
The polyadenylation site of Mimivirus transcripts obeys a stringent ‘hairpin rule’
- Deborah Byrne1,
- Renata Grzela1,
- Audrey Lartigue1,
- Stéphane Audic,
- Sabine Chenivesse,
- Stéphanie Encinas,
- Jean-Michel Claverie2 and
- Chantal Abergel2
- Structural and Genomic Information Laboratory, CNRS-UPR 2589, IFR-88, Aix-Marseille University, Parc Scientifique de Luminy, Case 934, 13288 Marseille Cedex 9, France
-
↵1 These authors contributed equally to this work.
Abstract
Mimivirus, a giant DNA virus infecting Acanthamoeba, is revealing an increasing list of unique features such as a 1.2-Mb genome with numerous genes not found in other viruses, a uniquely conserved promoter signal, and a particle of unmatched complexity using two distinct portals for genome delivery and packaging. Herein, we contribute a further Mimivirus distinctive feature discovered by sequencing a panel of viral cDNAs produced for probing the structure of Mimivirus transcripts. All Mimivirus mRNAs are polyadenylated at a site coinciding exactly with unrelated, but strongly palindromic, genomic sequences. The analysis of 454 Life Sciences (Roche) FLX cDNA tags (150,651) confirmed this finding for all Mimivirus genes independent of their transcription timings and expression levels. The absence of a suitable palindromic signal between adjacent genes results in transcripts encompassing multiple ORFs in the same or even in opposite orientations. Surprisingly, Mimivirus tRNAs are expressed as polyadenylated messengers, including an ORF/tRNA composite mRNA. To our knowledge, both the nature and the stringency of the “hairpin rule” defining the location of polyadenylation sites are unique, raising once more the question of Mimivirus's evolutionary origin. The precise molecular mechanisms implementing the hairpin rule into the 3′-end processing of Mimivirus pre-mRNAs remain to be elucidated.
Acanthamoeba polyphaga Mimivirus, a double-stranded DNA virus infecting several Acanthamoeba species, is the largest and most complex virus isolated to date (Raoult et al. 2004; Claverie et al. 2008). It is the prototype member of the new family “mimiviridae” and belongs to the generic category of Nucleo-Cytoplasmic Large DNA viruses (NCLDVs). This superfamily was introduced to highlight the probable common evolutionary origin of four diverse families of eukaryotic DNA viruses: poxviruses, asfarviruses, iridoviruses, and phycodnaviruses (Iyer et al. 2006). A phylogenetic analysis of the set of core genes shared among these diverse viruses firmly anchored Mimivirus within the NCLDVs, closer to the phycodnaviruses (Raoult et al. 2004; Claverie et al. 2008). However, the complete transcription apparatus encoded by Mimivirus, as well as the complex proteome of its particle embarking most of the Mimivirus-encoded transcription proteins, is more reminiscent of large poxviruses (Lefkowitz et al. 2006; Renesto et al. 2006; Claverie et al. 2008).
Besides its record genome size (1.2 Mb) and gene content (911 encoded proteins), Mimivirus exhibits an increasing number of unique and intriguing features. The finding of many Mimivirus genes not found in other viruses, such as aminoacyl-tRNA synthetases (Abergel et al. 2007), revived the debate about the evolutionary origin of large nucleo-cytoplasmic DNA viruses (Claverie 2006; Forterre 2006; Iyer et al. 2006; Claverie et al. 2008). Among other oddities, half of Mimivirus genes present an identically conserved promoter signal AAAATTGA (Suhre et al. 2005). The icosahedral Mimivirus particle is made of at least 114 proteins (Renesto et al. 2006) and, among all known double-stranded DNA viruses, is the only one to possess two distinct portals—one for genome delivery, the other for packaging (Zauberman et al. 2008). The content of Mimivirus particles is delivered to the host-cell cytoplasm by the fusion of an internal viral membrane with the cell membrane, following the simultaneous opening of five icosahedral faces at a unique vertex of the capsid, coined the “stargate” (Zauberman et al. 2008). More recently, La Scola et al. (2008) reported the isolation of a new strain of Mimivirus, associated with a new type of satellite DNA virus they called a “virophage.”
With the exception of structural/functional studies on individual gene products (Jeudy et al. 2005; Benarroch and Shuman 2006; Benarroch et al. 2006, 2008; Abergel et al. 2007; Monné et al. 2007; Thai et al. 2008), little attention has yet been devoted to the basic molecular processes at work during the Mimivirus replication cycle. As a first step in that direction, we set out to probe the structure of Mimivirus transcripts using an old-fashioned gene-by-gene approach. Eukaryotes (and their viruses) attach a poly(A) tail to the 3′-ends of most nuclear-encoded mRNAs. Models of polyadenylation signals involving a simple sequence consensus (such as AAUAAA) have progressively shown their limits; current models now include a large number of other signals with three types of sequence elements (cis-elements): far upstream elements, near upstream elements (e.g., AAUAAA in mammals), and cleavage sites, all of them located near the pre-mRNA 3′-end. Paradoxically, although this 3′-end processing is essential for the maturation of pre-mRNA among eukaryotes, the corresponding signals differ widely among yeast (Mandel et al. 2008), plants (Loke et al. 2005), algae (Wodniok et al. 2007), amoebae (Lopez-Camarillo et al. 2005), or animals (Mandel et al. 2008). For instance, the near upstream element AAUAAA, highly conserved in mammals, is less present in plants and is nowhere to be found in yeast or green algae. This contrasts with the evolutionary conservation of the process as a whole and its biochemical simplicity (RNA cleavage and polyadenylation). Tackling the notoriously difficult problem of predicting the 3′-end of mRNA in plants in absence of well defined cis-elements, Loke et al. (2005) proposed that RNA secondary structures rather than conserved sequences might play a significant role in guiding the 3′-end processing of pre-mRNAs.
