. Fifty-one percent to 53% had a normalized coverage depth of 0.5-fold, the average depth of coverage (Supplemental Fig. 2). These data match a uniformity recently reported for a solution-phase capture experiment combined with Illumina NGS technology for a comparable, discontiguous exon target (Gnirke et al. 2009). The availability of long-read platforms like the Roche/454 instrument and the continuing increase of read lengths of the Illumina Genome Analyzer and the ABI SOLiD system raise the question how this might impact the coverage characteristics of the method when applied to these systems. We anticipate that longer read lengths might further improve uniformity and consensus coverages, since regions with lower coverage could be rescued by reads from fragments captured at more distant sites.
We next questioned how individual sequence contexts impact the capture performance for the specific regions. Analysis of the correlation between average depth of coverage and GC-content of the 1000 regions for NA18561 revealed that 99.9% of all regions with a moderate GC-content of 40%–60% were covered >20-fold and 98.8% even >50-fold (Supplemental Fig. 3). This suggests considerable potential to even improve the observed capture performance by simple alterations in probe design.
Regional coverage distribution
The design of the dbSNP loci capture experiment with nonoverlapping regions of identical size and targeted with identical numbers of capture probes allows a facile statistical analysis of the average spatial distribution of coverage depth over all 1000 ROIs.
It is important to evaluate which fraction of coverage falls into the HR. Since library molecules can extend into the adjacent region within range of the fragment size distribution of the library, sequencing reads can be generated for this noninformative part of the ROI. This effectively decreases the achievable fraction of desired data in the NGS instruments sequence output. Previous microarray studies indicate that the fraction of reads falling into a probe region follows a binomial pattern and depends on the sizes of these regions and the length of the library fragments. The larger the probe region and the shorter the fragment size are, the lower the overlap and the lower the content of noninformative sequence tend to be (Hodges et al. 2007).
In a recent publication, there is further supporting evidence for the notion that longer capture probes could also increase the fraction of noninformative reads. In this study (Gnirke et al. 2009), 170mer probes were used, exceeding the 120-bp median length of human exons. Since library fragments preferentially hybridize with a maximal part of the probe sequence, this leads to considerable overlap into surrounding regions and only a small fraction of 47% in the informative regions. This diminishes the practical use of this enrichment approach for Illumina end sequencing with standard read length.
Analysis of spatial coverage depth distribution for our experiment (NA18561) revealed a binomial pattern with maximal coverage depths in the middle of the HR and relatively low representation of reads falling into noninformative regions (Fig. 3). Coverage depth was thereby highly uniform with only approximately twofold higher depth for the center compared with the edges of the probe regions. Overall, 81% of total coverage was obtained for the targeted HR.
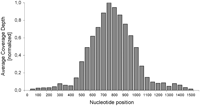
Average spatial distribution of coverage depths for ROI of 1000 human 500-bp dbSNP loci obtained from mapping analysis after sequence enrichment from a human genomic DNA sample and Illumina GAII sequencing. The x-axis shows the nucleotide positions of the ROI, consisting of the core region covered by capture probes for array-based sequence enrichment (HR, nucleotide positions 501–1000) with flanking regions of ±500 nucleotides. The y-axis shows the coverage depth for all 1000 loci of sample NA18561 averaged for each 50-bp segment and normalized to the maximal depth of coverage.
SNP calling accuracy
To assess the applicability of the approach for SNP detection, we analyzed the nucleotide representations of the 1000 captured dbSNP positions. Six hundred of these SNPs were chosen from chromosome 1 and have previously been genotyped in the HapMap project; 400 additional HapMap SNPs were chosen from ENCODE regions on several different chromosomes (dbSNP IDs can be found in Supplemental Table 1). SNPs were thereby selected to have an increased content of 50% heterozygous genotypes within the HapMap CHB population. This allows a balanced analysis of homo- and heterozygous positions and imposes a higher challenge to the process owing to higher coverage requirements and potential bias in nucleotide representation for heterozygous positions. We first filtered the regions for SNP coverage depths of 20-fold or higher as a stringent and pre-established criterion for reliable base calling (Bentley et al. 2008; Wang et al. 2008). Of 1000 SNPs, 913 SNPs fullfilled this criterion, with 449 being homozygous and 464 being heterozygous in the reference data (sample NA18561, Supplemental Table 2). Nucleotide analysis and comparison with HapMap reference data (data from HapMap project phases 1 and 2) revealed an overall concordance of 98.6% for all SNPs. Notably, concordance was significantly higher for homozygous positions (99.1%) than for heterozygous positions (98.1%), which suggests that combined call rates for both allele types would be higher for regions that are not enriched for heterozygous occurrences. Analysis of all 464 heterozygous SNP positions revealed an allelic ratio of 0.49, indicating a well-balanced enrichment of both alleles.
