
Genomic mapping of binding regions for the Ecdysone receptor protein complex
- Zareen Gauhar1,2,5,6,
- Ling V. Sun2,3,5,7,
- Sujun Hua1,3,5,
- Christopher E. Mason3,
- Florian Fuchs4,
- Tong-Ruei Li3,
- Michael Boutros4 and
- Kevin P. White1,8
- 1 Institute for Genomics and Systems Biology, Departments of Human Genetics and Ecology and Evolution, The University of Chicago, Chicago, Illinois 60637, USA;
- 2 Department of Molecular, Cellular, and Developmental Biology, Yale University School of Medicine, New Haven, Connecticut 06520, USA;
- 3 Department of Genetics, Yale University School of Medicine, New Haven, Connecticut 06520, USA;
- 4 German Cancer Research Center, Im Neuenheimer Feld 580, D-69120 Heidelberg, Germany
-
↵5 These authors contributed equally to this work.
Abstract
We determined the physical locations of the heterodimeric Ecdysone receptor/Ultraspiracle (ECR/USP) nuclear hormone receptor complex throughout the entire nonrepetitive genome of Drosophila melanogaster using a cell line (Kc167) that differentiates in response to 20-hydroxyecdysone (20-HE). 20-HE, the natural ligand of this complex, controls major aspects of insect development, including molting, metamorphosis, and reproduction. Direct gene targets of 20-HE signaling were identified by combining this physical binding-site profiling with gene expression profiling after treatment with 20-HE. We found 502 significant regions of ECR/USP binding throughout the genome. Only 42% of these regions are nearby genes that are 20-HE responsive in these cells. However, at least three quarters of the remaining ECR/USP regions are near 20-HE-regulated genes in other tissue and cell types during metamorphosis, suggesting that binding at many regulatory elements in the genome is largely noncell-type specific. The majority (21/26) of the early targets of 20-HE encode transcriptional regulatory factors. To determine whether any of these targets are required for the morphological differentiation of these cells, we used RNAi to reduce the expression of each of the 26 early genes. Accordingly, we found that three direct targets of ECR/USP—hairy, vrille, and Hr4—are required for cellular differentiation in response to the hormone. Initial mutational analysis of vrille in vivo reveals that it is required for metamorphosis.
Hormone-regulated nuclear receptors (NRs) are crucial for the coordination of biological processes underlying reproduction, metabolism, and development in animals (Richards 1981). NRs are self-contained signal-transduction modules that consist of several key domains—a highly conserved DNA-binding domain that targets the receptor to specific DNA sequences of hormone response elements, a less-conserved C-terminal ligand-binding domain, and one or more transcriptional activation domains (Mangelsdorf et al. 1995). The molecular mechanisms underlying the transduction of a hormonal signal into a transcriptional response via NRs have been well studied (Mangelsdorf and Evans 1995; Chawla et al. 2001; McKenna and O'Malley 2002). Steroid hormone nuclear receptors, such as the thyroid receptor (TR) and the Ecdysone receptor (ECR), function by heterodimerizing with the retinoid X receptor (RXR) (King-Jones and Thummel 2005) to regulate the transcription of target genes. These receptors can function as repressors in the absence of a ligand molecule and maintain target gene repression by using corepressor complexes. In the presence of hormones, coactivator proteins are recruited and corepressors are displaced, resulting in the activation of the target genes (Mangelsdorf and Evans 1995; Chawla et al. 2001; King-Jones and Thummel 2005). Due to the ability to change the functional state of nuclear receptors by adding lipophilic hormones, these nuclear receptors are intensively studied in transcriptional regulation studies and provide important insights into the regulation of eukaryotic gene expression (King-Jones and Thummel 2005). However, these studies have been limited to the action of NRs at only a small fraction of the regulatory elements that might exist in the genome. Here, we use DNA microarrays that contain sequences tiled throughout the entire nonrepetitive genome of Drosophila to systematically map the binding regions of one of the best-characterized NR complexes—the Ecdysone receptor protein complex (ECR-C).
In Drosophila, ecdysteroids (predominately 20-HE) are the only known physiologically active steroid hormones. 20-HE triggers the major postembryonic developmental transitions during metamorphosis from the larval form to the adult fly. The ECR-C contains two nuclear receptors: the Ecdysone receptor (ECR) (Koelle et al. 1991) that binds directly to 20-HE and its heterodimeric partner Ultraspiracle (USP), which is an ortholog of vertebrate RXRs (Yao et al. 1993). The ECR-C interacts with both transcriptional coactivators and corepressors in a manner that is thought to depend on DNA sequence or cellular context (Arbeitman and Hogness 2000).