In this study, we present the analysis of several cDNA sequences, in a search for the signals governing the polyadenylation of Mimivirus transcripts. This first analysis of the Mimivirus replication cycle at the cellular level revealed an original, stringent, and well-conserved signal defining the polyadenylation site, placing Mimivirus once more as an outsider among known microorganisms.
Results
Forty-two out of 45 individually sequenced mRNAs terminate within a high-scoring palindrome
The panel of tested genes (Table 1) was selected to include representatives of various gene types such as putative late-early genes (preceded by the AAAATTGA early promoter upstream element) (Suhre et al. 2005), genes encoding proteins associated with the particle (Renesto et al. 2006; Claverie et al. 2008), genes encoding proteins of unknown functions, genes unique to Mimivirus (e.g., aminoacyl-tRNA synthetases), or typical NCLDV genes (e.g., RNA polymerase). Total RNA was isolated from Acanthamoeba castellanii cells 30 min to 12 h post-infection by Mimivirus, and double-stranded cDNAs were produced using an optimized protocol. Individual cDNA were amplified using 3′-oligo(dT) primers and internal primers specific for each selected gene, and the resulting amplicons were sequenced. Throughout this paper, the last nucleotide at the 3′-end of the cDNA sequences that could be unambiguously mapped on the Mimivirus genome (i.e., non A) will be referred to as the polyadenylation site, in the absence of information about the detailed enzymatic mechanisms generating the mature mRNA 3′-ends. Once precisely defined, the (usually) short 3′-UTR sequences (Table 1) and adjacent genomic sequences were scrutinized in search of any conserved and statistically significant sequence motif that may serve as a termination/polyadenylation signal. Standard methods (for review, see Tompa et al. 2005) failed to identify a significant consensus motif of linear sequence within the 3′-UTR sequences we initially determined. Instead, by aligning the 3′-end of the experimentally sequenced cDNA onto the Mimivirus genome, the beginning of the poly(A) tail was found to be precisely located within a putative secondary structure element corresponding to the top-scoring palindrome immediately following (and sometimes overlapping) the ORF stop codon (Table 1).
Mapping of the 3′-polyadenylation sites of a panel of selected genes
Mimivirus genome exhibits a strong bias in the distribution of palindromic sequences
As shown in Table 1, the experimentally validated palindromes vary in length (total hairpin length from 26 to 46, including the hairpin stem and loop) and number of mismatches (from 0 to 3). They bear no significant sequence similarity with each other, except that they are mostly composed of A or T (A+T = 86%). The statistical significance of these palindromes (and putative associated RNA hairpin structures) was explored using various approaches. We first examined the frequency of putative hairpin structures (scoring over a given threshold as computed by RNAMotif) (Macke et al. 2001) in the annotated coding regions (CDS) versus their 3′-downstream intergenic regions (3′-IR). For a score threshold of 13 (e.g., a stem of 15 nucleotide [nt] with two mismatches), and a descriptor (see Methods) allowing loops of 0 up to 5 nt, 565 3′-IRs versus 127 CDSs were found to contain at least one occurrence of putative hairpin. Such a bias is highly significant, given that 82% of the Mimivirus genome corresponds to CDSs (P < 0.1%, 2 × 2 χ2 assuming an equal density of these motifs in CDS and 3′-IR as a null hypothesis).
As CDS are under different compositional (e.g., codon usage) constraints than 3′-IRs, the above CDS versus 3′-IR bias was further validated by computing the frequency of these putative hairpin motifs within randomly shuffled 3′-IR sequences (Supplemental Table S1). Using the same threshold as above, 565 3′-IRs exhibited the putative hairpin motif, while only 36 (±5.3) were found in the randomized 3′-IRs (Z-score ≈ 99, P ≪ 10−10). Even though such a simple shuffling of DNA sequences is known to overestimate the statistical significance of motifs (by neglecting higher-order Markov chain structure), these results indicate that the higher frequency of putative hairpin sequences in Mimivirus 3′-IRs is not a mere consequence of their highly biased (A+T-rich) composition. This is further confirmed by the fact that viral genomes with even higher A+T contents (Melanoplus sanguinipes entomopoxvirus, 81.7% A+T; Amsacta moorei entomopoxvirus, 82.2% A+T) are not enriched in palindromic sequences (Supplemental Table S1).
Figure 1 illustrates the sharp transition of the frequency of putative hairpin structures before and after the stop codon of each ORF. This graph also indicates that a sizeable number of these putative termination/polyadenylation signals overlap with the stop codon, thus predicting very short 3′-UTRs. This prediction was experimentally tested and validated for two genes (Table 1: L39, L46) with 3′-UTRs of lengths 1 nt and 6 nt, respectively.