Interestingly, very similar concordance (98.8–99.1%, depending on mapping algorithm) was previously reported for nontargeted whole-genome sequencing using Illumina technology and comparison to HapMap reference data of the same project phases (Bentley et al. 2008; Wang et al. 2008). This indicates that the HybSelect process does not interfere with the accuracy of SNP calling and provides a useful tool for resequencing studies.
Conclusion
Sequence enrichment performance
Although several approaches for enrichment of genomic sequences have been reported, no method so far has shown an enrichment performance allowing for reliable SNP calling over the full target region. This has previously been highlighted as the main challenge for hybridization-based sequence enrichment and severely impairs the actual power of NGS technologies (Garber 2008).
Our data show that enrichment factors, consensus coverage, and average depth of coverage for target regions can be multiplied by applying two instead of one enrichment cycle. Compared with two recent studies reporting targeted enrichment using Illumina NGS technology, this resulted in superior enrichment performance and excellent consensus coverages for all targeted regions. Importantly, our calculation of enrichment factors does not include a prefiltering of raw reads for reads uniquely mapping to the human genome. This can reduce the fraction of usable raw reads by a factor of ∼0.4–0.5 (Gnirke et al. 2009), whereas the number of unique reads mapping to the target should not be altered. Since this affects the ratio of on-target reads vs. total reads and thus the calculation of enrichment factors and the fraction of on-target reads, we believe that our actual process performance is even better in terms of these parameters than reported here.
Furthermore, this performance was achieved with standard short-read end-sequencing and should further improve with increasing read lengths. Average coverage depths in our experiments exceed those in other studies using this sequencing mode by up to more than one order of magnitude. Uniformity of coverage thereby matches comparable experiments as reported previously.
Uniqueness of NGS reads received after sequence enrichment has not been analyzed in previous studies and consequently the actual value of published coverage depths remains unclear. In contrast, our data show that no significant representation bias is observed in libraries after the HybSelect process, which indicates that no PCR duplicates account for the observed performance. We further showed that the process does not interfere with SNP calling and allows for efficient resequencing of large fractions of the targeted regions with accuracies typically observed for Illumina NGS technology with nonenriched samples.
Advantages of microfluidic biochip architecture
Previous approaches for sequence enrichment employed hybridization steps of >60 h and multiple manual washing and elution steps resulting in long processing times (Albert et al. 2007; Hodges et al. 2007; Okou et al. 2007; Gnirke et al. 2009).
Microfluidic array architecture with associated short hybridization times and a high level of automation throughout the HybSelect procedure enables fast processing and easy handling, despite the use of two enrichment cycles. The total process time starting with a sequencing library and resulting in an enriched, purified, and quantified library ready for Illumina sequencing is less than 60 h, shorter than the hybridization step of any previously reported approach alone.
The used biochips are scalable between one and eight samples and/or 230 kb and >1.8 Mb ROI (125 kb–1 Mb HR) with only 1.5 μg of Illumina library needed per array. This scalability facilitates adjustment of an experiment to different target sizes and can significantly reduce per sample cost for small targets. Further quantitative analyses suggest that biochips can be reused within the two-cycle protocol with typical enrichment performances, which would reduce cost of the approach.
We believe that further improvements in probe design and process optimization will allow us to reach depths of coverage that will enable efficient multiplexing of pooled samples. The general strategy to apply iterative cycles of sequence enrichment might thereby not only facilitate efficient targeted NGS for human genomic subsets. It might also enable analysis of much more complex samples that demand enrichment factors far beyond the possible limit of a single-cycle experiment, e.g., for environmental samples, low abundance cancer cells, or pathogens in a human background. We are therefore convinced that the HybSelect enrichment method will find wide application for large-scale, targeted genomics studies.
Methods
Microarray design and synthesis
Light-activated in situ oligonucleotide synthesis on Geniom biochips (febit biomed gmbh, Heidelberg, Germany) was performed as described previously (Baum et al. 2003). One biochip contains eight individual, microfluidic channels each containing an array of >15,000 individual DNA probe features.
For the enrichment of the two human genes BRCA1 and TP53, 50mer probes were tiled across the target regions with a density of 8 bp, corresponding to a total ROI of 100 kb or a capacity of >1.8 Mb per biochip. Probes were allowed to have a maximal content of 25 low-complexity bases in a row and a maximal total content of low-complexity bases of 80% according to the Hg18 annotation. This resulted in 6700 probes and a reduction of the ROI to the actual probe region (Hybselected region [HR]) of 54 kb, corresponding to a total capacity per biochip of >1 Mb HR.