Initial evidence for a 20-HE pathway was elucidated from an ex vivo culture system that used puffing patterns in salivary gland polytene chromosomes to reveal a gene expression hierarchy in response to 20-HE (Ashburner 1974). A late larval and pupal hormone pulse leads to the direct induction of overlapping, but nonidentical sets of a few “early genes” that repress their own activity and induce hundreds of “late genes” (Burtis et al. 1990; Segraves and Hogness 1990). The late genes are effectors as they contribute directly to the developmental changes in the larval–prepupal and the prepupal–pupal transition (Ashburner 1975). Molecular cloning of the early genes has shown that many of them encode transcription factors (Guay and Guild 1991; Koelle et al. 1992; Fletcher and Thummel 1995). Molecular genetic analysis of these transcription factors has revealed that a complex regulatory network exists to coordinate metamorphosis (Fletcher and Thummel 1995; Lam et al. 1997; White et al. 1997; King-Jones and Thummel 2005). While the interactions between the known early transcription factors activated by the ECR-C have been fairly well studied at the molecular level, it is not known how many targets exist genome-wide. Additionally, with the exception of 20-HE-induced programmed cell death of the larval salivary glands (Jiang et al. 2000), little is known about which direct targets are required for the cellular responses activated by 20-HE.
A number of studies have used microarrays to identify genes that respond to 20-HE as well as genes that are dependent on ECR for their regulation (White et al. 1999; Li and White 2003; Beckstead et al. 2005). Although several 20-HE primary response genes that require ECR were identified, the genomic regions to which ECR and USP physically bind to and their direct targets across the whole genome remained unidentified. Our list of 20-HE-regulated direct targets of ECR/USP contains many genes that were identified in earlier studies, as well as the puffing studies, indicating that we identified bona fide targets. In order to fully define the ecdysone network and understand how ECR and USP function together for 20-HE-regulated events, a thorough mapping of the direct targets of these two transcription factors in concert is necessary.
Using high-density oligonucleotide microarrays, we identified the binding regions of ECR and USP across the whole genome to identify direct targets of ECR/USP and as a first step in building a 20-HE network map. We identify many new targets of ECR/USP that are part of the 20E regulatory network and define roles for this complex in the regulation of transcription at the onset of metamorphosis. We also determined which of the ECR-C-binding regions are functionally relevant by comparing with expression data from cells responding to 20HE. Finally, we identified genes that are direct targets of the ECR/USP and that encode products required for 20-HE stimulated cellular differentiation.
Results
All experiments were carried out in the Kc167 cell line established from early 6–12-h Drosophila embryos, possibly from neural or glial origin (Echalier and Ohanessian 1969). Kc167 cells are not transformed, and most of these cells are diploid. They also retain many characteristics of original insect tissues, such as responding to ecdysteroids, making them suitable for this study. Before ecdysteroid treatment, Kc cells are relatively round, but they strongly respond to ecdysteroid treatment by arresting in G2 stage and ceasing to proliferate within 1–2 d. They also become spindle shaped, elongate, and emit long pseudopodia within 2–3 d after hormone treatment. Kc cells contain a single functional copy of ECR, and ecdysteroid treatment leads to rapid and substantial increases in the relative synthesis of ecdysteroid-inducible polypeptides, mediated by ECR (Cherbas et al. 1988).
We mapped the binding regions of ECR and USP across the genome using the DNA adenine methyltransferase IDentification (DamID) technique (van Steensel et al. 2001). This technique involves fusing the Dam enzyme to a chromatin-associated protein, resulting in the methylation of genomic DNA nearby binding regions. Methylated DNA is purified and hybridized to a DNA microarray to reveal the coordinates of the fusion protein binding regions in the genome. Fusions of the Dam enzyme were made to both the N- and C-terminal portions of ECR and USP. Dam-ECR and ECR-Dam fusion proteins can transduce the 20-HE signal in Kc cells missing a wild-type copy of the endogenous EcR gene, showing that the fusion proteins are functional (Fig. 1A). To detect genomic binding regions for ECR and USP, we used a high-density oligonucleotide array that contains 36-mer probes genome-wide (Methods; Stolc et al. 2004). Results with N- and C-terminal fusions were significantly correlated for both ECR and USP fusions; however, there is a low correlation between enriched probes (Supplemental Fig. 1).
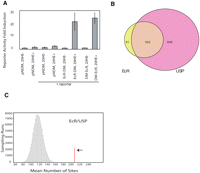
(A) ECR-DamMyc(DM) and DamMyc-ECR fusion proteins transduce the 20-HE signal in Kc cells (L57-3-11) using a wild-type copy of the endogenous EcR gene, showing that both fusion proteins are functional. pnDM (pnDamMyc) is the vector containing the N terminus Dam protein, and the lacZ reporter was used to measure beta-galactidose activity. (B) Overlap of significantly bound regions by ECR and USP at P < 0.001. (C) Enrichment of computationally predicted ECR/USP-binding sites in ECR/USP-bound regions. The red line indicates the number of computationally identified ECR/USP sites within the binding regions (P < 0.0001). The histogram shows the distribution (sampled from 100,000 runs) of putative ECR/USP sites number within the same number of randomly selected 4-kb windows.