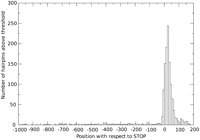
Histogram of the initial positions of the palindromes scoring above threshold with respect to the stop codons. X = 0 is the position of the downstream stop codon of each Mimivirus gene. Palindrome positions refer to their 5′ extremities. A bin width of 10 nt was used. The presence of the palindromes strongly correlates with the beginning of the 3′-UTR, just after the stop codon. Forty palindromes overlap the stop codon, leading to extremely short 3′-UTRs (Table 1).
The same palindrome can be used for two convergent transcripts
By definition, a perfect palindrome corresponds to a sequence that reads the same on the two opposite strands. One thus expects that genes that are transcribed in a convergent manner (i.e., share a common 3′-IR) might use a common palindrome, read from their respective strand, to define the polyadenylation site of their mature mRNAs (Fig. 2B). This was experimentally tested by sequencing the cDNAs of four pairs of predicted convergent transcripts: R257/L258, R453/L454, R497/L498, and R528/L529. As expected, all pairs of transcripts were found to be polyadenylated within the same palindrome read from the two opposite strands (Table 1B). However, none of these pairs of transcripts ended at the exact same (or symmetrical) position in the putative hairpins. This might result from their slightly different folding energies when read from the opposite strands (stem mismatches, nonidentical loops sequences). The unrelated 3′-UTR sequences flanking the palindromic region could also influence the hairpin formation and recognition process.
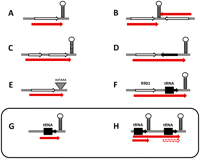
Experimentally validated Mimivirus polyadenylated transcript structures. Palindromic sequences are represented by hairpins; ORFs are indicated by open arrows; and transcripts by red arrows. (A) Single ORF followed by a palindromic polyadenylation signal; ∼70% of transcripts might fall in this category; 27 have been experimentally validated through a gene-by-gene analysis. (B) Convergent ORFs sharing the same palindromic polyadenylation signal; four such pairs have been validated (R257/ L258, R453/ L454, R497/L498, R528/L529). (C) “Polycistronic” transcripts, wherein polyadenylation occurs within the first palindromic signal encountered. Two such transcripts have been validated (L416-417, R882-883). (D) “Pseudo-polycistronic” messenger, wherein the 3′-UTR encompasses a neighboring ORF in the reverse orientation. Two such cases have been validated (L356-R355, L778-R777). (E) Infrequent cases of polyadenylation occurring within a low-scoring palindrome (e.g., L164) or following an AATAAA motif (e.g., R502, L532, polycistronic R463-465). (F) A unique case of a polyadenylated bi-cistronic transcript mixing the R901 ORF and a downstream neighboring tRNALeu. (G) Polyadenylated transcripts of individual tRNA genes (tRNATrp, tRNAHis). (H) Polyadenylated transcripts encompassing two adjacent tRNA genes (tRNAHis + tRNACys) coexisting with a single gene polyadenylated transcript (tRNAHis).
The absence of palindromic signal leads to polycistronic transcripts
The above analysis guided the further exploration of the 3′-UTR structure of Mimivirus transcripts, focusing on genomic intervals not showing the canonical one gene–one palindrome organization. To our surprise, these transcripts turned out to be more difficult to amplify and, in successful cases, resulted in amplicons of much larger sizes than expected. Successive rounds of sequence walking using specific primers finally established that these genes were expressed as polycistronic transcripts. We encountered two different cases (Fig. 2C,D), albeit presumably obeying the same rule: The transcription of a gene lacking a significant palindrome in its 3′-IR proceeds through the following gene until a downstream palindrome of sufficient quality is encountered (usually in the next 3′-IR). These bona fide polycistronic messengers encompass a succession of genes in the same orientation (Fig. 2C). Two such predicted transcripts were experimentally characterized: L416-417 and R882-883 (Table 1B). The absence of suitable palindromes between adjacent genes transcribed from the opposite strands (e.g., convergent genes) should result in an even more complex messenger structure that we refer to as “pseudo” polycistronic (Fig. 2D). In such transcripts, the 3′-UTR of one gene could totally contain a neighboring ORF in the reverse orientation. Two such predicted cases were experimentally validated (L356-R355, L778-R777). Presumably, only the first gene can be translated from this type of mRNA, while the other ORF is transcribed from the antisense strand. However, it is tempting to speculate that such a 3′-UTR might (negatively) interfere with the expression of the corresponding ORF by pairing with its sense messenger.
Given the computed distribution of palindromes in 3′-IR (>70%), only a small fraction of polycistronic messengers is predicted to encompass more than two genes. Following the hairpin rule, the longest polycistronic messenger was predicted to encompass the R463 to R467 genes. The longest polyadenylated transcript we could identify in this region corresponds to a messenger containing the three ORFs, R463-R464-R465. This transcript belongs to the rare exceptions (3/45) not ending within a hairpin (Fig. 2E; Table 1A).