For enrichment of the 500-bp dbSNP loci, 1000 nonoverlapping regions from high-complexity sequence context throughout the human genome were chosen containing a central dbSNP position. A total of 57,000 50mer probes were designed with a tiling density of 8 bp and synthesized on four array channels again resulting in a capacity of >1 Mb HR per biochip. For all experiments, array designs for the two enrichment cycles were identical.
DNA sample preparation
Human genomic DNA samples NA18558 and NA18561 were obtained from Coriell Repositories. DNA samples for enrichment of BRCA1 and TP53 were purchased from Promega. Five micrograms of human genomic DNA were dissolved in 190 μL of water and fragmented for 30 min by sonication at high intensity (Bioruptor, Diagenode). Preparation of the paired-end adaptor-ligated gDNA library ready for sequencing on an Illumina Genome Analyzer II (Illumina) was performed according to the manufacturer's standard protocol including excision of the size fraction of 300–400 bp from an agarose gel. The sample was analyzed by a Bioanalyzer experiment (Agilent), quantified by UV measurement (Nanodrop 1000, Thermo Scientific), and stored in water at −20°C until use.
Hybridization and elution
For each array, 1.5 μg of an adaptor-ligated gDNA library were dissolved in febit Hybmix-4 or -5, heated to 95°C for 5 min, and placed on ice. The sample mixture was injected into the microfluidic arrays of the biochip and hybridization was performed for 16 h at 45°C or 50°C with active movement of the sample using a febit active mixing device. After hybridization, each array was automatically washed with 6× SSPE at room temperature and 0.5× SSPE at 45°C within the Geniom One instrument (febit biomed gmbh). Each array was subsequently washed with SSPE-based febit stringent wash buffers 1 and 2 at room temperature. For elution of the enriched samples, arrays were each filled with 10 μL of febit elution reagent in a febit hybridization holder and incubated at 70°C for 30 min. Solution was manually transferred into an Eppendorf tube and dried by vacuum centrifugation in a Speed-Vac at 65°C. After an amplification step according to the Illumina library preparation procedure using paired-end primers for 18–35 cycles, the sample was treated like the original library and subjected to a second round of enrichment under the same conditions as before. After enrichment, hundreds of picograms of DNA library are typically recovered from each array depending on the array template as judged by qPCR using the Illumina adaptor primers and SYBRgreen quantitation (data not shown).
NGS using Illumina technology
Eluted samples were subjected to 10 cycles of PCR according to Illumina paired-end library preparation kit and purified by a MinElute PCR purification column (Qiagen). Quantification of samples was done by the Quant-It Picogreen assay (Invitrogen) using the Nanodrop 3300 instrument. Sequencing was performed using an Illumina GAII system using the paired-end mode and read lengths of 36 bp according to the manufacturer's protocol.
Data analysis
Paired-end sequencing reads were first filtered by removing reads with ambiguous nucleotide calls (three or more N) and reads with 34 or more A (or T or C or G). Reads from File 1 and File 2 of the two paired-end sequencing runs were aligned to target genes by using RazerS (Weese et al. 2009), which is part of SeqAn, an open-source C++ library of efficient algorithms and data structures for the analysis of biological sequences (Doring et al. 2008). The parameters used were “-gn 1 -f -r -i 94 -rr 100 -m 10,” which allows up to two mismatches. The output alignment files were matched for each pair of reads: The two reads were mapped to opposite strands and in correct orientation and the length between the two reads (inclusive) was within 100–500 bp. The paired reads were matched to the ROI to obtain the reads for analysis of coverage depth. For the 1000 SNP loci experiment, the HR (being all loci of 500 bp) with extensions of ±500 bp for each locus was defined as ROI. The fold coverage for each base within the probe regions was calculated. For unique amplicon analysis, each pair of read sequences was counted only once, and duplicates were ignored. For visualization, reads on the HR obtained by paired-end mapping were mapped with the CLC genomics workbench using single-end mode and default conditions. For SNP analyses, base representations for each target position were calculated in percent. For positions with one base represented >90%, position was called homozygous. If no position was represented >90%, but two bases were represented >10%, position was called heterozygous for these two bases.
Acknowledgments
We thank Jack Leonard and Sonja Vorwerk for helpful discussions and critically reading the manuscript. We thank Andreas Keller for his assistance in setting up razerS for efficient alignment. We thank Anthony Caruso and Marcel Kränzle for assistance in data analysis.
Footnotes
-
↵4 Corresponding author.
E-mail daniel.summerer{at}febit.de; fax +49-6221-6510-390.
-
[Supplemental material is available online at http://www.genome.org. The sequence data from this study have been submitted to the NCBI Short Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi) under accession no. SRA009002.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.091942.109.
-
- Received February 5, 2009.
- Accepted June 18, 2009.
-
Freely available online through the Genome Research Open Access option.
- Copyright © 2009 by Cold Spring Harbor Laboratory Press