To identify probes corresponding to significantly bound genomic DNA fragments (P < 0.001), we used the Limma algorithm that employs an empirical Bayesian approach through the use of a moderated t-test statistic (Smyth 2004). Genome-wide, there were 1279 significant probes identified in the ECR experiments and 3188 significant probes identified in the USP experiments. However, because the domains of methylation typically spread 4–5 kb along the genomic DNA, many of these significant probes are clustered together (van Steensel and Henikoff 2000). We therefore identified regions of genomic binding for ECR and USP fusion proteins by analyzing 4-kb windows using a combined P-value test (Fisher 1950a). There are 593 significant ECR windows (P < 0.0001) and 1450 significant USP windows (P < 0.0001). We find that 84.6% of significant regions bound by ECR (502 out of 593 regions) are in common with USP-bound regions (Fig. 1B) (chromosomal locations available in Supplemental Table 1). If we decrease the threshold to P < 0.01 for USP probes, the overlap increases to 95.4% (Supplemental Fig. 2). This shows that ECR colocalizes with USP, as expected. USP has been shown to have other dimerization partners such as HR38 (also known as DHR38), which has been implicated in an ECR-independent 20-HE signal-transduction pathway (Baker et al. 2000). Therefore, the larger superset of USP-binding regions is also expected.
The 502 ECR/USP regions are highly enriched for the consensus-binding motif of this heterodimer. To identify putative motifs, a genome-wide scan was carried out by using a positional weight matrix based on a training set of 10 known ECR/USP-binding sites (Hertz and Stormo 1999). A total of 5638 putative motifs were identified across the genome. The number of putative ECR/USP binding motifs in the 502 ECR/USP-bound regions was calculated (Fig. 1C) and 208 ECR/USP-binding motifs are found. We carried out a control background estimation (100,000 sampling runs) to see how many putative motifs are located in same numbers and same lengths of randomly selected genomic regions for ECR/USP, resulting in a mean of 130. Thus, we determined that the observed overlap of ECR/USP-bound regions and computationally derived ECR/USP sites is highly significant (P < 1.0 × 10−16). Nonetheless, fewer than half of the 502 ECR/USP-binding regions we identified using microarrays contain computationally defined ECR/USP-binding motifs. This result may reflect the limitations of the computational approach due to nonconsensus binding sites for ECR/USP, or it could be partly due to ECR/USP interactions with other proteins that are directly bound to DNA. It may also reflect false positives from the chromatin profiling experiments, although the high degree of overlap between the ECR-bound regions with the USP-bound regions argues against a high false positive rate.
To determine which of the ECR/USP-binding regions might be functional in this cell type, we carried out a time course of 20-HE treatment. 20-HE was added to cells and total RNA was isolated after 1, 3, 6, 12, 24, and 48 h. Total RNA from each time point was reverse transcribed and hybridized against the control sample of 0 h to a DNA microarray containing fragments from over 13,000 predicted genes (White et al. 1999). 20-HE responsive genes for each time point were identified using the Limma t-test (P < 0.05 and 1.5-fold cutoff) (Gene Lists in Supplemental Table 2; Wettenhall and Smyth 2004). Using this procedure we identified a total of 818 genes that are 20-HE responsive. We identified 113 genes that are up-regulated in response to 20-HE at any early time point (1, 3, and 6 h) and 192 genes that are increased in expression at any of the later time points (12, 24, and 48 h). Likewise, there are 59 unique early responding genes and 470 unique late responding genes that show decreased expression levels during the time course (Supplemental Table 3). Only 16 genes were expressed in opposite directions at the early and late time points (Supplemental Table 2). The number of 20-HE-responsive genes increases across the time course, with the earliest responsive genes much more likely to be induced than reduced in expression levels (Fig. 2A). Later in the time course, genes reduced in expression predominate. This bias for early induced genes may be due to initial exposure to 20-HE, resulting in activation of genes by the ECR-C, or the detection of fewer early down-regulated transcripts could result from low turnover of transcripts that correspond to genes directly repressed by the ECR-C (Levine et al. 2003).
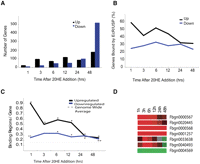
Binding profiles of 20-HE-responsive genes. (A) Total number of up-regulated or down-regulated genes 1–48 h after 20-HE addition. (B) The number of up-regulated direct targets decreases steadily with time; however, the number of down-regulated direct targets stays steady across the time course. (C) The number of binding regions per up-regulated 20-HE-responsive gene (br/g) decreases with time; however, the number of binding sites near down-regulated direct targets stays ∼0.24 throughout the time course. Dotted line is the average binding regions/gene (br/g) across the genome. P-values are shown ([*] <0.001, [**] <0.01) from a distribution of br/g generated from permutation tests of randomly sampled genes. (D) Expression profile of several early up-regulated (red) and down-regulated (green) direct targets of ECR/USP.