Mimivirus tRNA genes are expressed as polyadenylated messengers
The Mimivirus genome includes tRNAs as found in Chlorella viruses (Nishida et al. 1999), another family of NCLDVs. In eukaryotes, tRNA transcripts are synthesized in the cell nucleus by RNA polymerase III without being polyadenylated and are exported to the cytoplasm after full maturation and an initial round of aminoacylation (Hopper and Shaheen 2008). Mimivirus encodes its own RNA Pol II machinery that is found in the particle (Renesto et al. 2006; Claverie et al. 2008), and like poxviruses, most (if not all) of its transcriptional activity occurs outside of the host nucleus. The absence of Mimivirus-encoded RNA Pol III homologs prompted us to investigate the existence and structure of Mimivirus tRNA transcripts. Interestingly, we found that all six tRNA genes were closely followed by a suitable palindrome, suggesting that they could be transcribed by the same machinery as protein genes, possibly leading to polyadenylated tRNA messengers. Even more interesting, one tRNALeu gene is located downstream from the R901 ORF, the 3′-IR of which does not contain a suitable palindrome. The application of the hairpin rule predicted that the R901 ORF could be transcribed as a hybrid protein/tRNA messenger. This predicted transcript was identified experimentally, and its 3′-UTR was shown to precisely end in the palindrome immediately following the tRNALeu gene (Table 1B). The existence of four other polyadenylated tRNA transcripts was experimentally demonstrated (Fig. 2; Table 1), including one encompassing two adjacent tRNA genes (tRNAHis and tRNACys). The latter finding indicates that hairpin “read through,” although probably rare, may add some variants to the repertoire of basic messenger structures implied by the “hairpin rule.” It is not yet known if Mimivirus-encoded tRNAs are functional, as they are in phycodnaviruses (Nishida et al. 1999). No mechanism is known by which the 5′ and 3′ of those tRNA messengers might be trimmed to lead to mature tRNA molecules, but the Mimivirus genome encodes numerous proteins with various RNA interactions or helicase domains that may be involved in this process.
Confirmation of the above results by the analysis of 150,651 cDNA tags
The above analysis led to a larger-scale transcriptomic study of the Mimivirus infectious cycle in its host A. castellanii. 454 Life Science (Roche) FLX pyrosequencing was used to generate a total of 257,477 sequences tags from four cDNA libraries made from A. castellanii cells 30 min, 3 h, 6 h, and 9 h post-infection by Mimivirus. Out of these sequence tags (average length ≈ 240), 150,651 matched the Mimivirus genome, the rest presumably corresponding to transcripts from the amoebal host. While the detailed analysis of this data set is still in progress, it has already confirmed the results obtained on the initial 45-gene panel in two ways. First, sequences corresponding to each annotated gene in the Mimivirus genome were identified, indicating that all Mimivirus protein-encoding genes are expressed as polyadenylated transcripts, as is typical of eukaryotic systems. Second, out of the 581 Mimivirus genes for which the 3′-UTR ends are mapped by a 454 FLX tag, 473 (81.4%) correspond to 3′-IRs containing a palindrome satisfying our initial descriptor.
A visual inspection of the residual cases often revealed palindromes slightly beyond our descriptor constraints, such as loops longer by 1 nt. Taking into account the higher noise level inherent to large-scale cDNA sequencing approaches [such as oligo(dT) priming on internal A-rich mRNA sequences leading to false 3′-UTR ends], and the simple signal descriptor we used, these results further suggest that the “hairpin rule” defines the polyadenylation site of the vast majority of Mimivirus transcripts.
Finally, we found an AATAAA motif in 30% of the 3′-UTRs of hairpin-containing genes and 32% of the 3′-UTRs without hairpin. In contrast with the precise localization of the polyadenylation site within the hairpin, the AATAAA occurrences are uniformly distributed throughout the 3′-UTR (Supplemental Fig. S4), which strongly suggests that the AATAAA motif is not used as a polyadenylation signal for the genes without hairpins.
Discussion
An important common feature shared by Mimivirus and other NCLDVs such as poxviruses and asfarvirus is that they remain in the host-cell cytoplasm for the duration of the infectious cycle. This implies that these viruses evolved a high level of independence for processes normally taking place in the cell nucleus such as DNA replication, transcription, and pre-mRNA maturation (3′-end processing and 5′-end capping). Accordingly, their genomes appear to encode a complete replication and transcription apparatus, most of the latter being found in the infectious particles (Renesto et al. 2006; Yoder et al. 2006). Among NCLDVs, transcription has been studied in detail in only a few viruses and for only a few genes of specific interest, most often focusing on the initiation steps in relation with promoter structures. Only for poxviruses (in particular, vaccinia virus), which took 40 yr of research, is there a global picture of the transcription process, including the 3′-end processing of the pre-mRNA of early, intermediate, and late genes (Broyles 2003).