We analyzed the overlap between 20-HE-regulated genes and the genes nearby in ECR/USP-binding regions to find direct targets of the ECR-C. The exact physical distance over which transcription factors can regulate their targets in the Drosophila genome can range from tens of base pairs to tens of thousands of base pairs. However, most genes are probably regulated by enhancers that are within 10 kb (Levine et al. 2003), so we determined which of the 502 significantly bound ECR/USP regions are within 10 kb of the 20-HE-regulated genes at each time point. We found that only 42% (209 regions; 51 of these also contain a computationally derived site) of the ECR/USP-bound regions are nearby at least one 20-HE-responsive gene in Kc167 cells. A total of 228 20-HE-regulated genes (46% induced and 54% reduced in expression levels) are found nearby the 209 ECR/USP-binding regions. These numbers are highly significant when considering the probability of finding associations between the 20-HE-regulated genes we identified and randomly chosen 502 genomic regions: We sampled 502 blocks in the genome using 1,000,000 permutations and never observed 228 or greater nearby 20-HE-responsive genes (average, 88.9; StDev, 10.04; P = 6.63 × 10−44). Additionally, we observed that the earliest responders to 20-HE are more likely to have ECR/USP-bound regions nearby than genes that respond to the hormone at later time points. Across the time course, the proportion of 20-HE-regulated genes bound by ECR/USP is greater during the early hours after ecdysone treatment—almost 60% after 1 h of hormone exposure, but decreases with time after several hours of exposure (Fig. 2B).
Interestingly, there is a greater average number of ECR/USP-binding regions associated with up-regulated genes at early time points than at later time points (Fig. 2C). Furthermore, genes that are induced most rapidly in response to 20-HE have more binding regions on average than late-induced genes or genes with decreasing levels (Fig. 2C). However, we do not detect multiple binding sites for down-regulated direct targets, which retain an average number of 0.24 binding regions per 20-HE-regulated gene throughout the time course. These results may indicate that down-regulated genes are more likely to be indirect targets than the rapidly induced, up-regulated genes. It is also possible that many down-regulated genes are controlled via nontranscriptional mechanisms. However, both of the down-regulated genes have a significant enrichment for ECR/USP-binding regions nearby. Figure 2C shows that all observed values of binding regions nearby 20-HE-regulated genes are significant except for down-regulated genes at 3 h. Therefore, these results suggest that ECR/USP may have inductive or repressive effects in the presence of hormone, dependent upon context. The expression profile of several representative early ECR/USP-bound genes across the time course is shown in Figure 2D.
Since only 42% of ECR/USP-binding regions are nearby 20-HE-regulated genes in Kc167 cells, we wondered whether the remaining 58% (293) of ECR/USP-bound regions might be nearby genes that are not 20-HE responsive in Kc167 cells, but are 20-HE responsive in other cell types. To test this idea, we determined whether these 293 ECR/USP-bound regions are instead nearby putative 20-HE-regulated genes in several tissues that were previously examined for expression changes during the onset of metamorphosis—when there is a dramatic increase in 20-HE concentrations in the animal (Li and White 2003). We found that 158 (53%) of those regions are within 10 kb of genes that are normally modulated in other tissue and cell types during metamorphosis (Fig. 3; Supplemental Fig. S3). To further assess the functional significance of the bound regions, we used a compilation of gene expression data from ECR mutants and mutants deficient in 20-HE biosynthesis. From the 293 ECR/USP-binding regions, we found 66 additional ECR/USP regions that are within 10 k of the genes that are up-regulated or down-regulated in null mutant ECR animals (M. Davis and K.P. White, in prep.). In sum, we found 224 additional direct targets of ECR/USP from in vivo data in 20-HE-responsive tissues during metamorphosis, as well as ECR and 20-HE mutant whole animals (Fig. 3). Our data thus indicate that 86% (433/502) of the ECR/USP-bound regions in Kc cells are nearby 20-HE-responsive genes in either Kc cells or in vivo (209 from Kc cells and 224 from in vivo data). This indicates that while many ECR/USP-binding regions appear to be nontissue specific, functional activation of the ECR-C likely occurs in a tissue-specific manner. Mapping direct targets in different tissues will be imperative for understanding how the genomic response to a single hormone diverges and confers specificity.

ECR/USP-bound regions near 20-HE genes in different tissue types. A total of 209 regions (42%) are near 20-HE genes in Kc167 cells, an additional 224 (44%) are near 20-HE genes identified in vivo, and 69 (14%) did not have targets in tested tissues. A total of 86% of ECR/USP-bound regions have 20-HE-responsive targets in either Kc cells or in vivo data.