We investigated the 3′-end mRNA structures of 45 selected Mimivirus genes by sequencing individual cDNAs, and generalized our initial findings using 150,651 cDNA tags generated by 454 FLX sequencing. First, we showed that all Mimivirus protein genes are expressed as polyadenylated transcripts, as expected for a virus infecting a eukaryotic host. Second, more than 80% of the analyzed 3′-UTRs were found to end within a large variety of palindromic sequences allowing the perfect pairing of at least 13 successive nucleotides (not including G-U pairs) into hairpin-like structures. Finally, in contrast to cellular systems, we found that Mimivirus-encoded tRNAs are expressed as polyadenylated transcripts, suggesting that these tRNA genes are transcribed by the same transcription machinery (including the virus-encoded RNA polymerase II) as protein-encoding genes. Our results pointed out several fundamental differences between Mimivirus and the well-studied poxviruses. For instance, the termination of vaccinia virus early genes occurs in response to the sequence element TTTTTNT, presumably read by the capping enzyme in interaction with the RNA polymerase (Broyles 2003). Such a motif did not emerge as a significant sequence signal from our analysis. Indeed, the sequence of the Mimivirus capping enzyme (R382) is only remotely related to the one in poxviruses, despite sharing global functional features (Benarroch et al. 2008). In addition, the 3′-end processing of poxvirus transcripts seems to markedly differ between early, intermediate, and late genes. Genes of the latter categories show extremely heterogeneous 3′-termini, compared to early genes (Broyles 2003). In contrast, the Mimivirus “hairpin rule” appears to apply equally well to early, intermediate, or late gene transcripts. For instance, the major capsid protein gene (L425), a typical late transcript, does not exhibit a heterogeneous 3′-end (Supplemental Fig. S2). Furthermore, no correlation was observed between the presence of the early promoter element AAAATTGA and the presence/absence of a 3′-end palindrome (Table 1). Altogether, these results suggest that the same termination/polyadenylation machinery is used for all types of genes in Mimivirus throughout the 12 h of its replication cycle, despite the visible changes occurring in the shape and size of its cytoplasmic virus factories (Suzan-Monti et al. 2007; Claverie et al. 2008).
The hairpin rule, as well as the stringency of its implementation, is presently unique to Mimivirus among known NCLDVs. Is it possible that these features were, in fact, acquired from the Mimivirus host, Acanthamoeba? Unfortunately, the termination/polyadenylation signals at work in this amoeba have not been studied in detail, and only a handful of A. castellanii transcripts have been mapped to date. We verified that none of them ended in the proximity of a significant palindrome. Another indication is that gene expression vectors integrating the HSV-TK polyadenylation signal (AAUAAA) (NCBI# NC_001806) have been successfully used in A. castellanii (Peng et al. 2005). This suggests that this Acanthamoeba species polyadenylates its transcripts in response to “regular” eukaryotic signals. It is thus tempting to propose that the palindromic signal so systematically exhibited at the end of Mimivirus genes is recognized by a pre-mRNA 3′-end processing complex encoded by the Mimivirus genome.
With a single hairpin signal replacing the multiple low-consensus cis-elements used in cellular systems, one could expect the pre-mRNA 3′-end processing complex of Mimivirus to be very different, possibly much simpler than its cellular counterparts involving more than 20 proteins (Mandel et al. 2008). Accordingly, a thorough bioinformatics analysis (including sequence threading in 3D protein models) could not identify any convincing Mimivirus-encoded homologs of the pre-mRNA 3′-end processing factors found in yeast, mammals, or Entamoeba histolytica (Lopez-Camarillo et al. 2005; Mandel et al. 2008), with the noticeable exception of a poly(A) polymerase catalytic domain in the N-terminal region (210 residues) of ORF R341 (584 total residues in length). However, the 300-residue-long C-terminal domain of the Mimivirus R341-encoded protein remained unmatched (except to a handful of environmental sequences). This extra domain may thus be responsible for the specific step in the 3′-end processing of Mimivirus pre-mRNAs. We also noticed that the nearby R343 gene (like R341 featuring the promoter element AAAATTGA associated with early transcripts) encodes a tandem repeat of a double-stranded RNA specific ribonuclease domain (e.g., ribonuclease III-like). This protein (quite divergent in sequence and apparently unique in associating two ribonuclease III-like domains) could be part of the minimal 3′-end pre-mRNA processing complex of Mimivirus, responsible for the recognition and cleavage of the 3′-UTR final palindrome in a region that almost always corresponds to a double-stranded RNA stem in the predicted secondary structure (Table 1; Supplemental Figs. S1 and S3).
The statistical analysis of 40 DNA virus genomes larger than 200 kb (Supplemental Fig. S1) indicated that none of them come close to Mimivirus in terms of palindrome frequency in their 3′-IRs, with the exception of Chilo iridescent virus (IIv6). In this iridovirus infecting insects, putative hairpins are found in 109 of the 178 3′-IRs (61%), while only 7.3 ± 2.4 are found in the corresponding randomized sequences (Z-score ≈ 42, P < 10–6). It is thus tempting to propose that the hairpin rule may be at work for a subset of the IIv6 genes. Coincidentally, it was previously noticed that, among NCLDVs, IIv6 had the highest proportion (17%) of genes with the AAAATTGA promoter element (Suhre et al. 2005; Nalçacioglu et al. 2007), a trademark of Mimivirus early genes, suggesting some similarity at the transcription level between these two viruses.
The involvement of a short hairpin structure in determining the polyadenylation site of Mimivirus transcripts is obviously reminiscent of the so-called Rho-independent or intrinsic transcription termination process described in prokaryotes (Wilson and von Hippel 1995). Here, transcription stops when the newly synthesized RNA molecule forms a short G-C-rich hairpin loop followed by a run of Us, which makes it detach from the DNA template. However, there are plenty of differences between the two processes: (1) Mimivirus mRNAs end up thoroughly polyadenylated, while bacterial transcripts are not; (2) Mimivirus hairpins are mostly made of A and T, while they are GC-rich in bacteria; (3) there is no trace of poly(T) tracks following palindromes in the genome of Mimivirus. Finally, the Rho-independent transcription termination strategy coexists with Rho-dependent processes to a variable extent in different bacteria (Kingsford et al. 2007), while the Mimivirus's hairpin rule applies to nearly 100% of its genes. These numerous differences invalidate the tempting but simplistic view that Mimivirus could be intermediate between prokaryotes and eukaryotes.