To further determine whether the ECR/USP-binding regions we mapped in Kc cells are relevant to 20-HE controlled gene expression during metamorphosis, we used Gene Ontology (GO) annotation for the biological process and confirmed that 20-HE-induced direct targets of ECR/USP are enriched in genes involved in developmental processes associated with metamorphosis such as salivary gland cell death, whereas down-regulated direct targets of ECR/USP are enriched in genes involved in metabolism (P < 0.01; Supplemental Tables 4, 5; Boyle et al. 2004). Early molecular cloning experiments revealed that many of the known 20-HE-induced early genes encode transcription factors (Fletcher and Thummel 1995; Fletcher et al. 1997; Lam et al. 1997). Several newly reported transcription factor genes such as vrille were also shown to require ECR for their expression in a 20-HE profiling experiment using RNAi to knock down the expression of ECR (Beckstead et al. 2005). However, it is not known how many total transcription factors are early response genes and whether transcription factors are in fact the major genomic targets of the early 20-HE response via direct binding by ECR/USP. In order to see whether ECR/USP binding is associated with genes encoding transcription factors, we used GO annotation for molecular function (P < 0.01; Supplemental Table 6). There are 24 annotated transcription factors (TFs) that are directly bound by ECR/USP and are also 20-HE regulated in Kc cells. We found that genome-wide, 47 genes categorized in GO annotation “RNA polymerase II transcription factor activity” were within 10 kb of ECR/USP localization as well as 135 genes associated with GO term “transcriptional regulator activity” (P < 0.05, Supplemental Table 7). In addition to the known TFs such as EIP75B (also known as E75), CROL, and EIP74EF (also known as E74), we identified over several TFs that are directly regulated by ECR/USP, including E2F, SOX14, Hairy, Vrille, and BRAT. Intriguingly, mutations in some of these genes such as brat and vrille are pupal lethals (Beckstead et al. 2005; FlyBase; T.R. Li and K.P. White, unpubl.).
We also found that ECR/USP targets are enriched for genes classified as signal-transduction factors and catalytic activity (P < 0.05). Our results are consistent with the idea that the 20-HE signal is amplified using a wide range of transcription factors and signaling pathways (Li and White 2003). Binding near targets that are annotated with catalytic activity indicates that ECR/USP may also operate at the level of directly regulating catalytic processes such as dehydrogenase and transferase activity by genes such as endoA and Mdh.
Finally, we wished to determine whether any of the newly identified direct targets activated by ECR/USP are required for cellular differentiation. Kc167 cells that are treated with 20-HE undergo a morphological change and show a halt in cell proliferation at G2-M phase. Cells respond to 20-HE treatment by generating neuronal-like projections and undergoing cell-cycle arrest (Berger et al. 1978; Cherbas 1981; Besson et al. 1987). We tested which of the early targets disrupts the clearly discernable projection phenotype induced by 20-HE. We tested 15 early response genes that contain regions bound by ECR/USP within 10 kb upstream or downstream of their gene regions, seven other genes that are early response genes at a less conservative P-value, and four genes that are not early responders, but have nearby ECR/USP regions (Table 1). These 26 genes encode 21 different transcription factors. The knockdown by RNAi of these 26 candidates revealed three transcription factors that were direct targets: Hairy, Hr4, and Vrille, as well as ECR, a positive control, that caused a reversal of the phenotype, that is, no projections in the presence of 20-HE (Table 2). These results indicate that these key transcription factors are directly bound by ECR and USP and are playing a direct role in cellular differentiation.
Partial list of 20-HE-responsive genes
Total numbers of round and elongated cells seen after knockdown of direct targets of ECR/USP and controls from four biological replicate experiments
Since vrille (vri) was recently implicated as a 20-HE-induced, ECR-dependent gene (Beckstead et al. 2005), and our results, which show that it is a direct target of ECR/USP, we further investigated the role of vrille in 20-HE-controlled cellular differentiation. The Vrille protein is closely related to bZIP transcription factors involved in growth or cell death. Loss of function and overexpression analyses reveals several cellular phenotypes. vri clones in the adult cuticle contain smaller cells with atrophic bristles. Also, overexpression of Vrille is antiproliferative in embryonic dorsal epidermis, induces apoptosis in imaginal discs, and gives rise to smaller cells and organs in salivary glands (Szuplewski et al. 2003). We used two vri mutant alleles, vri1 and vri7, which represent antimorph alleles: vri1 involves a stop codon at position 924 upstream of the bZIP domain, and vri7 contains a Pf insertion that maps to position 838 within the first intron (George and Terracol 1997). We observe a reduction in cellular proliferation phenotype in vri1/vri7 mutant flies at the third larval instar stage. vri1/vri7 flies show pupal lethality and do not develop beyond the prepupal stage (Supplemental Fig. 4). The two major types of cells in the larval midgut, larval epidermal cells and midgut imaginal islands, respond in opposite ways to ecdysone. The larval epidermal cells initiate the process of programmed cell death, while the imaginal cells proliferate and form the adult midgut (Li and White 2003). We also find that large cell clusters of imaginal islands in the midgut are absent in the vrille mutants, which could be because they either failed to form or failed to proliferate (Supplemental Fig. 4). This phenotype is consistent with the hypothesis that vrille is required for proliferation at the late third instar larval stage, where 20-HE levels are highest.
Discussion
The biological effects of 20-HE activity in Drosophila unfold from a complex chain of events beginning with the secretion of the steroid, its binding to a nuclear receptor, and the subsequent regulation of target genes in a time-and-space-specific manner.
Our results identify direct targets of the Ecdysone receptor complex across the entire genome in a specific cell type. However, many genes involved in metamorphic processes such as salivary gland death and imaginal disc development are identified, although the Kc167 cells we used represent neither of these cell types. This result indicates that ECR/USP binds to a large proportion of the genomic targets in this cell type, but the cells are not competent to respond. The identity of ECR/USP targets is also revealing. Traditional representation of the ecdysone regulatory cascade involves a small set of regulatory molecules (most of which are transcription factors) serving as direct targets, which in turn amplify the signal by regulating a large number of downstream effectors. Our results indeed indicate that ECR/USP transduces the ecdysone signal by directly using other regulators such as transcription factors to pass on the signal. However, ECR/USP appears to also regulate a large number of effector genes such as those encoding enzymes and cell cycle machinery.
We observe that 20-HE-responsive direct targets are more likely to be up-regulated and to contain more ECR/USP binding regions associated with it at early time points than down-regulated target genes. The 19 transcription factors that are 20-HE responsive or direct targets of ECR/USP are all up-regulated within 3 h after 20-HE addition. The presence of multiple binding sites for the ECR/USP might allow for the complex spatial-temporal expression patterns of such target genes, as different cis-regulatory modules (CRMs) for the same gene may be activated in different cell types. Several of the ECR/USP transcription factor targets such as EIP75B are known to encode multiple transcripts in different tissues. This hypothesis to explain the presence of multiple ECR/USP-binding regions in these regulatory factor-encoding genes remains to be proven, but it is consistent with other studies that have shown that expression of such genes is likely to be controlled by multiple cis-regulatory elements with spatial and temporal specificity (Davidson et al. 2001).
The combination of genome-wide expression data with genome-wide location analysis constitutes a powerful tool not only in verifying predicted interactions, but also in elucidating transcriptional networks involved in pathways controlling cellular morphology and differentiation. Since the ecdysone pathway is critical for fly development, the recruitment of transcription factors with pleiotropic effects reflects an important evolutionary aspect of the genetic architecture of the ecdysone network. We identify two targets that are specifically involved in 20-HE-regulated cellular differentiation—hairy and vrille—that were previously shown to be primary response genes (Beckstead et al. 2005). A third target, Hr4, is required for the cellular phenotypes induced by ecdysone and has previously been shown to be a primary response gene (King-Jones and Thummel 2005). A binding-site study on targets of Hairy identified 59 putative targets in Kc167 cells and suggests roles for Hairy in cell cycle, cell growth, and morphogenesis (Bianchi-Frias et al. 2004). Specifically, three targets involved in cell proliferation were identified (20-HE-inducible gene ImpL2, string, and Idgf2). Since dividing cells cannot undergo cell shape changes simultaneously, it is plausible that Hairy could be responsible for regulating cell cycle genes and transiently pausing the cell cycle. This could explain why in its absence, this “check” is removed and cells cannot undergo shape changes, and therefore lose their projections. The Hr4 gene is expressed specifically at onset of metamorphosis and acts as both a repressor of the early 20-HE-induced regulatory genes and an inducer of the beta-FTZ-F1 mid-prepupal competence factor (King-Jones and Thummel 2005). Hr4 might be acting in a manner similar to Hairy, since its absence would remove the repression of early genes, which themselves might be regulating cellular differentiation. A surprising discovery was that Vrille, a bZIP transcription factor whose expression is controlled by the biological clock of adult flies (Blau and Young 1999) is also required for cellular responses to 20-HE both in cell culture and in vivo. In summary, the systematic identification of targets of the ECR-C reveals the architecture at the top of the 20-HE-regulated hierarchical network and sets a foundation for relating this multilayered transcriptional network to the phenotypes it controls.