This study revealed that the polyadenylation of Mimivirus pre-mRNA systematically occurs within a high-scoring palindrome in 3′-IR sequences, presumably recognized as a secondary structure signal. Very few exceptions to this hairpin rule were found, in contrast to the fuzziness that usually characterizes the bioinformatics definition and/or the biological recognition of sequence signals governing the transcription process in eukaryotes. Following the unprecedented level of conservation of its AAAATTGA promoter element, Mimivirus is now found to possess a uniquely simple and stringent signal at the 3′-end of its mature mRNAs. We can only speculate on the reasons why Mimivirus is maintaining two such simple signals at the extremities of its transcription units. One explanation would be that the minimal virus-encoded transcription machinery cannot accommodate the signal flexibility seen in cellular organisms. Yet, the question remains whether the simplified gene recognition system unique to Mimivirus is ancestral and pre-dates the more sophisticated cis-elements at work in extant eukaryotic organisms or, at the opposite, if it emerged in Mimivirus as a result of reductive evolution, in the context of an increasingly A+T-rich genome rending the recognition of AT-rich cis-elements progressively impossible.
More studies on the structure of the Mimivirus transcription complex and on the detailed mechanisms of its 3′-end pre-mRNA processing are needed to resolve this issue.
Methods
Virus production and purification
A. castellanii Neff was purchased from the American Type Culture Collection (ATCC # 30871) and cultured in PPYG medium. A. castellanii was grown to confluence and infected with Acanthamoeba polyphaga Mimivirus with a multiplicity of infection (MOI) of 1. After several days, almost all cells were lysed, and the virus released into the medium was recovered and clarified by centrifugation at 250g for 5 min to remove cellular debris. The supernatant was incubated with 0.2 mg/mL DNase at 25°C overnight, followed by 1 h of incubation in 1 M NaCl at 4°C, and then centrifuged at 250g for 5 min to remove further debris. The virus was pelleted by 10 min of centrifugation at 15,000g at 4°C and resuspended in PBS with CsCl to obtain a density of 1.15. Twenty milliliters of the suspension was layered onto a discontinuous gradient of CsCl (1.45/1.35/1.25) and centrifuged at 18,000g for 30 min. The virus band was collected and washed/centrifuged five times with 50 mM Tris (pH 8.0) storage buffer. An endpoint titration assay (TCID50) was performed to measure the virus titer in infectious units per milliliter (Dulbecco and Vogt 1954; Hierholzer and Killington 1996). Briefly, A. castellanii cells were seeded in a 96-well microtiter plate at a density of ∼0.5 × 105 cells per well in PPYG medium. The supernatant was then replaced by 50 μL of fresh PPYG containing serial dilutions of the virus from 10−6 to 10−13. Each dilution was repeated 10 times, and the cell-virus cultures were maintained for 7 d at 37°C (total volume 50 μL/well). Lysis of the cells was monitored daily, and TCID50/mL values were calculated according to the Spearman–Kärber method (Dulbecco and Vogt 1954; Hierholzer and Killington 1996).
A. castellanii infection by Mimivirus
A total of 2.5 × 108 adherent cells in 167 mL of culture medium were recovered, centrifuged at 300g, and re-suspended in 100 mL of Page's amoeba saline (PAS) (2.5 mM NaCl, 1 mM KH2PO4, 0.5 mM Na2HPO4, 40 mM CaCl2 · 6H2O and 20 mM MgSO4 · 7H2O). Cells were infected by Mimivirus with an MOI of 1000. After 30 min of incubation at 30°C under gentle stirring (150 rpm), infected cells were centrifuged (300g for 5 min), and the supernatant containing excess virus was discarded. The cell pellet was washed once with PAS medium, once with PPYG medium (100 mL each time), and distributed to 16 flasks (1.25 × 107 cells/ flask of 175 cm2) containing 25 mL of PPYG medium. Four flasks (5 × 107) were recovered at 3, 6, 9, and 12 h post-infection, respectively, and harvested by centrifugation at 500g. An extra 5 × 107 cells were kept as a T0 culture control (30 min post-infection).
RNA extraction
RNA was extracted using the RNeasy Midi kit (Cat No: 75144 QIAGEN) using the manufacturer's protocol. Briefly, cells were re-suspended in the provided buffer and disrupted by subsequent −80°C freezing and thawing in a water bath for 5 min at 37°C. Total RNA was eluted with ∼200 μL of DEPC-treated water.
RNA quantification and quality control
RNA was quantified using the Qubit fluorometer and the Quant-iT RNA Assay Kit (Q3285, Q32852 Invitrogen). DNA contamination was assessed using the Quant-iT dsDNA HS Assay Kit (Q32851 Invitrogen). The integrity of the RNA sample was assessed using the Experion Automated Electrophoresis System with RNA StdSens chips and reagents (700-7153, 700-7154, Bio-Rad). A. castellanii RNA extracts contained the expected peaks corresponding to the 18S small subunit and the 28S large subunit. A third peak corresponding to the 16S mitochondrial RNA was also visible. Our results were comparable to the ones obtained for the A. polyphaga rRNA (Grant et al. 2006).
cDNA production (first strand)
First-strand cDNA poly(A) synthesis was performed using the SuperScript III First-Strand kit (18080-051, Invitrogen). One microgram of total RNA was reverse-transcribed using the oligo(dT)20 primer provided by the kit in a reaction volume of 20 μL. A control reaction was also performed without the Superscript reverse transcriptase enzyme to monitor genomic contaminations.
cDNA production (second strand)
Full-length cDNA synthesis
First-strand cDNA synthesis was performed with the PrimeScript Reverse Transcriptase (Clontech Laboratries) using the SMART (Switching Mechanism at 5′ end of RNA Transcript) PCR technology (Clontech Laboratories), but following a modified protocol suggested by Roche Diagnostics to optimize sequencing using the 454 FLX Sequencing technology. Total RNA was reverse-transcribed using a modified CDS III oligo(dT) (Invitrogen Life Technologies): 5′-TAGAGACCGAGGCGGCCGACATGTTTTGTTTTTTTTTCTTTTTTTTTTN-3′ (N corresponds to the mix of A, G, or C).
At the 5′-terminus of the template, a poly(C) tail is added to the cDNA using the terminal transferase activity of the reverse transcriptase. The SMART V oligonucleotide provided with the kit hybridizes to the poly(C) tail to form an RNA/DNA hybrid: 5′-AAGCAGTGGTATCAACGCAGAGTGGCCATTACGGCCGGG-3′.
Full-length LD PCR (long distance polymerase chain reaction)
For the LD PCR reaction, we used the Advantage 2 PCR Kit (Clontech Laboratries). Only sscDNAs that have the 5′-SMART anchor can be used as a template for the LD PCR reaction, thus ruling out eventual genomic DNA contamination. For each time course (30 min, 3 h, 6 h, and 9 h), we performed 5 × 100 μL PCR reactions. To each 100-μL mix, we added 2 μL of first-strand cDNA reaction, 10 μL of Advantage Buffer10×, 2 μL of 10 mM dNTP mix, 2 μL of 10 μM 5′-SMART PCR primer, 2 μL of 10 μM Modified CDSIII 3′-PCR Primer, 2 μL of Advantage Polymerase mix 50×. The reaction volume was completed with 80 μL of RNase/DNase-free H2O. PCR were performed as follows: 1 min at 95°C (20 sec at 95°C, 6 min at 68°C) for 23 cycles. The optimal number of cycles was determined previously as 23 cycles by comparing different numbers of cycles, that is, 20, 23, 26, and 29 cycles. To assess the quality of the dscDNA sample, we loaded 5 μL of each sample onto a 1.1% agarose gel, resulting in a smear from 100 bp to 3.5 kb. The dscDNA was cleaned and concentrated using the PureLink PCR Purification Kit (Invitrogen Life Technologies). Samples were eluted with 40 μL of H2O. cDNA purity was measured using the 260/280 nM ratio against 10 mM Tris (pH 7.5).
LD PCR primers
5′-SMART V PCR: 5′-AAGCAGTGGTATCAACGCAGAGT-3′; Modified CDSIII 3′PCR (N corresponds to the mixture of A, C or G): 5′-TAGAGGCCGAGGCGGCCGACATGTTTTGTCTTTTGTTCTGTTTCTTTTN-3′.
Gene-specific cDNA amplification
One microliter of either first-strand cDNA or dscDNA was used in a final volume of 50 μL. One unit of Phusion High-Fidelity DNA Polymerase (F-530S, Finnzymes) was used for the PCR reaction. The forward primers used were gene specific (Supplemental Table S2).
Reverse anchoring on first-strand cDNA (Supplemental Table S3) was done using a mix of 5′-7G(dT)24V3 (V corresponds to the mix of 3′ A, G, or C). For most of the selected genes chosen, touchdown PCR was performed:
-
Denaturation: 30 sec at 98°C, 10 sec at 98°C.
-
Annealing and amplification (touchdown): 40 sec at 60°C over 10 cycles with a −1°C decrease for each cycle with an elongation time of 4 min at 72°C. Twenty-five more cycles are added with an annealing temperature of 50°C.
Reverse anchoring on ds-cDNA was done using the modified CDSIII 3′-PCR. In that case, PCR was performed as follows:
-
Denaturation: 30 sec at 98°C.
-
Annealing and amplification: (10 sec at 95°C, 40 sec at 57°C to 62°C) for 20 cycles.
-
Elongation: 30 sec to 3 min at 72°C, depending on the expected length of the transcript.
-
Last step: 5 min at 72°C.
PCR products were analyzed on 2% agarose gels, and when enough material was obtained, the various length products were purified using the e-gel technology (G6618-08, Invitrogen) and sent for sequencing (Cogenics). PCR product concentrations were ∼6 ng/μL in 40 μL.
454 cDNA tag sequencing
cDNA tag sequencing was performed on the French National sequencing platform (“Genoscope”) according to the manufacturer's protocol using 4 μg of ds-cDNA (260/280 absorbance ratio >1.6) prepared as described above from Mimivirus-infected A. castellanii cells 30 min, 3 h, 6 h, and 9 h post-infection. The sequencing from a single 454 plate (divided into four sectors) generated a total of 257,477 usable tags corresponding to 67,323, 78,265, 74,316, and 37,573 tags for times 30 min, 3 h, 6 h, and 9 h, respectively. The corresponding numbers of sequence tags mapped on the Mimivirus genome sequence for these various times are: 34,399, 27,704, 59,206, and 29,313. More infectious time points will be performed and a detailed analysis of these data will be presented elsewhere.