Methods
Cell culture and transfection
Drosophila Kc167 cells are kindly provided by Dr. Lucy Cherbas (Indiana University, Bloomington) and Dr. Amy Kiger (Univ. of California, San Diego). Drosophila L57-3-11 cells are also kindly provided by Dr. Lucy Cherbas as ECRB1 knock-out Kc cells, which are ecdysone resistant. Kc167 cells from Dr. Cherbas and L57-3-11 cells were grown in M3 medium (Sigma) supplemented with yeast extract and bacto-peptone containing 5% fetal bovine serum (Invitrogen) at 24°C (Cherbas et al. 1994). Kc167 cells from Dr. Kiger were grown largely as previously described in Schneider's Drosophila medium (Invitrogen) containing 10% fetal bovine serum (Invitrogen) at 24°C (Bashirullah et al. 2003). Electroporation with the Gene Pulser II system (Bio-Rad) was used to transfect cells for transient expression of proteins in cells mainly as previously described (van Steensel and Henikoff 2000). Cells were seeded at 0.5 × 106 cells/mL, cultured for 48 h at 24°C, and collected by centrifugation at 2000 rpm for 3 min. Cells were washed twice with serum-free medium, resuspended using 0.8 mL of serum-free medium, mixed with 20 μg of plasmid, and electroporated using settings at 960 μF and 250 V. Cells were transferred to fresh medium for transient expression of gene. The proteins produced were detected by an immunofluorescence experiment using anti-myc antibody (9E10, Santa Cruz Biotechnology), anti-ECRB1 antibody (DDA2.7), and anti-USP antibody (AB11), respectively.
DamID and microarray
DamID experiments were carried out with Kc167 cells from Dr. Cherbas. The DamID technique was carried out by fusing DNA adenine methyltransferase (Dam) to full-length coding sequences for ECRB1 and USP (two fusion constructs for each gene of interest with Dam on 5′ terminal and 3′ terminal of the fusion, respectively) and expressed as a transgene (van Steensel et al. 2001; Sun et al. 2003). The fusion protein binds DNA sequences normally bound by the endogenous protein and locally methylates genomic DNA within a 3–10-kb range. Cells transfected with a fusion construct were recovered for 24 h, genomic DNA was isolated from cells and digested with a restriction enzyme (DpnI) that only cuts the methylated DNA and was ligated to an adapter for linearized amplification as previously reported (van Steensel et al. 2001; Sun et al. 2003). After amplification, the amplified methylated DNA near the binding site is digested into small fragments, labeled, and hybridized to high-density oligonucleotide microarray essentially as previously described with modification on probe labeling (Stolc et al. 2004). The probe was labeled using a BD Atlas PowerScript Fluorescent Labeling Kit (BD Biosciences). Instead of the BD PowerScript reverse transcriptase and 5× first-strand buffer, Klenow fragment and 2.5× random primers solution from BioPrime DNA Labeling System (Invitrogen) was used to incorporate aminoallyl-dUTP into the probe with DNA templates. Three replicates were carried out for ECRB1:Dam fusion protein, two for ECRB1 on the 5′side, and one for ECRB1 on the 3′ side of corresponding fusion protein. Similar replicates were executed for USP:Dam fusion protein. The control in these experiments is transfection with Dam alone. We used high-density oligonucleotide microarrays with probes for each predicted exon and noncoding probe tiled throughout the predicted intronic and intergenic regions of the genome (Stolc et al. 2004).
Identification of ECR/USP-binding regions
Fisher's combined probability test (Fisher 1950b) was used to identify significant ECR/USP-binding regions with length 4 kb, and overlapping regions were merged together.
Based on Fisher's method, a χ2 test statistic was calculated based on logarithmic transformations of the P-values (Limma t-test) of each noncoding probe within a given 4-kb region:
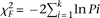
Here, k is the total number of probes within a given 4-kb region and Pi is the Limma P-value for each probe. If all the null hypothesis in Limma t-test were true, the χF2 will follow a χ2 distribution with 2 k degrees of freedom.
We first estimated the false positive rate of enriched probe identification. The intensities for all probes on the tiling arrays were randomly shuffled, and the same Limma analysis was then performed. A total of 1000 runs for each data set (ECR or USP) were tested. We found that the mean of “significant probe” number for ECR is 12.72 and the mean of “significant probe” number for USP is 10.97. We estimated that, at the probe level, the false positive rates for ECR and USP are 0.0099% and 0.0034%, respectively.
At the window level, we tested a total of 62,086 4-kb windows with 2 kb overlapped between two neighboring windows. The overlapped significantly bound windows were finally merged. If N is the number of significant enriched probes, the maximum number of significant 4-kb windows will be 2N, where each probe could represent a significant 4-kb window and no probes are close to each other. There are 593 significant ECR windows (P < 0.0001) and 1450 significant USP windows (P < 0.0001). Thus, we estimated the false positive rate for significant 4-kb regions is around 2*12.72/593 = 4.2% for ECR and 2*10.97/1450 = 1.5% for USP.