Bioinformatics tools and analysis
Search for significantly enriched conserved linear motifs in the 3′-UTRs
The intergenic regions encompassing the experimentally delineated 3′-UTR sequences were analyzed in search of a conserved linear motif (with a minimal length of 4 nt) using standard methods such as “word counting” (Claverie et al. 1990) or a more sophisticated algorithm such as MEME (Bailey et al. 2006) and Improbizer (Ao et al. 2004). No statistically significant signal could be extracted from these sequences.
Identification of the palindromic signal
The palindromic segments were identified within the genomic sequence using the RNA motif program (Macke et al. 2001), with the following descriptor:
This descriptor extracts hairpins with a stem of at least 13 nt, with at most three mismatches, and a loop between 0 and 5 nt. For each pattern, the score is equal to the length of the stem minus the number of mismatches, and sequence segments with a score of at least 13 are output. Those parameters were identified by a succession of trials and are optimal in maximizing the palindrome distribution bias between ORFs and intergenic regions. This descriptor matches all genes listed in Table 1, except for L164, for which the length of the loop had to be extended.
Palindrome statistics
We analyzed the 200-nt region after each ORF in the Mimivirus GenBank RefSeq entry (NC_006450) and extracted only those matching the above descriptor (on the direct strand, as our pattern is symmetric). This resulted in 565 3′-regions exhibiting a suitable palindrome. On the other hand, 127 ORFs were found to exhibit a palindrome matching the above descriptor. The same 3′-sequences were then shuffled, and the procedure was repeated 100 times. When performed on Mimivirus, only 36 randomized intergenic regions (SD = 5.34) exhibited a match to the above descriptor. The Z-score of the observed number of occurrences (565) is thus 99 (two-tailed probability ≪ 10−6). We thus observed many more occurrences of the palindrome described above than expected by chance in a random sequence of the same nucleotide composition. A similar procedure was followed for analyzing the virus genomes listed in Supplemental Table S1. The contingency χ2 test was also used to demonstrate the preferential occurrence of palindromes in 3′-IRs. 3′-IRs encompass 18% of the Mimivirus genome, while ORFs represent 82%. If we assume an unbiased distribution of palindromes between these two genomic regions, the total number of palindrome occurrences (692) should be distributed as 553 in ORFs, and 139 in 3′-IRs (Table 2). Table 2 has a χ2 value of 1630 (two-tailed P-value ≪ 10-3). The enrichment of palindromes in 3′-IRs and their avoidance in ORFs is thus highly statistically significant. We could then safely conclude that the motif (as defined by the above descriptor) is not equally distributed among 3′-IRs and ORFs. This, in turn, suggests that these palindromes might be used as signals for the 3′-end processing of Mimivirus pre-mRNAs.
Palindrome statistics in Mimivirus genome
We investigated possible cases of internal priming by computing the hybridization energy of the duplex formed between the modified CDS III oligo(dT) primer and the cognate genomic sequence (i.e., 48 nt downstream from the transcript end). Hybridization energies were calculated using the hybrid-min program from the UNAFold package (Markham and Zuker 2008). We predicted possible internal priming for free energy lower than 11 kcal/mol. Of the 46 experimentally validated transcripts (Table 1), 43 exhibit a palindromic sequence, of which 40 are predicted without internal priming. The three possible cases of internal priming are for L244, R512, and tRNAHis (352154-224).
cDNA sequence mapping
The cDNA sequences were mapped on the Mimivirus genome sequence using the multiple alignment tools provided by the PACA-Bioinformatics web servers (www.igs.cnrs-mrs.fr including www.giantvirus.org and www.phylogeny.fr).
Acknowledgments
We thank Jean Weissenbach for providing a rapid access to the Genoscope 454 FLX platform, and Julie Poulain for performing the 454 FLX sequencing. We thank Bruce Roe for advice on cDNA library preparation, Olivier Poirot for data management and preliminary statistical analyses, Mathieu Legendre for his help in generating Supplemental Figure S4, Garry Duncan and Pascal Hingamp for reading the manuscripts, and Laurent Kodjabachian for helpful discussions. We acknowledge the use of the IFR-88 transcriptomic platform, of the PACA-Bioinformatic platform, and of the phylogeny.fr server. This work was funded by the Centre National de la Recherche Scientifique, a grant from the French Genopole National network (RNG) to J-M.C., and a grant from the Agence Nationale de la Recherche (ANR-BLAN08-0089).
Footnotes
-
↵2 Corresponding authors.
E-mail Jean-Michel.Claverie{at}univmed.fr; fax 33-4-91825421.
E-mail Chantal.Abergel{at}igs.cnrs-mrs.fr; fax 33-4-91825421.
-
[Supplemental material is available online at www.genome.org. The mRNA sequence data from this study have been submitted to EMBL (http://www.ebi.ac.uk/embl/) under accession nos. FM992033–FM992076, FM992105–FM992106.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.091561.109.
-
- Received January 24, 2009.
- Accepted April 15, 2009.
- Copyright © 2009 by Cold Spring Harbor Laboratory Press