Time course experiment
For the time course study, Kc167 cells provided by Dr. Cherbas were used. Cells were plated at 0.5 × 106/mL and grown at 23.5°C for 48 h They were treated with 20-HE (Sigma) at a final concentration of 5 × 10−7 M. The reference sample was total RNA isolated at 0 h after treatment. The experimental samples were total RNA isolated from cells treated for 1, 3, 6, 12, 24, and 48 h. Total mRNA was extracted with OligodT (Ambion) and reverse transcribed in the presence of oligodeothymidine and random hexamers and labeled to the in-house cDNA microarray (five replicates) (White et al. 1999). We used the Limma algorithm that employs an empirical Bayesian approach through use of a moderated t-test statistic (P < 0.05 and 1.5-fold cutoff) to identify probes. We calculated the false positive rate for the gene expression experiments by randomly shuffling M-values (=log2[probe intensity for experiments/probe intensity for controls]) for the probes on the array. For example, we estimated the false positive rate for 48 h is 1.8% (12.73/689 where 12.73 is the mean of significant probe numbers and 689 is the observed number of probes).
RNA interference assay
dsRNA production and conditions for RNAi in Kc cells were carried out as in Clemens et al. (2000). Individual DNA fragments ∼500 bp in length containing coding sequences for the proteins to be “knocked out” were amplified by using PCR, and the PCR products were purified by using the Qiaquick PCR Purification Kit or gel purification kit (Qiagen Sciences). dsRNA was obtained by in vitro transcription as previously described (Li and White 2003). RNAi experiments were carried out in 6-well plates. In each well, 1 mL of Drosophila culture cells diluted to a final concentration of 1 × 106 cells/mL in Schnieder's serum-free medium (Gibco #11720-034) was added. The cells were incubated for 30 min at 25°C, followed by addition of 2 mL of Schneider's medium/10% FBS. 20-HE was added after 24 h at a concentration of 5 × 10−7 M. The projection phenotype was scored after 2 d. A complete loss of projections was scored as a successful phenotype. Primer sequences used to generate specific dsRNAs (CHI set) are shown in Supplemental Table 8. For further validation of target genes that showed a phenotype and negative hit Eip74EF, we used an additional independent set of dsRNAs (BKN set). The second set of dsRNAs was designed using a probe-finding algorithm, and the sequences can be retrieved by using the BKN ID associated with the genes (http://www.dkfz.de/signaling2/rnai/ernai_probes.php). The BKN IDs for the second set of dsRNA probes are EcR (BKN27721), vrille (BKN28168), Hr4 (BKN30718), as well as the gene Eip74 (BKN29288), which did not show a phenotype. For hairy, we used an additional dsRNA from the DRSC library with RNAi probeID HFA1135 (Supplemental Table 9).
Motif analysis
To identify putative ECR/USP-binding sites, a genome-wide scan was carried out with Patser (Hertz and Stormo 1999) using a positional weight matrix based on a training set of 10 known ECR/USP sites. Putative sites were filtered using the cutoff Patser score ≥8.0 (Hertz and Stormo 1999). The following sequences are known binding sites for ECR_USP (Morgan et al. 1986; Scholnick et al. 1986; Riddihough and Pelham 1987; Cherbas et al. 1991; Antoniewski et al. 1993; Vogtli et al. 1998; Petersen et al. 2003): 5′-GGTTGAATGAATT-3′, 5′-CGGTCATTGGCCG-3′, 5′-AGTTCGAGGCACC-3′, 5′-GTTTCAGTGAAAG-3′, 5′-GGGTCAATAGCCG-3′, 5′-CGTTGAATCAATG-3′, 5′-ATTTCTTTGAATT-3′, 5′-GGTTCAATGCACT-3′, 5′-GGGTTCATGCACT-3′, and 5′-AGTTCAGCGGCTG-3′.
Acknowledgments
We thank Bas Van Steensel for critical reading of the manuscript, Guoneng Zhong for help with programs, Kerstin Spirohn for technical help with RNAi experiments, and R. Terracol for the donation of vrille mutant alleles.
Footnotes
-
↵6 Present addresses: Department of Human Genetics, Massachusetts General Hospital, Boston, MA 02114, USA;
-
↵7 Institute of Developmental Biology and Molecular Medicine, Fudan University, Shanghai 200433, China.
-
↵8 Corresponding author.
E-mail kpwhite{at}uchicago.edu; fax (773) 834-2877.
-
[Supplemental material is available online at www.genome.org. The DNA-binding site data is available using Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) accession no. GSE9156, and the time course data is available using GSE11625.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.081349.108.
-
- Received May 27, 2008.
- Accepted February 10, 2009.
- Copyright © 2009 by Cold Spring Harbor Laboratory Press








